
Left-Sided Heart Defects and Laterality Disturbance in Hypoplastic Left Heart Syndrome


Abstract
1. Introduction
2. Co-Occurrences of Laterality Defects with HLHS
3. Association of HLHS with Genes Involved in the Specification of Left–Right Asymmetry
4. Hippo Signaling and LV Hypoplasia in HLHS
5. Sap130/Sin3A in Heart Organ Size Regulation and Left–Right Patterning in HLHS
6. Left–Right Patterning Integral to Heart Development and the Pathogenesis of CHD
7. Summary and Clinical Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siffel, C.; Riehle-Colarusso, T.; Oster, M.E.; Correa, A. Survival of Children With Hypoplastic Left Heart Syndrome. Pediatrics 2015, 136, e864–e870. [Google Scholar] [CrossRef]
- Yabrodi, M.; Mastropietro, C.W. Hypoplastic left heart syndrome: From comfort care to long-term survival. Pediatr. Res. 2016, 81, 142–149. [Google Scholar] [CrossRef]
- Burch, P.T.; Gerstenberger, E.; Ravishankar, C.; Hehir, D.A.; Davies, R.R.; Colan, S.D.; Sleeper, L.A.; Newburger, J.W.; Clabby, M.L.; Williams, I.A.; et al. Longitudinal Assessment of Growth in Hypoplastic Left Heart Syndrome: Results From the Single Ventricle Reconstruction Trial. J. Am. Heart Assoc. 2014, 3, e000079. [Google Scholar] [CrossRef]
- Goldberg, C.S.; Mussatto, K.; Licht, D.; Wernovsky, G. Neurodevelopment and quality of life for children with hypoplastic left heart syndrome: Current knowns and unknowns. Cardiol. Young 2011, 21, 88–92. [Google Scholar] [CrossRef]
- Donofrio, M.T.; Duplessis, A.J.; Limperopoulos, C. Impact of congenital heart disease on fetal brain development and injury. Curr. Opin. Pediatr. 2011, 23, 502–511. [Google Scholar] [CrossRef]
- Morton, P.D.; Ishibashi, N.; Jonas, R.A. Neurodevelopmental Abnormalities and Congenital Heart Disease. Circ. Res. 2017, 120, 960–977. [Google Scholar] [CrossRef]
- Volpe, J.J. Encephalopathy of Congenital Heart Disease– Destructive and Developmental Effects Intertwined. J. Pediatr. 2014, 164, 962–965. [Google Scholar] [CrossRef]
- Newburger, J.W.; Sleeper, L.A.; Frommelt, P.C.; Pearson, G.D.; Mahle, W.T.; Chen, S.; Dunbar-Masterson, C.; Mital, S.; Williams, I.A.; Ghanayem, N.S.; et al. Transplantation-Free Survival and Interventions at 3 Years in the Single Ventricle Reconstruction Trial. Circulation 2014, 129, 2013–2020. [Google Scholar] [CrossRef]
- Hoang, T.T.; Goldmuntz, E.; Roberts, A.E.; Chung, W.K.; Kline, J.K.; Deanfield, J.E.; Giardini, A.; Aleman, A.; Gelb, B.D.; Mac Neal, M.; et al. The Congenital Heart Disease Genetic Network Study: Cohort description. PLoS ONE 2018, 13, e0191319. [Google Scholar] [CrossRef]
- Ravishankar, C.; Zak, V.; Williams, I.A.; Bellinger, D.C.; Gaynor, J.W.; Ghanayem, N.S.; Krawczeski, C.D.; Licht, D.J.; Mahony, L.; Newburger, J.W.; et al. Association of Impaired Linear Growth and Worse Neurodevelopmental Outcome in Infants with Single Ventricle Physiology: A Report from the Pediatric Heart Network Infant Single Ventricle Trial. J. Pediatr. 2012, 162, 250–256.e2. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.J.; Offord, K.P.; Feldt, R.H.; Schaff, H.V.; Puga, F.J.; Danielson, G.K. Five- to fifteen-year follow-up after Fontan operation. Circulation 1992, 85, 469–496. [Google Scholar] [CrossRef] [PubMed]
- Gentles, T.L.; Gauvreau, K.; Mayer, J.E.; Fishberger, S.B.; Burnetta, J.; Colan, S.D.; Newburger, J.W.; Wernovsky, G. Functional outcome after the Fontan operation: Factors influencing late morbidity. J. Thorac. Cardiovasc. Surg. 1997, 114, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Firek, M.; Zakhary, B.; Brenner, M.; Hildebrand, F.; Coimbra, R. Severe Pelvic Fracture in the Elderly: High Morbidity, Mortality, and Resource Utilization. Am. Surg. 2020, 86, 1401–1406. [Google Scholar] [CrossRef]
- Tweddell, J.S.; Hoffman, G.M.; Mussatto, K.A.; Fedderly, R.T.; Berger, S.; Jaquiss, R.D.B.; Ghanayem, N.S.; Frisbee, S.J.; Litwin, S.B. Improved Survival of Patients Undergoing Palliation of Hypoplastic Left Heart Syndrome: Lessons Learned From 115 Consecutive Patients. Circulation 2002, 106, I-82. [Google Scholar] [CrossRef]
- Alsoufi, B.; Mahle, W.T.; Manlhiot, C.; Deshpande, S.; Kogon, B.; McCrindle, B.W.; Kanter, K. Outcomes of heart transplantation in children with hypoplastic left heart syndrome previously palliated with the Norwood procedure. J. Thorac. Cardiovasc. Surg. 2015, 151, 167–175.e2. [Google Scholar] [CrossRef] [PubMed]
- Cleves, M.A.; Ghaffar, S.; Zhao, W.; Mosley, B.S.; Hobbs, C.A. First-year survival of infants born with congenital heart defects in Arkansas (1993-1998): A survival analysis using registry data. Birth Defects Res. Part A Clin. Mol. Teratol. 2003, 67, 662–668. [Google Scholar] [CrossRef]
- Wilson, W.M.; Valente, A.M.; Hickey, E.J.; Clift, P.; Burchill, L.J.; Emmanuel, Y.; Gibson, P.; Greutmann, M.; Grewal, J.; Grigg, L.E.; et al. Outcomes of Patients With Hypoplastic Left Heart Syndrome Reaching Adulthood After Fontan Palliation. Circulation 2018, 137, 978–981. [Google Scholar] [CrossRef]
- Gabriel, G.C.; Lo, C.W. Left–right patterning in congenital heart disease beyond heterotaxy. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 90–96. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Sen, P.; Bhatt, S.S.; Storer, M.; Xia, Z.; Bejjani, B.A.; Ou, Z.; Wiszniewska, J.; Driscoll, D.J.; Bolivar, J.; et al. Genomic and Genic Deletions of the FOX Gene Cluster on 16q24.1 and Inactivating Mutations of FOXF1 Cause Alveolar Capillary Dysplasia and Other Malformations. Am. J. Hum. Genet. 2009, 84, 780–791. [Google Scholar] [CrossRef]
- Huseynova, R.A.; Bin Mahmoud, L.A.; Abdelrahim, A.; Alroiedy, M.A.; Huseynov, O. Polysplenia syndrome with complex heart disease and jejunal atresia with malrotation in neonate: A case report. Clin. Case Rep. 2020, 8, 848–851. [Google Scholar] [CrossRef]
- Becker, D.J.; Islam, S.; Geiger, J.D. Biliary atresia associated with hypoplastic left heart syndrome: A case report and review of the literature. J. Pediatr. Surg. 2004, 39, 1411–1413. [Google Scholar] [CrossRef]
- Lin, A.E.; Krikov, S.; Riehle-Colarusso, T.; Frías, J.L.; Belmont, J.; Anderka, M.; Geva, T.; Getz, K.D.; Botto, L.D.; The National Birth Defects Prevention Study. Laterality defects in the national birth defects prevention study (1998-2007): Birth prevalence and descriptive epidemiology. Am. J. Med. Genet. Part A 2014, 164, 2581–2591. [Google Scholar] [CrossRef]
- Pradat, P.; Francannet, C.; Harris, J.; Robert, E. The Epidemiology of Cardiovascular Defects, Part I: A Study Based on Data from Three Large Registries of Congenital Malformations. Pediatr. Cardiol. 2003, 24, 195–221. [Google Scholar] [CrossRef]
- Peeters, H.; Devriendt, K. Human laterality disorders. Eur. J. Med. Genet. 2006, 49, 349–362. [Google Scholar] [CrossRef]
- Narkewicz, M.R. Biliary atresia: An update on our understanding of the disorder. Curr. Opin. Pediatr. 2001, 13, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, D.H.; Shepherd, R.W. Extrahepatic biliary atresia: A disease or a phenotype? Hepatology 2002, 35, 1297–1304. [Google Scholar] [CrossRef]
- Schreiber, R.A.; Kleinman, R.E. Biliary Atresia. J. Craniofacial Surg. 2002, 35, S11–S16. [Google Scholar] [CrossRef] [PubMed]
- Lupo, P.J.; Isenburg, J.L.; Salemi, J.L.; Mai, C.T.; Liberman, R.F.; Canfield, M.A.; Copeland, G.; Haight, S.; Harpavat, S.; Hoyt, A.T.; et al. Population-based birth defects data in the United States, 2010-2014: A focus on gastrointestinal defects. Birth Defects Res. 2017, 109, 1504–1514. [Google Scholar] [CrossRef]
- Bates, M.D.; Bucuvalas, J.C.; Alonso, M.H.; Ryckman, F.C. Biliary Atresia: Pathogenesis and Treatment. Semin. Liver Dis. 1998, 18, 281–293. [Google Scholar] [CrossRef]
- Liu, S.; Wei, W.; Wang, P.; Liu, C.; Jiang, X.; Li, T.; Li, F.; Wu, Y.; Chen, S.; Sun, K.; et al. LOF variants identifying candidate genes of laterality defects patients with congenital heart disease. PLoS Genet. 2022, 18, e1010530. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Zender, G.A.; Fitzgerald-Butt, S.M.; Koehler, D.; Menesses-Diaz, A.; Fernbach, S.; Lee, K.; Towbin, J.A.; Leal, S.; Belmont, J.W. Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome). Eur. J. Hum. Genet. 2009, 17, 811–819. [Google Scholar] [CrossRef]
- Hinton, R.B.; Martin, L.J.; Rame-Gowda, S.; Tabangin, M.E.; Cripe, L.H.; Benson, D.W. Hypoplastic Left Heart Syndrome Links to Chromosomes 10q and 6q and Is Genetically Related to Bicuspid Aortic Valve. J. Am. Coll. Cardiol. 2009, 53, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.; Pignatelli, R.; Lewin, M.; Ho, T.; Fernbach, S.; Menesses, A.; Lam, W.; Leal, S.M.; Kaplan, N.; Schliekelman, P.; et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. Part A 2005, 134A, 180–186. [Google Scholar] [CrossRef]
- Hinton, R.B.; Martin, L.J.; Tabangin, M.E.; Mazwi, M.L.; Cripe, L.H.; Benson, D.W. Hypoplastic Left Heart Syndrome Is Heritable. J. Am. Coll. Cardiol. 2007, 50, 1590–1595. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.W.; Martin, L.J.; Lo, C.W. Genetics of Hypoplastic Left Heart Syndrome. J. Pediatr. 2016, 173, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.; Tariq, M.; Ware, S.M. Genetic and Functional Analyses of ZIC3 Variants in Congenital Heart Disease. Hum. Mutat. 2013, 35, 66–75. [Google Scholar] [CrossRef]
- Sempou, E.; Lakhani, O.A.; Amalraj, S.; Khokha, M.K. Candidate Heterotaxy Gene FGFR4 Is Essential for Patterning of the Left-Right Organizer in Xenopus. Front. Physiol. 2018, 9, 1705. [Google Scholar] [CrossRef]
- Hickey, E.J.; Caldarone, C.A.; McCrindle, B.W. Left Ventricular Hypoplasia: A Spectrum of Disease Involving the Left Ventricular Outflow Tract, Aortic Valve, and Aorta. J. Am. Coll. Cardiol. 2012, 59, S43–S54. [Google Scholar] [CrossRef]
- Reamon-Buettner, S.M.; Ciribilli, Y.; Traverso, I.; Kuhls, B.; Inga, A.; Borlak, J. A functional genetic study identifies HAND1 mutations in septation defects of the human heart. Hum. Mol. Genet. 2009, 18, 3567–3578. [Google Scholar] [CrossRef]
- Firulli, B.A.; Toolan, K.P.; Harkin, J.; Millar, H.; Pineda, S.; Firulli, A.B. The HAND1 frameshift A126FS mutation does not cause hypoplastic left heart syndrome in mice. Cardiovasc. Res. 2017, 113, 1732–1742. [Google Scholar] [CrossRef]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, I.; Ichikawa, H. Human Heterotaxy Syndrome. Circ. J. 2012, 76, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo signaling impedes adult heart regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef]
- Xu, X.; Jin, K.; Bais, A.S.; Zhu, W.; Yagi, H.; Feinstein, T.N.; Nguyen, P.K.; Criscione, J.D.; Liu, X.; Beutner, G.; et al. Uncompensated mitochondrial oxidative stress underlies heart failure in an iPSC-derived model of congenital heart disease. Cell Stem Cell 2022, 29, 840–855.e7. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, W.; Romanelli, L.; Atkins, M.; Hillen, H.; González-Blas, C.B.; Jacobs, J.; Xie, J.; Soheily, S.; Verboven, E.; Moya, I.M.; et al. Hippo signaling instructs ectopic but not normal organ growth. Science 2022, 378, eabg3679. [Google Scholar] [CrossRef]
- Saunders, A.; Huang, X.; Fidalgo, M.; Reimer, M.H.; Faiola, F.; Ding, J.; Sánchez-Priego, C.; Guallar, D.; Sáenz, C.; Li, D.; et al. The SIN3A/HDAC Corepressor Complex Functionally Cooperates with NANOG to Promote Pluripotency. Cell Rep. 2017, 18, 1713–1726. [Google Scholar] [CrossRef]
- van Oevelen, C.; Bowman, C.; Pellegrino, J.; Asp, P.; Cheng, J.; Parisi, F.; Micsinai, M.; Kluger, Y.; Chu, A.; Blais, A.; et al. The Mammalian Sin3 Proteins Are Required for Muscle Development and Sarcomere Specification. Mol. Cell. Biol. 2010, 30, 5686–5697. [Google Scholar] [CrossRef]
- Gao, Z.; Ure, K.; Ding, P.; Nashaat, M.; Yuan, L.; Ma, J.; Hammer, R.E.; Hsieh, J. The Master Negative Regulator REST/NRSF Controls Adult Neurogenesis by Restraining the Neurogenic Program in Quiescent Stem Cells. J. Neurosci. 2011, 31, 9772–9786. [Google Scholar] [CrossRef]
- Witteveen, J.S.; Willemsen, M.H.; Dombroski, T.C.D.; Van Bakel, N.H.M.; Nillesen, W.M.; Van Hulten, J.A.; Jansen, E.J.R.; Verkaik, D.; Veenstra-Knol, H.E.; Van Ravenswaaij-Arts, C.M.A.; et al. Haploinsufficiency of MeCP2-interacting transcriptional co-repressor SIN3A causes mild intellectual disability by affecting the development of cortical integrity. Nat. Genet. 2016, 48, 877–887. [Google Scholar] [CrossRef]
- Ota, M.; Sasaki, H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 2008, 135, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of Insulin-Like Growth Factor Signaling by Yap Governs Cardiomyocyte Proliferation and Embryonic Heart Size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef]
- Zhu, F.; Zhu, Q.; Ye, D.; Zhang, Q.; Yang, Y.; Guo, X.; Liu, Z.; Jiapaer, Z.; Wan, X.; Wang, G.; et al. Sin3a–Tet1 interaction activates gene transcription and is required for embryonic stem cell pluripotency. Nucleic Acids Res. 2018, 46, 6026–6040. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/Histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Ocaña, O.H.; Coskun, H.; Minguillón, C.; Murawala, P.; Tanaka, E.M.; Galceran, J.; Muñoz-Chápuli, R.; Nieto, M.A. A right-handed signalling pathway drives heart looping in vertebrates. Nature 2017, 549, 86–90. [Google Scholar] [CrossRef]
- Murray, S.A.; Gridley, T. Snail family genes are required for left–right asymmetry determination, but not neural crest formation, in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 10300–10304. [Google Scholar] [CrossRef] [PubMed]
- Logan, M.; Pagán-Westphal, S.M.; Smith, D.M.; Paganessi, L.; Tabin, C.J. The Transcription Factor Pitx2 Mediates Situs-Specific Morphogenesis in Response to Left-Right Asymmetric Signals. Cell 1998, 94, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, H.; Meno, C.; Koshiba, K.; Sugihara, M.; Itoh, H.; Ishimaru, Y.; Inoue, T.; Ohuchi, H.; Semina, E.V.; Murray, J.C.; et al. Pitx2, a Bicoid-Type Homeobox Gene, Is Involved in a Lefty-Signaling Pathway in Determination of Left-Right Asymmetry. Cell 1998, 94, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Li, E. Smad2 role in mesoderm formation, left–right patterning and craniofacial development. Nature 1998, 393, 786–790. [Google Scholar] [CrossRef]
- Gilljam, T.; McCrindle, B.W.; Smallhorn, J.F.; Williams, W.G.; Freedom, R.M. Outcomes of left atrial isomerism over a 28-year period at a single institution. J. Am. Coll. Cardiol. 2000, 36, 908–916. [Google Scholar] [CrossRef] [PubMed]



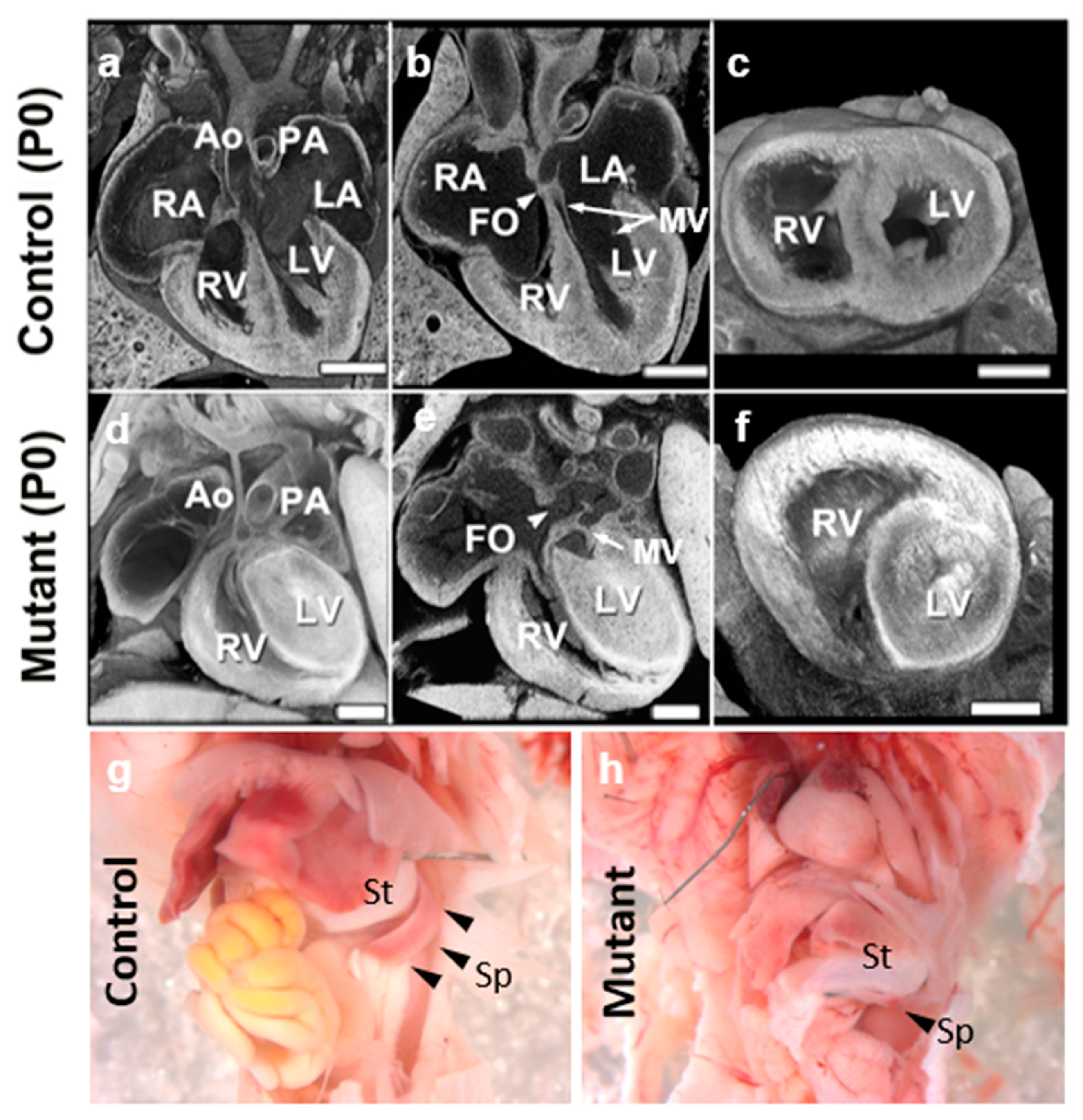{kind=link}
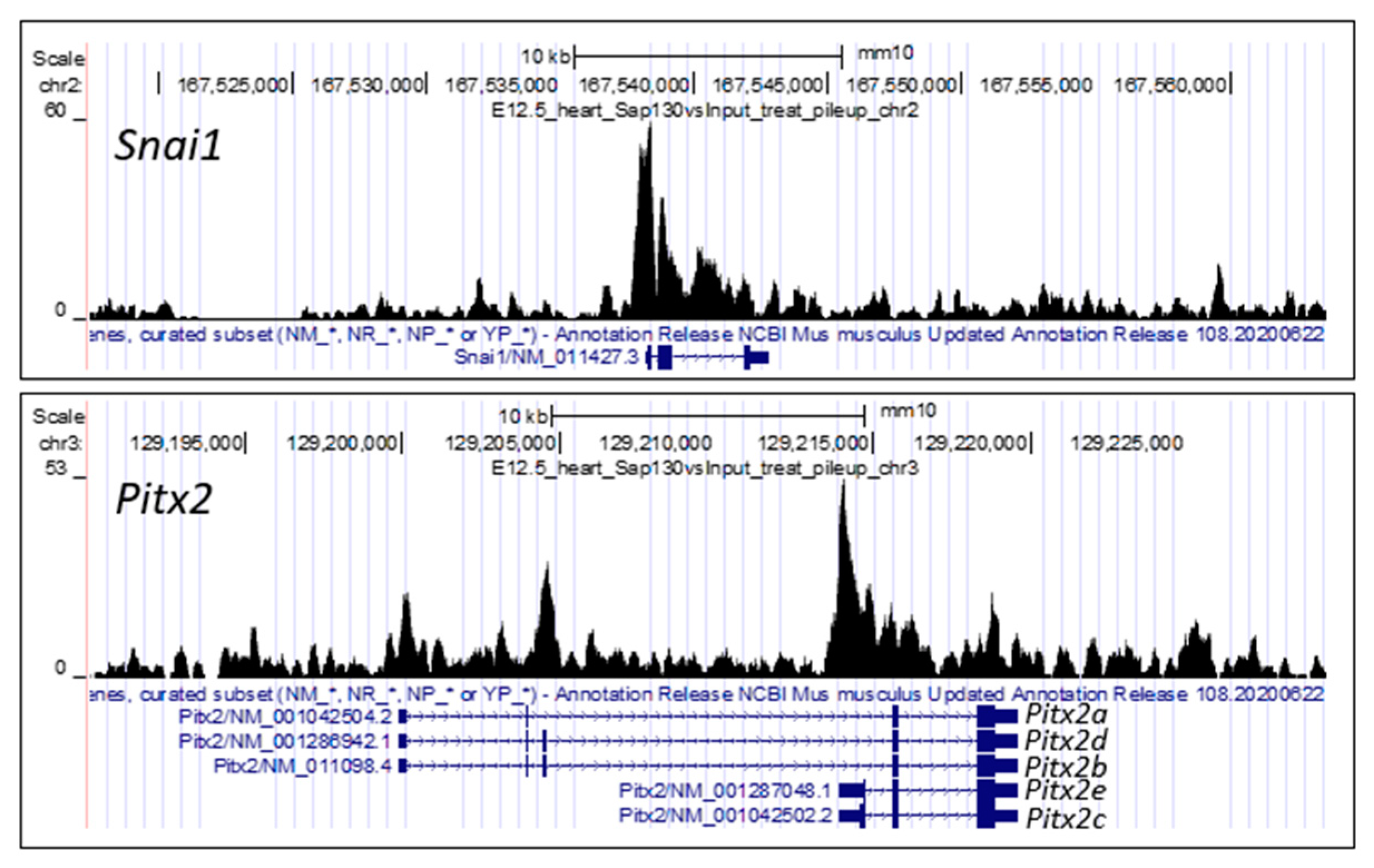{kind=link}
| HLHS | SV | TA | PTA | PA | TGA | DORV | AVSD/ ECD | TAPVR | TOF | EB | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A. CHD Associated with Heterotaxy and Situs Inversus in National Birth Defects Prevention Study by Lin et al. [22] | ||||||||||||
| Situs Inversus | 1.4% | 5.8% | 0.7% | 0% | 1.4% | 2.4% | 7.9% | 5.0% | 2.2% | 2.9% | 0.7% | 43.2% |
| Heterotaxy | 3.2% | 14.0% | 2.1% | 1.1% | 9.8% | 3.7% | 25.7% | 48.4% | 31.2% | 4.2% | 0.3% | 96.6% |
| All Laterality | 2.7% | 11.8% | 1.7% | 0.8% | 4.9% | 3.1% | 20.9% | 36.8% | 23.4% | 3.9% | 0.4% | 82.2% |
| B. Situs Anomalies Associated with CHD in Birth Defects Registry Study on Congenital Malformations by Pradat et al. [23] | ||||||||||||
| Situs Inversus | 4.7% | 17.6% | 0% | 1.0% | 9.8% | 5.7% | 13.6% | 9.4% | 4.8% | 3.7% | 5.0% | 5.6% |
| Gut Malrotation | 6.5% | 11.8% | 0% | 7.1% | 4.9% | 2.4% | 11.9% | 10.4% | 6.3% | 4.9% | 0% | 6.2% |
| Spleen Abnormal | 7.7% | 23.5% | 4.1% | 6.1% | 4.9% | 4.9% | 8.5% | 11.3% | 12.7% | 5.8% | 0% | 7.5% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yagi, H.; Lo, C.W. Left-Sided Heart Defects and Laterality Disturbance in Hypoplastic Left Heart Syndrome. J. Cardiovasc. Dev. Dis. 2023, 10, 99. https://doi.org/10.3390/jcdd10030099
Yagi H, Lo CW. Left-Sided Heart Defects and Laterality Disturbance in Hypoplastic Left Heart Syndrome. Journal of Cardiovascular Development and Disease. 2023; 10(3):99. https://doi.org/10.3390/jcdd10030099
Chicago/Turabian StyleYagi, Hisato, and Cecilia W. Lo. 2023. "Left-Sided Heart Defects and Laterality Disturbance in Hypoplastic Left Heart Syndrome" Journal of Cardiovascular Development and Disease 10, no. 3: 99. https://doi.org/10.3390/jcdd10030099
APA StyleYagi, H., & Lo, C. W. (2023). Left-Sided Heart Defects and Laterality Disturbance in Hypoplastic Left Heart Syndrome. Journal of Cardiovascular Development and Disease, 10(3), 99. https://doi.org/10.3390/jcdd10030099