
Breast Cancer Clinical Trial of Chemotherapy and Trastuzumab: Potential Tool to Identify Cardiac Modifying Variants of Dilated Cardiomyopathy

Abstract
:1. Introduction
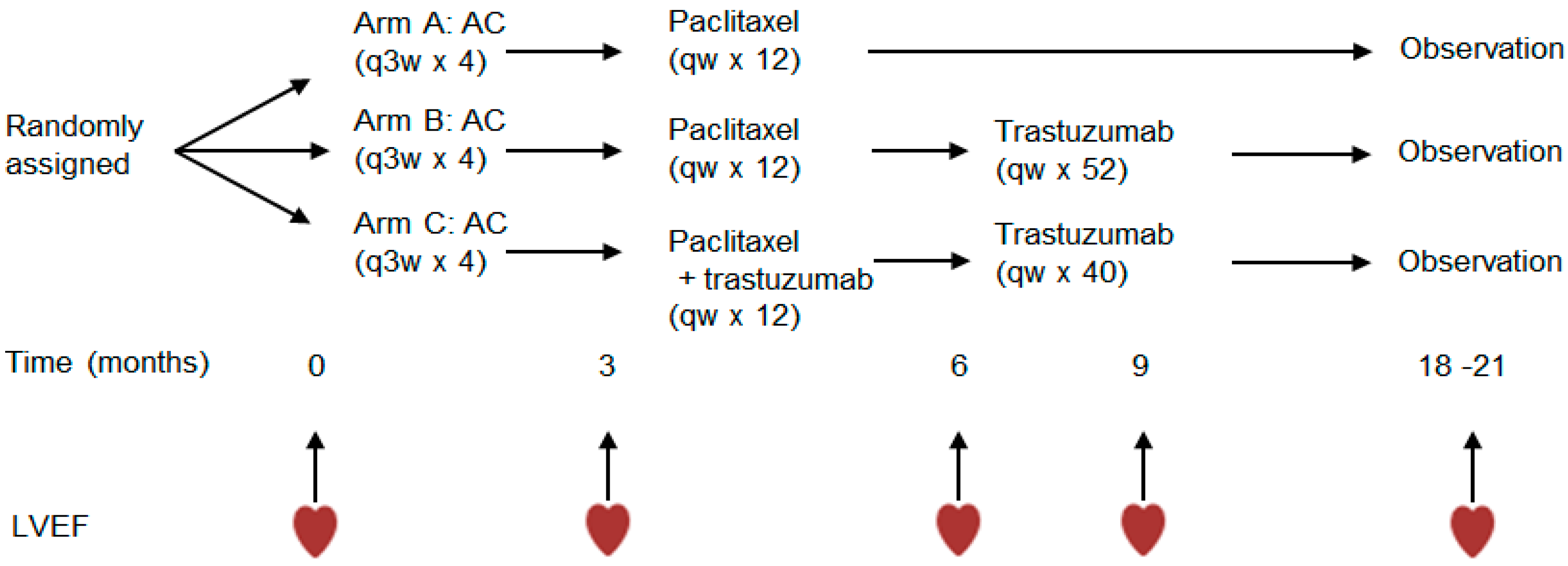
2. Materials and Methods
3. Results
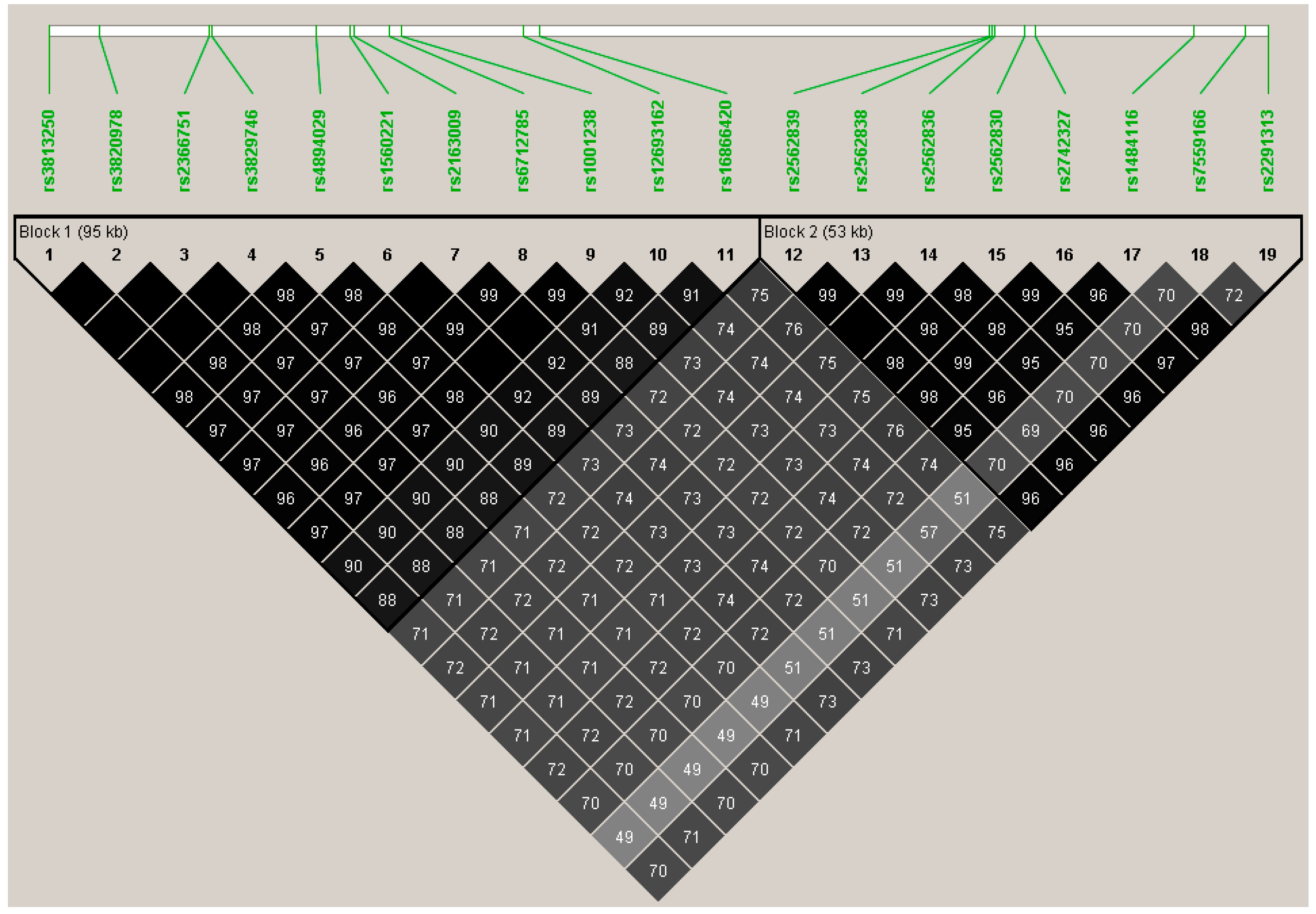
3.1. Single Marker Analyses of Common Variants
3.2. Gene-Based Analyses
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Norton, N.; Robertson, P.D.; Rieder, M.J.; Zuchner, S.; Rampersaud, E.; Martin, E.; Li, D.; Nickerson, D.A.; Hershberger, R.E. Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ. Cardiovasc. Genet. 2012, 5, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Norton, N.; Morales, A.; Li, D.; Siegfried, J.D.; Gonzalez-Quintana, J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Morales, A.; Hershberger, R.E. The Rationale and Timing of Molecular Genetic Testing for Dilated Cardiomyopathy. Can. J. Cardiol. 2015, 31, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Zuchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Crone, S.A.; Zhao, Y.Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 2002, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Romond, E.H.; Suman, V.J.; Jeong, J.H.; Davidson, N.E.; Geyer, C.E., Jr.; Martino, S.; Mamounas, E.P.; Kaufman, P.A.; Wolmark, N. Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2-positive breast cancer: Joint analysis of data from NCCTG N9831 and NSABP B-31. J. Clin. Oncol. 2011, 29, 3366–3373. [Google Scholar] [CrossRef] [PubMed]
- Necela, B.M.; Axenfeld, B.C.; Serie, D.J.; Kachergus, J.M.; Perez, E.A.; Thompson, E.A.; Norton, N. The antineoplastic drug, trastuzumab, dysregulates metabolism in iPSC-derived cardiomyocytes. Clin. Transl. Med. 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Plana, J.C.; Galderisi, M.; Barac, A.; Ewer, M.S.; Ky, B.; Scherrer-Crosbie, M.; Ganame, J.; Sebag, I.A.; Agler, D.A.; Badano, L.P.; et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: A report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 1063–1093. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M.P.; van Spaendonck-Zwarts, K.Y.; van Veldhuisen, D.J.; Gietema, J.A.; Postma, A.; van Tintelen, J.P. Familial dilated cardiomyopathy: Another risk factor for anthracycline-induced cardiotoxicity? Eur. J. Heart Fail. 2010, 12, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Wasielewski, M.; van Spaendonck-Zwarts, K.Y.; Westerink, N.D.; Jongbloed, J.D.; Postma, A.; Gietema, J.A.; van Tintelen, J.P.; van den Berg, M.P. Potential genetic predisposition for anthracycline-associated cardiomyopathy in families with dilated cardiomyopathy. Open Heart 2014, 1, e000116. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Romond, E.H.; Suman, V.J.; Jeong, J.H.; Sledge, G.; Geyer, C.E., Jr.; Martino, S.; Rastogi, P.; Gralow, J.; Swain, S.M.; et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: Planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J. Clin. Oncol. 2014, 32, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Advani, P.P.; Ballman, K.V.; Dockter, T.J.; Colon-Otero, G.; Perez, E.A. Long-Term Cardiac Safety Analysis of NCCTG N9831 (Alliance) Adjuvant Trastuzumab Trial. J. Clin. Oncol. 2016, 34, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.; Montgiraud, C.; Munster, A.B.; Copeland, O.; Choi, O.; Dos Remedios, C.; Messer, A.E.; Ehler, E.; Knoll, R. OBSCN Mutations Associated with Dilated Cardiomyopathy and Haploinsufficiency. PLoS ONE 2015, 10, e0138568. [Google Scholar] [CrossRef] [PubMed]
- Khouri, M.G.; Douglas, P.S.; Mackey, J.R.; Martin, M.; Scott, J.M.; Scherrer-Crosbie, M.; Jones, L.W. Cancer therapy-induced cardiac toxicity in early breast cancer: Addressing the unresolved issues. Circulation 2012, 126, 2749–2763. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Koehler, M.; Byrne, J.; Preston, A.J.; Rappold, E.; Ewer, M.S. Cardiac safety of lapatinib: Pooled analysis of 3689 patients enrolled in clinical trials. Mayo Clin. Proc. 2008, 83, 679–686. [Google Scholar] [CrossRef]
- Wu, M.C.; Lee, S.; Cai, T.; Li, Y.; Boehnke, M.; Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011, 89, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Emond, M.J.; Bamshad, M.J.; Barnes, K.C.; Rieder, M.J.; Nickerson, D.A.; Christiani, D.C.; Wurfel, M.M.; Lin, X. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012, 91, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Lee, S.; Makarov, V.; Buxbaum, J.D.; Lin, X. Sequence kernel association tests for the combined effect of rare and common variants. Am. J. Hum. Genet. 2013, 92, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Matsumoto, Y.; Okazaki, O.; Hayashi, T.; Takahashi, M.; Inagaki, N.; Hinohara, K.; Ashizawa, N.; Yano, K.; Kimura, A. Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2007, 362, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Li, D.; Rampersaud, E.; Morales, A.; Martin, E.R.; Zuchner, S.; Guo, S.; Gonzalez, M.; Hedges, D.J.; Robertson, P.D.; et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 144–153. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name |
|---|---|
| ABCC9 | ATP-binding cassette, sub-family C, member 9 |
| ACTC1 | Actin, Alpha, Cardiac Muscle 1 |
| ACTN2 | Actinin Alpha 2 |
| AKAP9 | A-Kinase Anchoring Protein 9 |
| ANK2 | Ankyrin 2 |
| ANKRD1 | Ankyrin Repeat Domain 1 |
| BAG3 | BCL2 Associated Athanogene 3 |
| CACNA1C | Calcium Voltage-Gated Channel Subunit Alpha 1 C |
| CAV3 | Caveolin 3 |
| CRYAB | Crystallin Alpha B |
| CSRP3 | Cysteine And Glycine Rich Protein 3 |
| DES | Desmin |
| DMD | Dystrophin |
| DSC2 | Desmocollin 2 |
| DSG2 | Desmoglein 2 |
| DSP | Desmoplakin |
| ELN | Elastin |
| EMD | Emerin |
| EYA4 | EYA Transcriptional Coactivator And Phosphatase 4 |
| FXN | Frataxin |
| GATA4 | GATA Binding Protein 4 |
| GLA | Galactosidase Alpha |
| ILK | Integrin Linked Kinase |
| JAG1 | Jagged 1 |
| KCNE1 | Potassium Voltage-Gated Channel Subfamily E Regulatory Subunit 1 |
| KCNE2 | Potassium Voltage-Gated Channel Subfamily E Regulatory Subunit 2 |
| KCNH2 | Potassium Voltage-Gated Channel Subfamily H Member 2 |
| KCNJ2 | Potassium Voltage-Gated Channel Subfamily J Member 2 |
| KCNJ5 | Potassium Voltage-Gated Channel Subfamily J Member 5 |
| KCNQ1 | Potassium Voltage-Gated Channel Subfamily Q Member 1 |
| LAMA4 | Laminin Subunit Alpha 4 |
| LAMP3 | Lysosomal Associated Membrane Protein 3 |
| LDB3 | LIM Domain Binding 3 |
| LMNA | Lamin A/C |
| MURC | Muscle Related Coiled-Coil Protein |
| MYBP3 | Myosin binding protein 3 |
| MYH6 | Myosin Heavy Chain 6 |
| MYH7 | Myosin Heavy Chain 7 |
| MYL2 | Myosin Light Chain 2 |
| MYL3 | Myosin Light Chain 3 |
| MYLK2 | Myosin Light Chain Kinase 2 |
| MYOZ2 | Myozenin 2 |
| MYPN | Myopalladin |
| NEBL | Nebulette |
| NEXN | Nexilin F-Actin Binding Protein |
| NKX2-5 | NK2 Homeobox 5 |
| NOTCH1 | Notch 1 |
| NOTCH2 | Notch 2 |
| OBSCN | Obscurin |
| PDLIM3 | PDZ And LIM Domain 3 |
| PKP2 | Plakophilin 2 |
| PLN | Phospholamban |
| PRKAG2 | Protein Kinase AMP-Activated Non-Catalytic Subunit Gamma 2 |
| PSEN1 | Presenilin 1 |
| PSEN2 | Presenilin 2 |
| RAF1 | Raf-1 Proto-Oncogene, Serine/Threonine Kinase |
| RBM20 | RNA Binding Motif Protein 20 |
| RYR2 | Ryanodine Receptor 2 |
| SCN4B | Sodium Voltage-Gated Channel Beta Subunit 4 |
| SCN5A | Sodium Voltage-Gated Channel Alpha Subunit 5 |
| SGCD | Sarcoglycan Delta |
| SNTA1 | Syntrophin Alpha 1 |
| TAZ | Tafazzin |
| TCAP | Titin-Cap |
| TMEM43 | Transmembrane Protein 43 |
| TMPO | Thymopoietin |
| TNNC1 | Troponin C1, Slow Skeletal And Cardiac Type |
| TNNI3 | Troponin I3, Cardiac Type |
| TNNT2 | Troponin T2, Cardiac Type |
| TPM1 | Tropomyosin 1 (Alpha) |
| TTN | Titin |
| VCL | Vinculin |
| Gene | Size (Kb) | Number SNPs | SNP ID | SNP Effect | Beta (95% CI) | MAF | p-Value |
|---|---|---|---|---|---|---|---|
| VCL | 122 | 105 | rs12250729 | intron | 0.84 (0.10 to 1.57) | 0.25 | 0.0259 |
| rs76974852 | intron | −3.34 (−5.52 to −1.15) | 0.02 | 0.0029 | |||
| rs111748583 | downstream | −3.66 (−6.22 to −1.11) | 0.02 | 0.0051 | |||
| DMD | 2220 | 432 | rs140820221 | intron | −1.94 (−3.37 to −0.52) | 0.05 | 0.0078 |
| rs1795571 | intron | −0.86 (−1.70 to −0.03) | 0.13 | 0.0438 | |||
| rs331317 | intron | 0.85 (0.21 to 1.49) | 0.47 | 0.0090 | |||
| rs72626080 | intron | 1.87 (0.76 to 2.97) | 0.09 | 0.0010 | |||
| rs2050074 | intron | 1.28 (0.34 to 2.22) | 0.13 | 0.0077 | |||
| rs2050076 | intron | 1.32 (0.37 to 2.27) | 0.13 | 0.0065 | |||
| rs12559939 | intron | 1.48 (0.65 to 2.30) | 0.19 | 0.0005 | |||
| rs141927233 | intron | 1.45 (0.63 to 2.28) | 0.19 | 0.0006 | |||
| rs73623943 | intron | 1.18 (0.44 to 1.93) | 0.24 | 0.0020 | |||
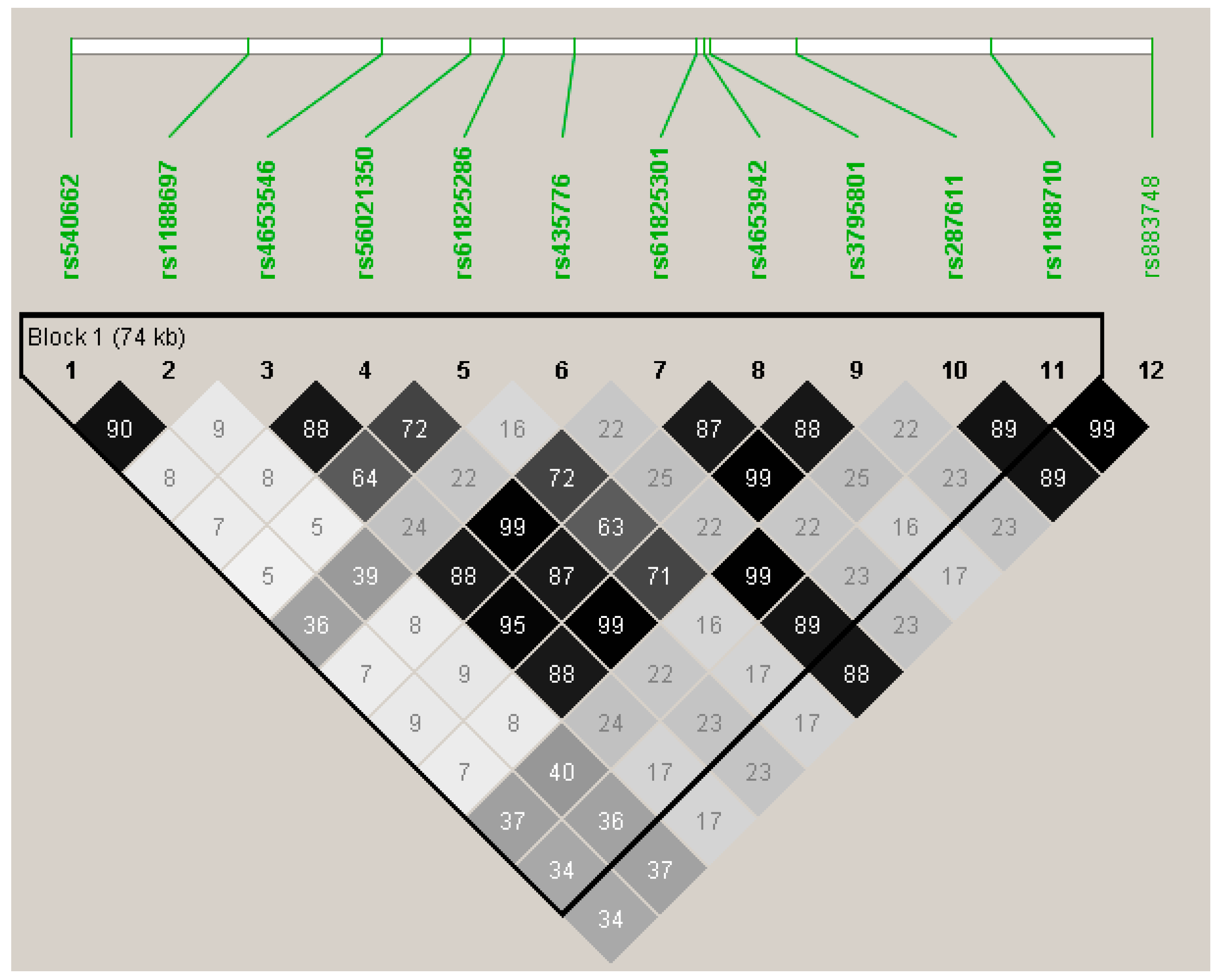
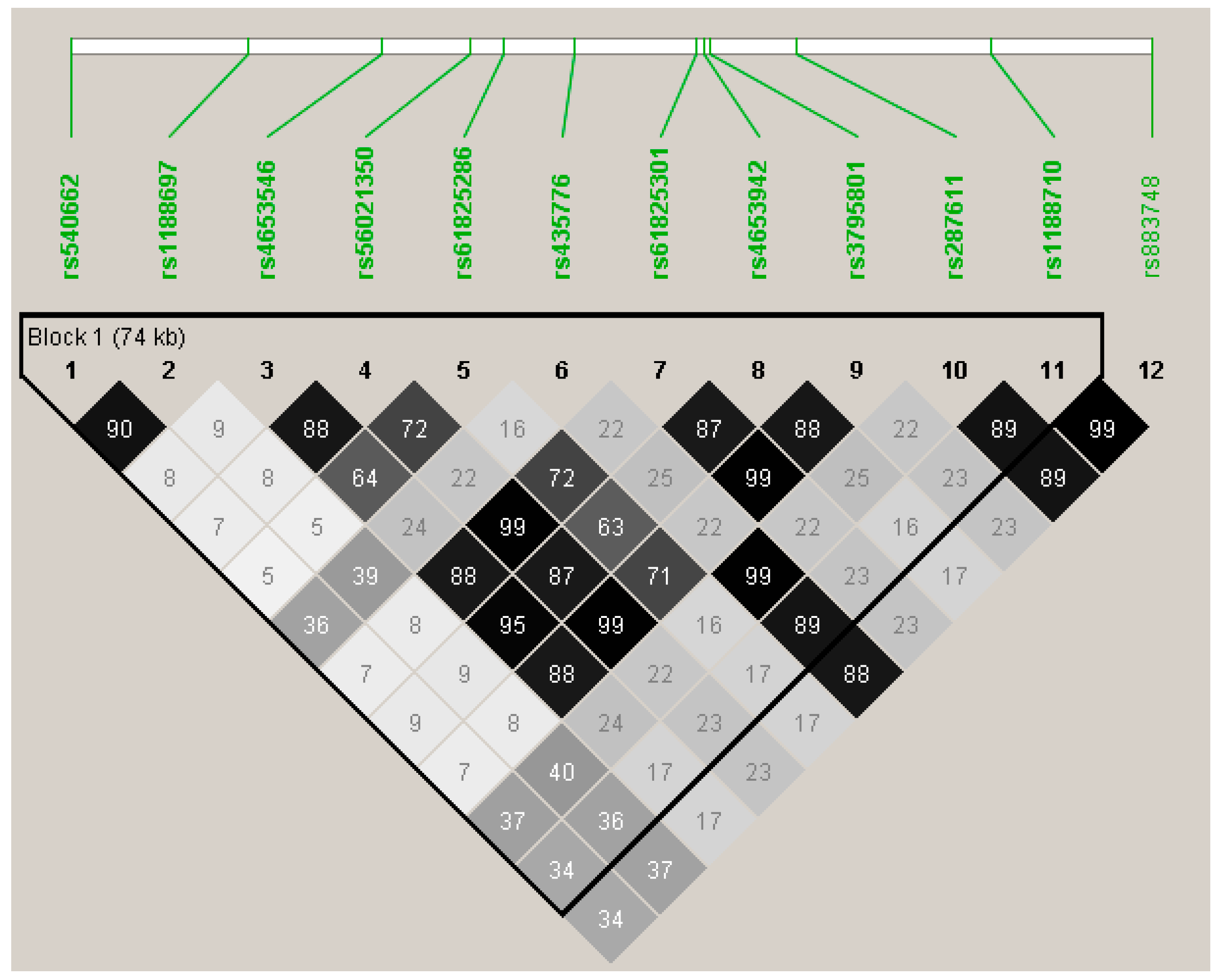
| OBSCN | 171 | 55 | rs540662 | intron | 1.05 (0.34 to 1.76) | 0.29 | 0.0038 |
| rs1188697 | Val2720Met | 1.02 (0.32 to 1.72) | 0.31 | 0.0045 | |||
| rs4653546 | intron | −1.20 (−2.03 to −0.38) | 0.19 | 0.0044 | |||
| rs56021350 | Thr4399Met | −1.42 (−2.26 to −0.59) | 0.18 | 0.0009 | |||
| rs61825286 | intron | −1.39 (−2.35 to −0.44) | 0.14 | 0.0044 | |||
| rs435776 | Gly4039Arg | −1.09 (−1.76 to −0.43) | 0.50 | 0.0014 | |||
| rs61825301 | His4489Gln | −1.41 (−2.25 to −0.58) | 0.18 | 0.0010 | |||
| rs4653942 | Arg4534His | −1.43 (−2.26 to −0.61) | 0.20 | 0.0007 | |||
| rs3795801 | Gly4666Ser | −1.39 (−2.23 to −0.56) | 0.18 | 0.0011 | |||
| rs287611 | intron | −1.10 (−1.76 to −0.43) | 0.49 | 0.0013 | |||
| rs1188710 | Gln5891Glu | −1.13 (−1.79 to −0.47) | 0.47 | 0.0008 | |||
| rs883748 | intron | −1.16 (-1.82 to −0.50) | 0.47 | 0.0006 | |||
| RYR2 | 792 | 273 | rs80107454 | intron | −2.89 (−4.59 to −1.19) | 0.04 | 0.0009 |
| rs2253083 | intron | 2.10 (0.55 to 3.65) | 0.04 | 0.0081 | |||
| TPM1 | 29 | 81 | rs5813188 | intron | 1.42 (0.49 to 2.34) | 0.13 | 0.0027 |
| rs8026502 | intron | 1.42 (0.49 to 2.34) | 0.13 | 0.0027 | |||
| rs79854225 | intron | 1.26 (0.28 to 2.24) | 0.12 | 0.0121 | |||
| rs57645645 | intron | 1.42 (0.49 to 2.34) | 0.13 | 0.0027 | |||
| rs73431508 | intron | 1.31 (0.38 to 2.23) | 0.14 | 0.0058 | |||
| rs12441488 | intron | 1.42 (0.49 to 2.34) | 0.13 | 0.0027 | |||
| KCNQ1 | 404 | 881 | rs80056995 | intron | −1.94 (−3.41 to −0.46) | 0.05 | 0.0102 |
| rs16928363 | intron | −1.84 (−3.30 to −0.37) | 0.05 | 0.0142 | |||
| rs2237868 | intron | −1.84 (−3.30 to −0.37) | 0.05 | 0.0142 | |||
| rs74392867 | intron | −1.92 (−3.39 to −0.45) | 0.05 | 0.0106 | |||
| rs79295543 | intron | −1.94 (−3.41 to −0.46) | 0.05 | 0.0102 | |||
| rs77059665 | intron | −2.29 (−3.90 to −0.69) | 0.04 | 0.0051 | |||
| rs28730663 | intron | −2.71 (−4.62 to −0.80) | 0.03 | 0.0056 | |||
| rs35237966 | intron | −0.94 (−1.61 to −0.26) | 0.39 | 0.0065 | |||
| rs72844252 | intron | −1.57 (−2.71 to −0.42) | 0.09 | 0.0074 | |||
| rs231880 | intron | −0.70 (−1.39 to −0.01) | 0.35 | 0.0466 | |||
| rs71476688 | intron | 2.59 (0.87 to 4.32) | 0.03 | 0.0033 | |||
| rs34861825 | intron | 1.38 (0.40 to 2.36) | 0.16 | 0.0061 | |||
| rs12419030 | intron | 0.89 (0.17 to 1.61) | 0.31 | 0.0160 | |||
| rs12419347 | intron | 0.91 (0.19 to 1.63) | 0.31 | 0.0139 | |||
| JAG1 | 36 | 18 | rs3748480 | intron | 1.52 (0.51 to 2.53) | 0.12 | 0.0033 |
| SGCD | 1060 | 1851 | rs6860238 | intron | 2.24 (0.71 to 3.78) | 0.05 | 0.0043 |
| SCN5A | 98 | 243 | rs11129796 | intron | −1.44 (−2.67 to −0.20) | 0.09 | 0.0228 |
| rs9832586 | intron | −1.28 (−2.35 to −0.21) | 0.10 | 0.0190 | |||
| rs7430407 | synonymous | −1.43 (−2.44 to −0.41) | 0.11 | 0.0060 | |||
| rs6790619 | intron | −1.44 (−2.70 to −0.17) | 0.07 | 0.0264 | |||
| rs7645173 | intron | −1.52 (−2.81 to −0.24) | 0.07 | 0.0206 | |||
| rs9311190 | intron | −1.56 (−2.85 to −0.27) | 0.07 | 0.0180 | |||
| rs11711097 | intron | −0.85 (−1.64 to −0.07) | 0.22 | 0.0340 | |||
| rs7432532 | intron | −1.45 (−2.47 to −0.43) | 0.11 | 0.0053 | |||
| rs7426433 | intron | −1.50 (−2.52 to −0.48) | 0.11 | 0.0040 | |||
| rs7433889 | intron | −1.13 (−2.10 to −0.17) | 0.13 | 0.0213 | |||
| rs6599214 | intron | −1.21 (−2.17 to −0.25) | 0.13 | 0.0142 | |||
| rs6599215 | intron | −1.27 (−2.23 to −0.31) | 0.13 | 0.0099 | |||
| rs6599216 | intron | −1.21 (−2.17 to −0.26) | 0.13 | 0.0126 | |||
| rs6599217 | intron | −1.20 (−2.15 to −0.25) | 0.13 | 0.0142 | |||
| rs6599218 | intron | −1.20 (−2.15 to −0.24) | 0.13 | 0.0142 | |||
| rs7613045 | intron | −1.36 (−2.33 to −0.40) | 0.12 | 0.0059 | |||
| rs63200660 | intron | −1.19 (−2.14 to −0.25) | 0.13 | 0.0137 | |||
| rs6599221 | intron | −1.30 (−2.59 to −0.01) | 0.07 | 0.0478 | |||
| rs7627488 | downstream | −2.99 (−5.55 to −0.43) | 0.02 | 0.0224 | |||
| rs7636280 | downstream | −2.96 (−5.68 to −0.23) | 0.01 | 0.0337 | |||
| RBM20 | 195 | 57 | rs7069694 | intron | −0.79 (−1.45 to −0.13) | 0.41 | 0.0198 |
| rs2181407 | intron | −0.76 (−1.47 to −0.06) | 0.28 | 0.0342 | |||
| rs17831429 | intron | −2.68 (−4.58 to −0.78) | 0.03 | 0.0058 | |||
| SCN4B | 20 | 8 | rs955917 | intron | 1.07 (0.22 to 1.93) | 0.17 | 0.0140 |
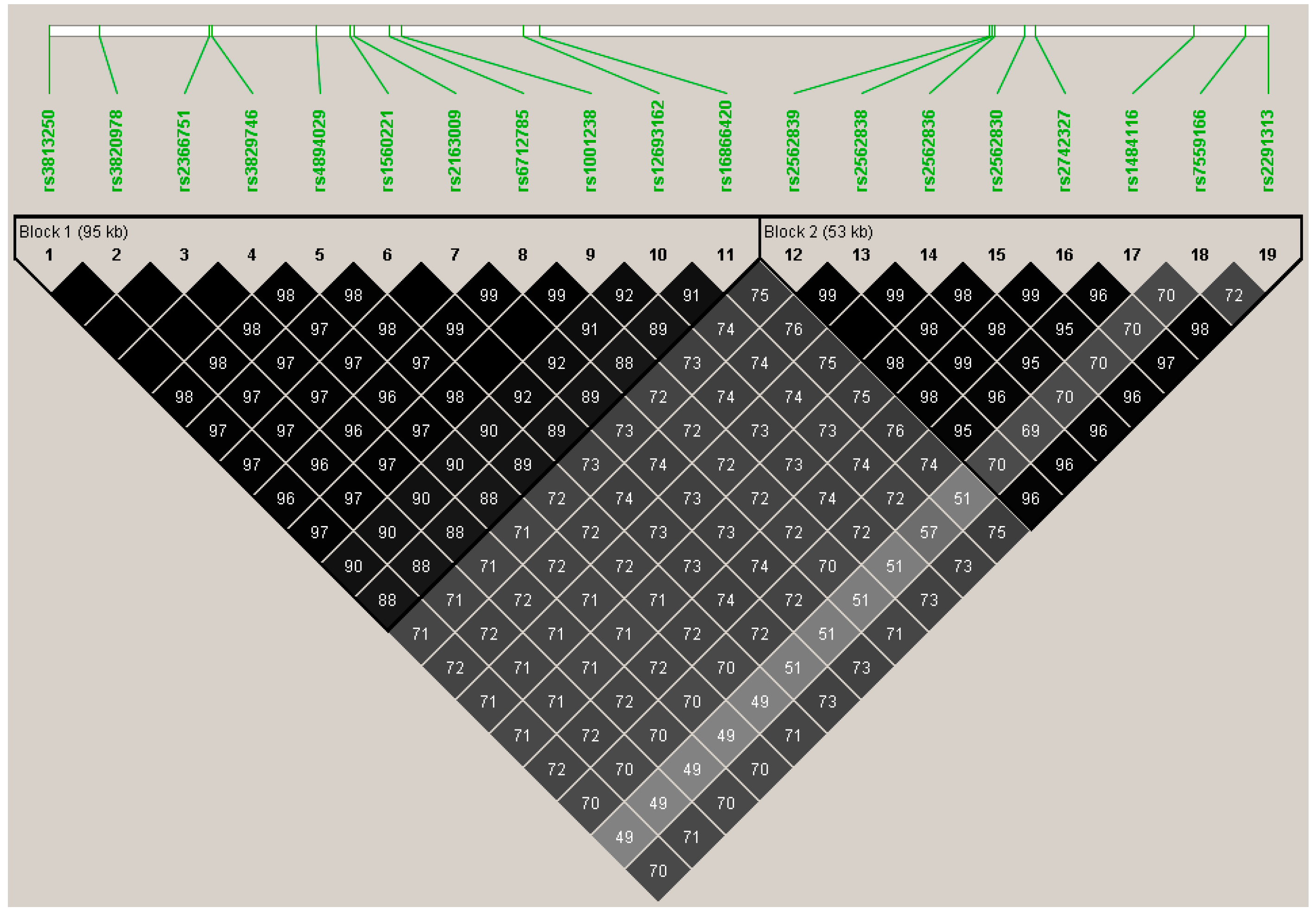
| TTN | 281 | 275 | rs3813250 | synonymous | 0.85 (0.06 to 1.63) | 0.22 | 0.0349 |
| rs3820978 | downstream | 0.79 (0.01 to 1.58) | 0.22 | 0.0486 | |||
| rs2366751 | synonymous | 0.81 (0.03 to 1.60) | 0.22 | 0.0432 | |||
| rs3829746 | Ile26134Val | 0.81 (0.03 to 1.60) | 0.22 | 0.0432 | |||
| rs4894029 | synonymous | 0.81 (0.02 to 1.59) | 0.22 | 0.0453 | |||
| rs1560221 | synonymous | 0.82 (0.04 to 1.60) | 0.23 | 0.0406 | |||
| rs2163009 | synonymous | 0.82 (0.04 to 1.61) | 0.23 | 0.0399 | |||
| rs6712785 | intron | 0.82 (0.03 to 1.60) | 0.22 | 0.0421 | |||
| rs1001238 | Asn17060Asp | 0.82 (0.04 to 1.61) | 0.23 | 0.0399 | |||
| rs12693162 | downstream | 0.86 (0.06 to 1.65) | 0.21 | 0.0344 | |||
| rs16866420 | intron | 0.82 (0.02 to 1.60) | 0.21 | 0.0437 | |||
| rs2562839 | synonymous | 0.83 (0.01 to 1.64) | 0.20 | 0.0467 | |||
| rs2562838 | synonymous | 0.83 (0.02 to 1.64) | 0.20 | 0.0449 | |||
| rs2562836 | synonymous | 0.83 (0.01 to 1.64) | 0.20 | 0.0467 | |||
| rs2562830 | intron | 0.82 (0.01 to 1.63) | 0.20 | 0.0472 | |||
| rs2742327 | intron | 0.82 (0.02 to 1.63) | 0.20 | 0.0460 | |||
| rs1484116 | intron | 0.88 (0.08 to 1.68) | 0.20 | 0.0314 | |||
| rs7559166 | intron | 1.08 (0.19 to 1.98) | 0.15 | 0.0191 | |||
| rs2291313 | intron | 0.86 (0.06 to 1.67) | 0.20 | 0.0366 | |||
| CACNA1C | 645 | 216 | rs1009281 | intron | −0.76 (−1.41 to −0.12) | 0.48 | 0.0204 |
| rs11832738 | intron | −0.81 (−1.49 to −0.12) | 0.30 | 0.0215 |
| Gene | N.Marker All | N.Marker Test | SKAT p-Value | SKAT-O p-Value | SKAT CR p-Value | SKAT CR N.Marker.Rare | SKAT CR N.Marker.Common |
|---|---|---|---|---|---|---|---|
| ILK | 40 | 31 | 0.032 | 0.011 | 0.070 | 2 | 29 |
| TCAP | 9 | 2 | 0.011 | 0.011 | 0.025 | 1 | 1 |
| DSC2 | 72 | 12 | 0.006 | 0.012 | 0.008 | 6 | 6 |
| VCL | 403 | 195 | 0.014 | 0.018 | 0.026 | 99 | 96 |
| DSG2 | 118 | 25 | 0.454 | 0.028 | 0.395 | 12 | 13 |
| FXN | 16 | 11 | 0.042 | 0.038 | 0.202 | 0 | 11 |
| DSP | 208 | 43 | 0.021 | 0.042 | 0.097 | 27 | 16 |
| KCNQ1 | 1795 | 1063 | 0.024 | 0.044 | 0.153 | 220 | 843 |
| NEXN | 9 | 8 | 0.065 | 0.110 | 0.044 | 4 | 4 |
| KCNJ2 | 49 | 2 | 0.159 | 0.222 | 0.031 | 1 | 1 |
| DMD | 460 | 443 | 0.267 | 0.444 | 0.009 | 16 | 427 |
| OBSCN | 136 | 82 | 0.428 | 0.632 | 0.019 | 38 | 44 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serie, D.J.; Crook, J.E.; Necela, B.M.; Axenfeld, B.C.; Dockter, T.J.; Colon-Otero, G.; Perez, E.A.; Thompson, E.A.; Norton, N. Breast Cancer Clinical Trial of Chemotherapy and Trastuzumab: Potential Tool to Identify Cardiac Modifying Variants of Dilated Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2017, 4, 6. https://doi.org/10.3390/jcdd4020006
Serie DJ, Crook JE, Necela BM, Axenfeld BC, Dockter TJ, Colon-Otero G, Perez EA, Thompson EA, Norton N. Breast Cancer Clinical Trial of Chemotherapy and Trastuzumab: Potential Tool to Identify Cardiac Modifying Variants of Dilated Cardiomyopathy. Journal of Cardiovascular Development and Disease. 2017; 4(2):6. https://doi.org/10.3390/jcdd4020006
Chicago/Turabian StyleSerie, Daniel J., Julia E. Crook, Brian M. Necela, Bianca C. Axenfeld, Travis J. Dockter, Gerardo Colon-Otero, Edith A. Perez, E. Aubrey Thompson, and Nadine Norton. 2017. "Breast Cancer Clinical Trial of Chemotherapy and Trastuzumab: Potential Tool to Identify Cardiac Modifying Variants of Dilated Cardiomyopathy" Journal of Cardiovascular Development and Disease 4, no. 2: 6. https://doi.org/10.3390/jcdd4020006
APA StyleSerie, D. J., Crook, J. E., Necela, B. M., Axenfeld, B. C., Dockter, T. J., Colon-Otero, G., Perez, E. A., Thompson, E. A., & Norton, N. (2017). Breast Cancer Clinical Trial of Chemotherapy and Trastuzumab: Potential Tool to Identify Cardiac Modifying Variants of Dilated Cardiomyopathy. Journal of Cardiovascular Development and Disease, 4(2), 6. https://doi.org/10.3390/jcdd4020006