
Why Don’t More Mitochondrial Diseases Exhibit Cardiomyopathy?


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M. Pediatric Mitochondrial Diseases and the Heart. Curr. Opin. Pediatr. 2017, 29, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Kothari, S. Cardiac Manifestations of Primary Mitochondrial Disorders. Int. J. Cardiol. 2014, 177, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Fosslien, E. Mitochondrial Medicine—Cardiomyopathy Caused by Defective Oxidative Phosphorylation. Ann. Clin. Lab. Sci. 2003, 33, 371–395. [Google Scholar]
- Frazier, A.E.; Thorburn, D.R.; Compton, A.G. Mitochondrial Energy Generation Disorders: Genes, Mechanisms, and Clues to Pathology. J. Biol. Chem. 2019, 294, 5386–5395. [Google Scholar] [CrossRef]
- Scaglia, F.; Towbin, J.A.; Craigen, W.J.; Belmont, J.W.; Smith, E.O.B.; Neish, S.R.; Ware, S.M.; Hunter, J.V.; Fernbach, S.D.; Vladutiu, G.D.; et al. Clinical Spectrum, Morbidity, and Mortality in 113 Pediatric Patients with Mitochondrial Disease. Pediatrics 2004, 114, 925–931. [Google Scholar] [CrossRef]
- Holmgren, D.; Wåhlander, H.; Eriksson, B.O.; Oldfors, A.; Holme, E.; Tulinius, M. Cardiomyopathy in Children with Mitochondrial Disease: Clinical Course and Cardiological Findings. Eur. Heart J. 2003, 24, 280–288. [Google Scholar] [CrossRef]
- Marchant, A.; Evans, K.O. Incomplete Penetrance. Encycl. Anim. Cogn. Behav. 2019, 1–2. [Google Scholar]
- Falk, M.J. (Ed.) Mitochondrial Disease Genes Compendium: From Genes to Clinical Manifestations; Academic Press: London, UK, 2020. [Google Scholar]
- Claypool, S.M.; Koehler, C.M. The Complexity of Cardiolipin in Health and Disease. Trends Biochem. Sci. 2012, 37, 32–41. [Google Scholar] [CrossRef]
- Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS Regulation of Proliferating Cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The Mitochondrial Respiratory Chain Is Essential for Haematopoietic Stem Cell Function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Proteins: From Biogenesis to Functional Networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Mastantuono, E.; Wolf, C.M.; Prokisch, H. Genetic Basis of Mitochondrial Cardiomyopathy; Springer: Cham, Switzerland, 2019; pp. 93–139. [Google Scholar]
- Picard, M.; Shirihai, O.S. Mitochondrial Signal Transduction. Cell Metab. 2022, 34, 1620–1653. [Google Scholar] [CrossRef]
- Hom, J.R.; Quintanilla, R.A.; Hoffman, D.L.; de Mesy Bentley, K.L.; Molkentin, J.D.; Sheu, S.-S.; Porter, G.A. The Permeability Transition Pore Controls Cardiac Mitochondrial Maturation and Myocyte Differentiation. Dev. Cell 2011, 21, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Porter, G.A., Jr.; Hom, J.R.; Hoffman, D.L.; Quintanilla, R.A.; de Mesy Bentley, K.L.; Sheu, S.-S. Bioenergetics, Mitochondria, and Cardiac Myocyte Differentiation. Prog. Pediatr. Cardiol. 2011, 31, 75–81. [Google Scholar] [CrossRef]
- Beutner, G.; Eliseev, R.A.; Porter, G.A. Initiation of Electron Transport Chain Activity in the Embryonic Heart Coincides with the Activation of Mitochondrial Complex 1 and the Formation of Supercomplexes. PLoS ONE 2014, 9, e113330. [Google Scholar] [CrossRef]
- Menendez-Montes, I.; Escobar, B.; Palacios, B.; Gómez, M.J.; Izquierdo-Garcia, J.L.; Flores, L.; Jiménez-Borreguero, L.J.; Aragones, J.; Ruiz-Cabello, J.; Torres, M.; et al. Myocardial VHL-HIF Signaling Controls an Embryonic Metabolic Switch Essential for Cardiac Maturation. Dev. Cell 2016, 39, 724–739. [Google Scholar] [CrossRef]
- Lingan, J.V.; Alanzalon, R.E.; Porter, G.A. Preventing Permeability Transition Pore Opening Increases Mitochondrial Maturation, Myocyte Differentiation and Cardiac Function in the Neonatal Mouse Heart. Pediatr. Res. 2017, 81, 932–941. [Google Scholar] [CrossRef]
- Dogan, S.A.; Cerutti, R.; Benincá, C.; Brea-Calvo, G.; Jacobs, H.T.; Zeviani, M.; Szibor, M.; Viscomi, C. Perturbed Redox Signaling Exacerbates a Mitochondrial Myopathy. Cell Metab. 2018, 28, 764–775.e5. [Google Scholar] [CrossRef]
- Smith, A.C.; Robinson, A.J. MitoMiner v3.1, an Update on the Mitochondrial Proteomics Database. Nucleic Acids Res. 2016, 44, D1258–D1261. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Robinson, A.J. MitoMiner 4.0. Available online: https://mitominer.mrc-mbu.cam.ac.uk/release-4.0/begin.do (accessed on 2 September 2020).
- Arroyo, J.D.; Jourdain, A.A.; Calvo, S.E.; Ballarano, C.A.; Doench, J.G.; Root, D.E.; Mootha, V.K. A Genome-Wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab. 2016, 24, 875–885. [Google Scholar] [CrossRef] [PubMed]
- McKusick-Nathans Institute of Genetic Medicine Online Mendelian Inheritance in Man, OMIM®. Available online: https://www.omim.org (accessed on 2 September 2020).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Computational Structural Biology Group SWISS-MODEL. Available online: https://swissmodel.expasy.org (accessed on 3 December 2020).
- BioGRID Team BioGRID. Available online: https://thebiogrid.org/ (accessed on 20 February 2023).
- Stark, C. BioGRID: A General Repository for Interaction Datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-Throughput Discovery of Novel Developmental Phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef]
- Groza, T.; Gomez, F.L.; Mashhadi, H.H.; Munoz-Fuentes, V.; Gunes, O.; Wilson, R.; Cacheiro, P.; Frost, A.; Keskivali-Bond, P.; Vardal, B.; et al. The International Mouse Phenotyping Consortium: Comprehensive Knockout Phenotyping Underpinning the Study of Human Disease. Nucleic Acids Res. 2023, 51, D1038–D1045. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Mirra, B.; Barretta, F.; Caiazza, M.; Lombardo, B.; Scudiero, O.; Tinto, N.; Limongelli, G.; Frisso, G. Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search among Mitochondrial and Nuclear Genes. Int. J. Mol. Sci. 2021, 22, 5742. [Google Scholar] [CrossRef]
- Yaplito-Lee, J.; Weintraub, R.; Jamsen, K.; Chow, C.W.; Thorburn, D.R.; Boneh, A. Cardiac Manifestations in Oxidative Phosphorylation Disorders of Childhood. J. Pediatr. 2007, 150, 407–411. [Google Scholar] [CrossRef]
- Popoiu, T.-A.; Dudek, J.; Maack, C.; Bertero, E. Cardiac Involvement in Mitochondrial Disorders. Curr. Heart Fail. Rep. 2023, 20, 76–87. [Google Scholar] [CrossRef]
- Agren, A.; Borg, K.; Brolin, S.E.; Carlman, J.; Lundqvist, G. Hydroxyacyl CoA Dehydrogenase, an Enzyme Important in Fat Metabolism in Different Cell Types in the Islets of Langerhans. Diabete Metab. 1977, 3, 169–172. [Google Scholar] [PubMed]
- Demirbilek, H.; Rahman, S.A.; Buyukyilmaz, G.G.; Hussain, K. Diagnosis and Treatment of Hyperinsulinaemic Hypoglycaemia and Its Implications for Paediatric Endocrinology. Int. J. Pediatr. Endocrinol. 2017, 2017, 9. [Google Scholar] [CrossRef]
- Hsu, Y.H.R.; Yogasundaram, H.; Parajuli, N.; Valtuille, L.; Sergi, C.; Oudit, G.Y. MELAS Syndrome and Cardiomyopathy: Linking Mitochondrial Function to Heart Failure Pathogenesis. Heart Fail. Rev. 2016, 21, 103–116. [Google Scholar] [CrossRef]
- Schiff, M.; Ogier de Baulny, H.; Lombès, A. Neonatal Cardiomyopathies and Metabolic Crises Due to Oxidative Phosphorylation Defects. Semin. Fetal Neonatal Med. 2011, 16, 216–221. [Google Scholar] [CrossRef]
- Ebihara, T.; Nagatomo, T.; Sugiyama, Y.; Tsuruoka, T.; Osone, Y.; Shimura, M.; Tajika, M.; Matsuhashi, T.; Ichimoto, K.; Matsunaga, A.; et al. Neonatal-Onset Mitochondrial Disease: Clinical Features, Molecular Diagnosis and Prognosis. Arch. Dis. Child. Fetal Neonatal Ed. 2022, 107, 329–334. [Google Scholar] [CrossRef]
- Williams, J.L.; Paudyal, A.; Awad, S.; Nicholson, J.; Grzesik, D.; Botta, J.; Meimaridou, E.; Maharaj, A.V.; Stewart, M.; Tinker, A.; et al. Mylk3 Null C57BL/6N Mice Develop Cardiomyopathy, Whereas Nnt Null C57BL/6J Mice Do Not. Life Sci. Alliance 2020, 3, e201900593. [Google Scholar] [CrossRef] [PubMed]
- Figueira, T.R. A Word of Caution Concerning the Use of Nnt -Mutated C57BL/6 Mice Substrains as Experimental Models to Study Metabolism and Mitochondrial Pathophysiology. Exp. Physiol. 2013, 98, 1643. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Barth Syndrome: Connecting Cardiolipin to Cardiomyopathy. Lipids 2017, 52, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Caudal, A.; Ren, L.; Tu, C.; Wu, J.C. Human Induced Pluripotent Stem Cells for Studying Mitochondrial Diseases in the Heart. FEBS Lett. 2022, 596, 1735–1745. [Google Scholar] [CrossRef]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the Mitochondrial Cardiomyopathy of Barth Syndrome with Induced Pluripotent Stem Cell and Heart-on-Chip Technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Shah, A.P.; Siedlecka, U.; Gandhi, A.; Navaratnarajah, M.; Abou Al-Saud, S.; Yacoub, M.H.; Terracciano, C.M. Genetic Background Affects Function and Intracellular Calcium Regulation of Mouse Hearts. Cardiovasc. Res. 2010, 87, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient Care Standards for Primary Mitochondrial Disease: A Consensus Statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 1380–1397. [Google Scholar] [CrossRef] [PubMed]
- Lioncino, M.; Monda, E.; Caiazza, M.; Fusco, A.; Cirillo, A.; Dongiglio, F.; Simonelli, V.; Sampaolo, S.; Ruggiero, L.; Scarano, G.; et al. Cardiovascular Involvement in MtDNA Disease. Heart Fail. Clin. 2022, 18, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Savvatis, K.; Vissing, C.R.; Klouvi, L.; Florian, A.; Rahman, M.; Behin, A.; Fayssoil, A.; Masingue, M.; Stojkovic, T.; Becane, H.M.; et al. Cardiac Outcomes in Adults With Mitochondrial Diseases. J. Am. Coll. Cardiol. 2022, 80, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Bougouin, W.; Béhin, A.; Stojkovic, T.; Bécane, H.M.; Jardel, C.; Berber, N.; Mochel, F.; Lombès, A.; Eymard, B.; et al. Long-Term Cardiac Prognosis and Risk Stratification in 260 Adults Presenting with Mitochondrial Diseases. Eur. Heart J. 2015, 36, 2886–2893. [Google Scholar] [CrossRef]


{kind=link}
| Cardiomyopathy (n = 107) | No Cardiomyopathy (n = 134) | p-Test | |
|---|---|---|---|
| Localization Category (%) | 0.511 | ||
| Gene Category (%) | <0.001 | ||
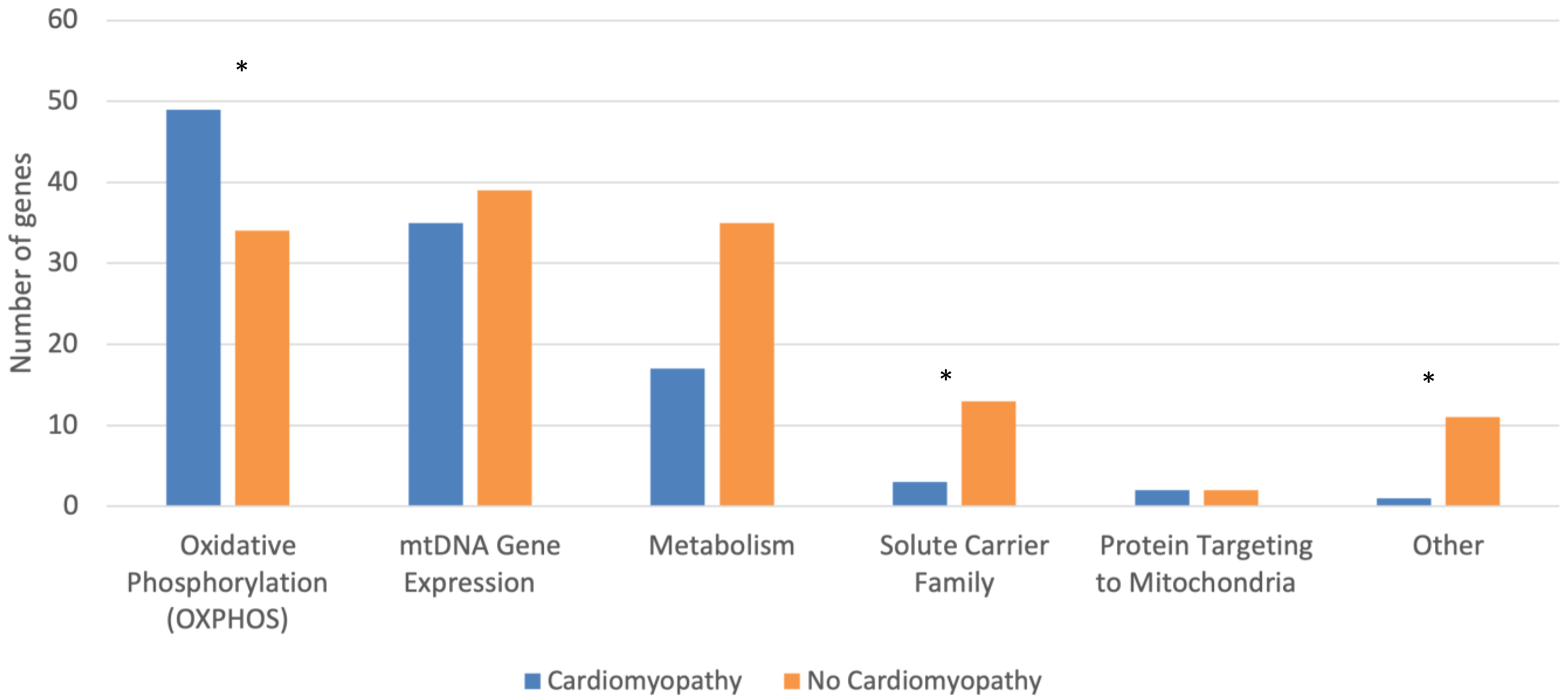
| Oxidative Phosphorylation (OXPHOS) | 49 (45.8) | 34 (25.4) | 0.001 |
| OXPHOS Complex (%) | 0.020 | ||
| I | 19 (38.8) | 17 (50.0) | 0.371 |
| II | 4 (8.2) | 1 (2.9) | 0.644 |
| III | 0 (0.0) | 5 (14.7) | 0.010 |
| IV | 8 (16.3) | 6 (17.6) | 1.000 |
| V | 4 (8.2) | 0 (0.0) | 0.141 |
| Accessory | 14 (28.6) | 5 (14.7) | 0.187 |
| mtDNA Gene Expression | 35 (32.7) | 39 (29.1) | 0.576 |
| mtDNA Gene Expression Subcategory (%) | 0.797 | ||
| tRNA | 10 (28.6) | 12 (30.8) | 1.000 |
| tRNA Synthetase | 6 (17.1) | 11 (28.2) | 0.284 |
| Ribosome Component | 4 (11.4) | 3 (7.7) | 0.701 |
| Nucleotide Modification | 5 (14.3) | 4 (10.3) | 0.727 |
| Other | 10 (28.6) | 9 (23.1) | 0.606 |
| Metabolism | 17 (15.9) | 35 (26.1) | 0.060 |
| Metabolism Subcategory (%) | 0.058 | ||
| Intermediary Metabolism | 6 (35.3) | 13 (37.1) | 1.000 |
| Mitochondrial Fatty Acid Metabolism | 7 (41.2) | 3 (8.6) | 0.009 |
| Amino Acid Metabolism | 1 (5.9) | 8 (22.9) | 0.241 |
| Phospholipid Metabolism | 2 (11.8) | 3 (8.6) | 1.000 |
| Krebs/TCA Cycle | 0 (0.0) | 4 (11.4) | 0.290 |
| Nucleotide Metabolism | 0 (0.0) | 3 (8.6) | 0.542 |
| Other | 1 (5.9) | 1 (2.9) | 1.000 |
| Solute Carrier Family | 3 (2.8) | 13 (9.7) | 0.038 |
| Other | 1 (0.9) | 11 (8.2) | 0.014 |
| Protein Targeting to Mitochondria | 2 (1.9) | 2 (1.5) | 1.000 |
| Genome Origin (%) | 0.188 | ||
| Nuclear Gene | 88 (82.2) | 116 (86.6) | 0.374 |
| Nervous System Manifestations | 101 (94.4) | 123 (91.8) | 0.462 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, N.; Ren, M.; Phoon, C.K.L. Why Don’t More Mitochondrial Diseases Exhibit Cardiomyopathy? J. Cardiovasc. Dev. Dis. 2023, 10, 154. https://doi.org/10.3390/jcdd10040154
Singh N, Ren M, Phoon CKL. Why Don’t More Mitochondrial Diseases Exhibit Cardiomyopathy? Journal of Cardiovascular Development and Disease. 2023; 10(4):154. https://doi.org/10.3390/jcdd10040154
Chicago/Turabian StyleSingh, Nina, Mindong Ren, and Colin K. L. Phoon. 2023. "Why Don’t More Mitochondrial Diseases Exhibit Cardiomyopathy?" Journal of Cardiovascular Development and Disease 10, no. 4: 154. https://doi.org/10.3390/jcdd10040154
APA StyleSingh, N., Ren, M., & Phoon, C. K. L. (2023). Why Don’t More Mitochondrial Diseases Exhibit Cardiomyopathy? Journal of Cardiovascular Development and Disease, 10(4), 154. https://doi.org/10.3390/jcdd10040154