
The Role of ncRNAs in Cardiac Infarction and Regeneration
 ,
,  , and
, and 
Abstract
1. Introduction
2. The Process of Inflammation during Cardiac Injury
3. The Process of Fibrosis during Cardiac Injury
4. Inductive and Transcriptional Regulatory Mechanisms Driving Cardiac Injury and Regeneration
5. The Emerging Impact of Non-Coding RNAs’ Regulation of Cardiac Injury and Regeneration
5.1. The Functional Roles of miRNAs in the Cardiac Inflammatory Response
5.1.1. The Role of miR-181
5.1.2. The Role of miR-155
5.1.3. The Role of miR-21
5.1.4. The Role of miR-146
5.1.5. The Role of miR-133
5.1.6. The Role of miR-130
5.1.7. The Role of miR-24
5.1.8. The Role of miR-223
5.1.9. The Role of miR-23
5.1.10. The Role of miR-22
5.1.11. The Role of miR-30
5.1.12. The Role of miR-499
5.1.13. The Role of miR-486
5.2. The Functional Role of miRNAs in Cardiac Fibrosis Response
5.2.1. The Role of miR-34
5.2.2. The Role of miR-145
5.2.3. The Role of miR-181
5.2.4. The Role of miR-155
5.2.5. The Role of miR-133
5.2.6. The Role of miR-22
5.2.7. The Role of miR-21
5.2.8. The Role of miR-26
5.2.9. The Role of miR-29
5.2.10. The Role of miR-30
5.2.11. The Role of miR-24
5.2.12. The Role of miR-433
5.2.13. The Role of miR-146
5.2.14. The Role of miR-486
5.2.15. The Role of miR-132
5.2.16. The Role of miR-130
5.2.17. The Role of miR-195
5.3. The Role of miRNAs in Other Biological Processes during MI
5.3.1. The Role of miR-145
5.3.2. The Role of miR-34
5.3.3. The Role of miR-23
5.3.4. The Role of miR-126
5.3.5. The Role of miR-146
5.3.6. The Role of miR-22
5.3.7. The Role of miR-21
5.3.8. The Role of miR-155
5.3.9. The Role of miR-24
5.3.10. The Role of miR-208
5.3.11. The Role of miR-590
5.4. The Role of miRNAs during Cardiac Regeneration
5.4.1. The Role of miR-195
5.4.2. The Role of miR-126 and miR-146
5.4.3. The Role of miR-98
5.4.4. The Role of miR-22
5.4.5. The Roles of miR-1 and miR-29
6. LncRNAs in Cardiac Infarction and Regeneration
6.1. The Role of lncRNAs in Cardiac Inflammation
6.2. The Role of lncRNAs in Cardiac Fibrosis
6.3. The Role of lncRNAs in Other Biological Processes of Cardiac Infarction
6.4. The Role of lncRNAs in Cardiac Regeneration
7. circRNAs in Cardiac Infarction and Regeneration
7.1. The Role of circRNAs in Cardiac Inflammation
7.2. The Role of circRNAs in Cardiac Fibrosis
7.3. The Role of circRNAs in Other Biological Processes of Cardiac Infarction
7.4. The Role of circRNAs in Cardiac Regeneration
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhatt, A.S.; Ambrosy, A.P.; Velazquez, E.J. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Curr. Cardiol. Rep. 2017, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef] [PubMed]
- Basara, G.; Bahcecioglu, G.; Ozcebe, S.G.; Ellis, B.W.; Ronan, G.; Zorlutuna, P. Myocardial infarction from a tissue engineering and regenerative medicine point of view: A comprehensive review on models and treatments Myocardial infarction from a tissue engineering and regenerative medicine point of view: A comprehensive review on m. Biophys. Rev. 2022, 3, 031305. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G.; Service, M.; Einstein, A. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar]
- McCoy, C.E.; Sheedy, F.J.; Qualls, J.E.; Doyle, S.L.; Quinn, S.R.; Murray, P.J.; O’Neill, L.A.J. IL-10 inhibits miR-155 induction by toll-like receptors. J. Biol. Chem. 2010, 285, 20492–20498. [Google Scholar] [CrossRef]
- Chen, Q.; Lv, J.; Yang, W.; Xu, B.; Wang, Z.; Yu, Z.; Wu, J.; Yang, Y.; Han, Y. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis. Theranostics 2019, 9, 6424–6442. [Google Scholar] [CrossRef]
- Kyoko, I.O.; Taisuke, K.; Minako, I.; Yoshimura, A. SOCS, inflammation, and cancer. JAKSTAT 2013, 2, 324053. [Google Scholar]
- Zhu1, M.; Goetsch, S.C.; Wang, Z.; Luo, R.; Hill, J.A.; Schneider, J.; Morris, S.M., Jr.; Liu, Z.-P. FoxO4 Promotes Early Inflammatory Response Upon Myocardial Infarction via Endothelial Arg1. Circ. Res. 2015, 117, 967–977. [Google Scholar] [CrossRef]
- Mouton, A.J.; Rivera, O.J.; Lindsey, M.L. Myocardial infarction remodeling that progresses to heart failure: A signaling misunderstanding. Am. J. Physiol. Circ. Physiol. 2018, 315, H71–H79. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Slava, E.; Lavine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Ansuman, T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Skrzeczyńska-Moncznik, J.; Bzowska, M.; Loseke, S.; Grage-Griebenow, E.; Zembala, M.; Pryjma, J. Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand. J. Immunol. 2008, 67, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Kanti Barman, P.; Kumar Thatoi, P.; Tripathy, R.; Kumar Das, B.; Ravindran, B. Non-Classical monocytes display inflammatory features: Validation in Sepsis and Systemic Lupus Erythematous. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Eigsti, R.L.; Sudan, B.; Wilson, M.E.; Graff, J.W. Regulation of activation-associated microRNA accumulation rates during monocyte-to-macrophage differentiation. J. Biol. Chem. 2014, 289, 28433–28447. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 1–13. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Liu, L.; Xi, A.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef]
- Yonggang, M.; Alan, J.M.; Merry, L.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar]
- Nikolaos, G.; Frangogiannis, M. Inflammation in cardiac injury, repair and regeneration Nikolaos. Curr. Opin. Cardiol. 2015, 30, 240–245. [Google Scholar]
- Hu, Y.; Zhang, H.; Lu, Y.; Bai, H.; Xu, Y.; Zhu, X.; Zhou, R.; Ben, J.; Xu, Y.; Chen, Q. Class A scavenger receptor attenuates myocardial infarction-induced cardiomyocyte necrosis through suppressing M1 macrophage subset polarization. Basic Res. Cardiol. 2011, 106, 1311–1328. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Begeman, I.J.; Kang, J. Transcriptional programs and regeneration enhancers underlying heart regeneration. J. Cardiovasc. Dev. Dis. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Quaife-Ryan, G.A.; Sim, C.B.; Ziemann, M.; Kaspi, A.; Rafehi, H.; Ramialison, M.; El-Osta, A.; Hudson, J.E.; Porrello, E.R. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation 2017, 136, 1123–1139. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Serpooshan, V.; Hurtado, C.; Diez-Cuñado, M.; Zhao, M.; Maruyama, S.; Zhu, W.; Fajardo, G.; Noseda, M.; Nakamura, K.; et al. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 2015, 525, 479–485. [Google Scholar] [CrossRef]
- Gemberling, M.; Karra, R.; Dickson, A.L.; Poss, K.D. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife 2015, 2015, 1–17. [Google Scholar] [CrossRef]
- Marinescu, M.C.; Lazar, A.L.; Marta, M.M.; Cozma, A.; Catana, C.S. Non-Coding RNAs: Prevention, Diagnosis, and Treatment in Myocardial Ischemia–Reperfusion Injury. Int. J. Mol. Sci. 2022, 23, 2728. [Google Scholar] [CrossRef]
- Garcia-padilla, C.; Lozano-velasco, E.; Garcia-lopez, V.; Aranega, A.; Franco, D.; Garcia-martinez, V.; Lopez-sanchez, C. Comparative Analysis of Non-Coding RNA Transcriptomics in Heart Failure. Biomedicines 2022, 10, 3076. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease Joshua. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef]
- Fiedler, J.; Thum, T. MicroRNAs in myocardial infarction. Arter. Thromb. Vasc. Biol. 2013, 33, 201–205. [Google Scholar] [CrossRef]
- Varzideh, F.; Kansakar, U.; Donkor, K.; Wilson, S.; Jankauskas, S.S.; Mone, P.; Wang, X.; Lombardi, A.; Santulli, G. Cardiac Remodeling After Myocardial Infarction: Functional Contribution of microRNAs to Inflammation and Fibrosis. Front. Cardiovasc. Med. 2022, 9, 44–46. [Google Scholar] [CrossRef]
- Collins, L.; Binder, P.; Chen, H.; Wang, X. Regulation of Long Non-coding RNAs and MicroRNAs in Heart Disease: Insight Into Mechanisms and Therapeutic Approaches. Front. Physiol. 2020, 11, 798. [Google Scholar] [CrossRef]
- Meng, Z.; Chen, C.; Cao, H.; Wang, J.; Shen, E. Whole transcriptome sequencing reveals biologically significant RNA markers and related regulating biological pathways in cardiomyocyte hypertrophy induced by high glucose. J. Cell. Biochem. 2019, 120, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Altesha, M.A.; Ni, T.; Khan, A.; Liu, K.; Zheng, X. Circular RNA in cardiovascular disease. J. Cell. Physiol. 2019, 234, 5588–5600. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.J.; Xin, H.; Wang, Y.C.; Liu, H.W.; Gao, Y.Y.; Zhang, Y.F. Emerging roles of circRNAs in the pathological process of myocardial infarction. Mol. Ther. Nucleic Acids 2021, 26, 828–848. [Google Scholar] [CrossRef]
- Wang, C.; Jing, Q. Non-coding RNAs as biomarkers for acute myocardial infarction review-article. Acta Pharmacol. Sin. 2018, 39, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Yu, L.F.; Zhou, T.G.; Wang, Y.D.; Sun, D.H.; Chen, H.R.; Hou, Y.F. Lipopolysaccharide-stimulated bone marrow mesenchymal stem cells-derived exosomes inhibit H2O2-induced cardiomyocyte inflammation and oxidative stress via regulating miR-181a-5p/ATF2 axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10069–10077. [Google Scholar]
- Wei, Z.; Qiao, S.; Zhao, J.; Yihai, L.; Qiaoling, L.; Zhonghai, W.; Qing, D.; Lina, K.; Biao, X. miRNA-181a over-expression in mesenchymal stem cell-derived exosomes influenced inflammatory response after myocardial ischemia-reperfusion injury. Life Sci. 2019, 232, 116632. [Google Scholar] [CrossRef]
- Xiao, S.H.; Wang, Y.; Cao, X.; Su, Z. Long non-coding RNA LUCAT1 inhibits myocardial oxidative stress and apoptosis after myocardial infarction via targeting microRNA-181a-5p. Bioengineered 2021, 12, 4546–4555. [Google Scholar] [CrossRef]
- Tan, J.K.; Ma, X.F.; Wang, G.N.; Jiang, C.R.; Gong, H.Q.; Liu, H. LncRNA MIAT knockdown alleviates oxygen-glucose deprivation-induced cardiomyocyte injury by regulating JAK2/STAT3 pathway via miR-181a-5p. J. Cardiol. 2021, 78, 586–597. [Google Scholar] [CrossRef]
- Cao, R.Y.; Li, Q.; Miao, Y.; Zhang, Y.; Yuan, W.; Fan, L.; Liu, G.; Mi, Q.; Yang, J. The emerging role of MicroRNA-155 in cardiovascular diseases. BioMed Res. Int. 2016, 2016, 9869208. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Arase, M.; Matsuyama, H.; Choi, Y.L.; Ueno, T.; Mano, H.; Sugimoto, K.; Miyazono, K. MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol. Cell. 2011, 44, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Jin, Z.; Kim, H.; Kolattukudy, P.E. MCP-1-induced protein attenuates post-infarct cardiac remodeling and dysfunction through mitigating NF-κB activation and suppressing inflammation-associated microRNA expression. Basic Res. Cardiol. 2015, 110, 26. [Google Scholar] [CrossRef]
- Agudelo, J.S.H.; Braga, T.T.; Amano, M.T.; Cenedeze, M.A.; Cavinato, R.A.; Peixoto-Santos, A.R.; Muscará, M.N.; Teixeira, S.A.; Cruz, M.C.; Castoldi, A.; et al. Mesenchymal stromal cell-derived microvesicles regulate an internal pro-inflammatory program in activated macrophages. Front. Immunol. 2017, 8, 881. [Google Scholar] [CrossRef]
- Essandoh, K.; Li, Y.; Huo, J.; Fan, G.C. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock 2016, 46, 122–131. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Wu, Y.; Han, Y.; Cui, W.; Jia, L.; Cai, L.; Cheng, J.; Li, H.; Du, J. Interleukin-12p35 deletion promotes CD4 T-cell-dependent macrophage differentiation and enhances angiotensin II-Induced cardiac fibrosis. Arter. Thromb. Vasc. Biol. 2012, 32, 1662–1674. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Cui, X.; Zhang, J.; Li, Y.; Li, J.; Zang, Y.; Li, Q.; Yang, Q.; Chen, Y.; Cai, W.; et al. Shenlian extract attenuates myocardial ischaemia-reperfusion injury via inhibiting M1 macrophage polarization by silencing miR-155. Pharm. Biol. 2022, 60, 2011–2024. [Google Scholar] [CrossRef]
- Matsushima, S.; Tsutsui, H.; Sadoshima, J. Physiological and Pathological Functions of NADPH Oxidases during Myocardial Ischemia-Reperfusion. Trends Cardiovasc. Med. 2014, 24, 202–205. [Google Scholar] [CrossRef]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate in Vivo and in Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, J.; Shi, J.; Zhou, W.; Wang, L.; Fang, W.; Zhong, Y.; Chen, X.; Chen, Y.; Sabri, A.; et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res. Cardiol. 2020, 115, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Li, X.; Tang, Z.; Wang, X.; Zhong, M.; Suo, Q.; Zhang, Y.; Lv, K. Silencing MicroRNA-155 Attenuates Cardiac Injury and Dysfunction in Viral Myocarditis via Promotion of M2 Phenotype Polarization of Macrophages. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef]
- Zidar, N.; Boštjančič, E.; Glavač, D.; Štajer, D. MicroRNAs, innate immunity and ventricular rupture in human myocardial infarction. Dis. Markers. 2011, 31, 259–265. [Google Scholar] [CrossRef]
- He, W.; Huang, H.; Xie, Q.; Wang, Z.; Fan, Y.; Kong, B.; Huang, D.; Xiao, Y. MIR-155 knockout in fibroblasts improves cardiac remodeling by targeting tumor protein p53-Inducible nuclear protein 1. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 423–435. [Google Scholar] [CrossRef]
- Yang, L.; Wang, B.; Zhou, Q.; Wang, Y.; Liu, X.; Liu, Z.; Zhan, Z. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Canfrán-Duque, A.; Rotllan, N.; Zhang, X.; Fernández-Fuertes, M.; Ramírez-Hidalgo, C.; Araldi, E.; Daimiel, L.; Busto, R.; Fernández-Hernando, C.; Suárez, Y. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol. Med. 2017, 9, 1244–1262. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, M.; Duan, S.; Zhao, Z.; Wu, X.; Chen, Y.; Yin, L. Macrophage-targeting gene silencing orchestrates myocardial microenvironment remodeling toward the anti-inflammatory treatment of ischemia-reperfusion (IR) injury. Bioact. Mater. 2022, 17, 320–333. [Google Scholar] [CrossRef]
- Bejerano, T.; Etzion, S.; Elyagon, S.; Etzion, Y.; Cohem, S. Nanoparticle Delivery of miRNA-21 mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Lett. 2018, 18, 5885–5891. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Jin, R.; Chen, L.; Dang, M.; Cao, H.; Dong, Y.; Cai, B.; Bai, G.; Justin Gooding, J.; et al. Injectable hydrogel with MSNs/microRNA-21-5p delivery enables both immunomodification and enhanced angiogenesis for myocardial infarction therapy in pigs. Sci. Adv. 2021, 7, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Tuccoli, A.; Mariani, L.; Evangelista, M.; Citti, L.; Woods, K.; Mercatanti, A.; Hammond, S.; Rainaldi, G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood 2006, 108, 3068–3071. [Google Scholar] [CrossRef]
- Sahoo, S.; Losordo, D.W. Exosomes and cardiac repair after myocardial infarction. Circ. Res. 2014, 114, 333–344. [Google Scholar] [CrossRef] [PubMed]
- de Jong, O.G.; van Balkom, B.W.M.; Schiffelers, R.M.; Bouten, C.V.C.; Verhaar, M.C. Extracellular vesicles: Potential roles in regenerative medicine. Front. Immunol. 2014, 5, 1–13. [Google Scholar] [CrossRef]
- Wei, Z.; Chen, Z.; Zhao, Y.; Fan, F.; Xiong, W.; Song, S.; Yin, Y.; Hu, J.; Yang, K.; Yang, L.; et al. Mononuclear phagocyte system blockade using extracellular vesicles modified with CD47 on membrane surface for myocardial infarction reperfusion injury treatment. Biomaterials 2021, 275, 121000. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dong, Y.; Chen, S.; Zhang, G.; Zhang, M.; Gong, Y.; Li, X. Circulating MicroRNA-146a and MicroRNA-21 Predict Left Ventricular Remodeling after ST-Elevation Myocardial Infarction. Cardiology 2015, 132, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Saba, R.; Sorensen, D.L.; Booth, S.A. MicroRNA-146a: A dominant, negative regulator of the innate immune response. Front. Immunol. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Liang, C.; Hua, J.; Zhang, B.; Liu, J.; Zhang, Y.; Wei, M.; Yu, X.; Xu, J.; Shi, S. A miR-146a-5p/TRAF6/NF-kB p65 axis regulates pancreatic cancer chemoresistance: Functional validation and clinical significance. Theranostics 2020, 10, 3967–3979. [Google Scholar] [CrossRef]
- Mukundan, L.; Bishop, G.A.; Head, K.Z.; Zhang, L.; Wahl, L.M.; Suttles, J. TNF receptor-assocaited factor 6 is an essential mediator of CD40-Activated Proinflammatory Pathways in Monocytes and Macrophages. J. Immunol. 2005, 174, 1081–1090. [Google Scholar] [CrossRef]
- Chen, J.; Chen, T.; Zhou, J.; Zhao, X.; Sheng, Q.; Lv, Z. MiR-146a-5p Mimic Inhibits NLRP3 Inflammasome Downstream Inflammatory Factors and CLIC4 in Neonatal Necrotizing Enterocolitis. Front. Cell Dev. Biol. 2021, 8, 594143. [Google Scholar] [CrossRef]
- Shimada, B.K.; Yang, Y.; Zhu, J.; Wang, S.; Suen, A.; Kronstadt, S.M.; Jeyaram, A.; Jay, S.M.; Zou, L.; Chao, W. Extracellular miR-146a-5p Induces Cardiac Innate Immune Response and Cardiomyocyte Dysfunction. ImmunoHorizons 2020, 4, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Ye, E.A.; Steinle, J.J. MiR-146a Attenuates Inflammatory Pathways Mediated by TLR4/NF-B and TNF α to Protect Primary Human Retinal Microvascular Endothelial Cells Grown in High Glucose. Mediat. Inflamm. 2016, 2016, 3958453. [Google Scholar] [CrossRef]
- Chen, C.; Cai, S.; Wu, M.; Wang, R.; Liu, M.; Cao, G.; Dong, M.; Yiu, K.H. Role of Cardiomyocyte-Derived Exosomal MicroRNA-146a-5p in Macrophage Polarization and Activation. Dis. Markers 2022, 2022, 2948578. [Google Scholar] [CrossRef]
- Ibrahim, A.G.E.; Cheng, K.; Marbán, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Li, H.; Cao, H.; Dong, Y.; Gao, L.; Liu, Z.; Ge, J.; Zhu, H. Therapeutic silencing miR-146b-5p improves cardiac remodeling in a porcine model of myocardial infarction by modulating the wound reparative phenotype. Protein Cell 2021, 12, 194–212. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczyk, E.; Eljaszewicz, A.; Zembko, P.; Tarasiuk, E.; Rusak, M.; Kulczynska-Przybik, A.; Lukaszewicz-Zajac, M.; Kaminski, K.; Mroczko, B.; Szmitkowski, M.; et al. The relationships among monocyte subsets, miRNAs and inflammatory cytokines in patients with acute myocardial infarction. Pharmacol. Rep. 2019, 71, 73–81. [Google Scholar] [CrossRef]
- Koga, Y.; Yasunaga, M.; Moriya, Y.; Akasu, T.; Fujita, S.; Yamamoto, S.; Matsumura, Y. Exosome can Prevent RNase from Degrading MicroRNA in Feces. J. Gastrointest. Oncol. 2011, 2, 215–222. [Google Scholar]
- Zhou, J.; Chaudhry, H.; Zhong, Y.; Ali, M.M.; Perkin, L.A.; Owens, W.B.; Morales, J.E.; McGuire, F.R.; Zumbrun, E.E.; Zhang, J.; et al. Dysregulation in microRNA Expression in Peripheral Blood Mononuclear Cells of Sepsis Patients is Associated with Immunopathology. Cytokine 2015, 71, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Tian, S.S.; Hang, P.Z.; Sun, C.; Guo, J.; Du, Z.M. Combination of microRNA-21 and microRNA-146a Attenuates Cardiac Dysfunction and Apoptosis During Acute Myocardial Infarction in Mice. Mol. Ther. Nucleic Acids 2016, 5, e296. [Google Scholar] [CrossRef]
- Zhu, Y.; Zou, C.; Jia, Y.; Zhang, H.; Ma, X.; Zhang, J. Knockdown of circular RNA circMAT2B reduces oxygen-glucose deprivation-induced inflammatory injury in H9c2 cells through up-regulating miR-133. Cell Cycle 2020, 19, 2622–2630. [Google Scholar] [CrossRef]
- Bian, Y.; Pang, P.; Li, X.; Yu, S.; Wang, X.; Liu, K.; Ju, J.; Wu, H.; Gao, Y.; Liu, Q.; et al. CircHelz activates NLRP3 inflammasome to promote myocardial injury by sponging miR-133a-3p in mouse ischemic heart. J. Mol. Cell. Cardiol. 2021, 158, 128–139. [Google Scholar] [CrossRef]
- Chu, X.; Wang, Y.; Pang, L.; Huang, J.; Sun, X.; Chen, X. miR-130 aggravates acute myocardial infarction-induced myocardial injury by targeting PPAR-γ. J. Cell. Biochem. 2018, 119, 7235–7244. [Google Scholar] [CrossRef]
- Xin, H.; Li, C.; Cai, T.; Cao, J.; Wang, M. LncRNA KCNQ1OT1 contributes to hydrogen peroxide-induced apoptosis, inflammation, and oxidative stress of cardiomyocytes via miR-130a-3p/ZNF791 axis. Cell Biol. Int. 2022, 46, 2018–2027. [Google Scholar] [CrossRef]
- Yang, J.; Xiang, Z.; Zhang, J.; Yang, J.; Zhai, Y. miR-24 Alleviates MI/RI by Blocking the S100A8/TLR4/MyD88/NF-kB Pathway. J. Cardiovasc. Pharmacol. 2021, 78, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; Shang, C.; Meng, L.R. Targeted silencing of S100A8 gene by miR-24 to increase chemotherapy sensitivity of endometrial carcinoma cells to paclitaxel. Med. Sci. Monit. 2016, 22, 1953–1958. [Google Scholar] [CrossRef]
- Pruenster, M.; Vogl, T.; Roth, J.; Sperandio, M. S100A8/A9: From basic science to clinical application. Pharmacol. Ther. 2016, 167, 120–131. [Google Scholar] [CrossRef]
- Qiao, S.; Zhang, W.; Yin, Y.; Wei, Z.; Chen, F.; Zhao, J.; Sun, X.; Mu, D.; Xie, J.; Xu, B. Extracellular vesicles derived from krüppel-Like factor 2-overexpressing endothelial cells attenuate myocardial ischemia-reperfusion injury by preventing Ly6Chigh monocyte recruitment. Theranostics 2020, 10, 11562–11579. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Wang, X.; Li, Y.; Yang, L.; Wang, R.; Peng, J.; Mu, K.E.X.; Peng, T.; Han, Q.; Yu, K.J.; et al. MicroRNA-223-5p and -3p Cooperatively suppress necroptosis in ischemic/reperfused hearts. J. Biol. Chem. 2016, 291, 20247–20259. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, J.; Guo, M.; Hao, M. MiR-223-3p affects myocardial inflammation and apoptosis following myocardial infarction via targeting FBXW7. J. Thorac. Dis. 2022, 14, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Kuai, X.; Li, L.; Chen, R.; Wang, K.; Chen, M.; Cui, B.; Zhang, Y.; Li, J.; Zhu, H.; Zhou, H.; et al. SCFFBXW7/GSK3β-mediated GFI1 degradation suppresses proliferation of gastric cancer cells. Cancer Res. 2019, 79, 4387–4398. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Cui, T.; Wang, Q.; Xu, Y.; Miao, C.; Liu, S. LncRNA FGD5-AS1 reduces cardiomyocyte apoptosis and inflammation by modulating Akt and miR-223-3p expression. Am. J. Transl. Res. 2022, 14, 6175–6186. [Google Scholar]
- Yao, J.; Shi, Z.; Ma, X.; Xu, D.; Ming, G. lncRNA GAS5/miR-223/NAMPT axis modulates the cell proliferation and senescence of endothelial progenitor cells through PI3K/AKT signaling. J. Cell. Biochem. 2019, 120, 14518–14530. [Google Scholar] [CrossRef]
- Kain, V.; Ingle, K.A.; Rajasekaran, N.S.; Halade, G.V. Activation of EP4 receptor limits transition of acute to chronic heart failure in lipoxygenase deficient mice. Theranostics 2021, 11, 2742–2754. [Google Scholar] [CrossRef]
- Yang, J.; Fan, Z.; Yang, J.; Ding, J.; Yang, C.; Chen, L. MicroRNA-22 attenuates myocardial ischemia-reperfusion injury via an anti-inflammatory mechanism in rats. Exp. Ther. Med. 2016, 12, 3249–3255. [Google Scholar] [CrossRef]
- Forini, F.; Kusmic, C.; Nicolini, G.; Mariani, L.; Zucchi, R.; Matteucci, M.; Iervasi, G.; Pitto, L. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology 2014, 155, 4581–4590. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem 2005, 280, 16891–16900. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Guo, M.; Ma, G.; Zhang, N.; Pan, F.; Fan, X.; Wang, R. MicroRNA-30c-5p protects against myocardial ischemia/reperfusion injury via regulation of Bach1/Nrf2. Toxicol. Appl. Pharmacol. 2021, 426, 115637. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, M.; Zhang, S.; Wu, J.; Xue, S. Rno-microRNA-30c-5p promotes myocardial ischemia reperfusion injury in rats through activating NF-κB pathway and targeting SIRT1. BMC Cardiovasc. Disord. 2020, 20, 240. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, M.; Ni, T.; Liu, Q. miR-214 mediates vascular inflammation and apoptosis via PTEN expression. Mol. Med. Rep. 2018, 18, 2229–2236. [Google Scholar] [CrossRef]
- Chen, Y.; Yin, Y.; Jiang, H. miR-30e-5p Alleviates Inflammation and Cardiac Dysfunction After Myocardial Infarction Through Targeting PTEN. Inflammation 2021, 44, 769–779. [Google Scholar] [CrossRef]
- Zhu, J.; Yao, K.; Wang, Q.; Guo, J.; Shi, H.; Ma, L.; Liu, H.; Gao, W.; Zou, Y.; Ge, J. Ischemic Postconditioning-Regulated miR-499 Protects the Rat Heart Against Ischemia/Reperfusion Injury by Inhibiting Apoptosis through PDCD4. Cell. Physiol. Biochem. 2016, 39, 2364–2380. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Huang, Z.; Li, Q.J.; Zhong, G.Q.; Meng, J.J.; Wang, D.X.; Tu, R.H. Ischemic postconditioning attenuates the inflammatory response in ischemia/reperfusion myocardium by upregulating miR-499 and inhibiting TLR2 activation. Mol. Med. Rep. 2020, 22, 209–218. [Google Scholar] [CrossRef]
- Ha, T.; Hu, Y.; Liu, L.; Lu, C.; McMullen, J.R.; Kelley, J.; Kao, R.L.; Williams, D.L.; Gao, X.; Li, C. TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Cardiovasc. Res. 2010, 87, 694–703. [Google Scholar] [CrossRef]
- Wang, Y.; Ge, P.; Yang, L.; Wu, C.; Zha, H.; Luo, T.; Zhu, Y. Protection of ischemic post conditioning against transient focal ischemia-induced brain damage is associated with inhibition of neuroinflammation via modulation of TLR2 and TLR4 pathways. J. Neuroinflammation 2014, 11, 15. [Google Scholar] [CrossRef]
- Zhou, R.; Huang, W.; Fan, X.; Liu, F.; Luo, L.; Yuan, H.; Jiang, Y.; Xiao, H.; Zhou, Z.; Deng, C.; et al. miR-499 released during myocardial infarction causes endothelial injury by targeting α7-nAchR. J. Cell. Mol. Med. 2019, 23, 6085–6097. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yu, Y. MiR-486 alleviates hypoxia/reoxygenation-induced H9c2 cell injury by regulating forkhead box D3. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 422–431. [Google Scholar] [PubMed]
- Huang, Y.; Qi, Y.; Du, J.Q.; Zhang, D.F. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin. Ther. Targets 2014, 18, 1355–1365. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Y.; Zhu, H.; Hu, J.; Xie, Z. MiR-34a/miR-93 target c-Ski to modulate the proliferaton of rat cardiac fibroblasts and extracellular matrix deposition in vivo and in vitro. Cell. Signal. 2018, 46, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, S.; Li, X.; Gong, M. LncRNA SNHG7 promotes cardiac remodeling by upregulating ROCK1 via sponging miR-34-5p. Aging 2020, 12, 10441–10456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Li, S.H.; Guo, J.; Mihic, A.; Wu, J.; Sun, L.; Davis, K.; Weisel, R.D.; Li, R.K. Role of miR-145 in cardiac myofibroblast differentiation. J. Mol. Cell. Cardiol. 2014, 66, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Song, H.F.; He, S.; Li, S.H.; Wu, J.; Yin, W.; Shao, Z.; Du, G.-Q.; Wu, J.; Li, J.; Weisel, R.D.; et al. Knock-out of MicroRNA 145 impairs cardiac fibroblast function and wound healing post-myocardial infarction. J. Cell. Mol. Med. 2020, 24, 9409–9419. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Liu, Z.; Tao, B.; Fan, S.; Pu, Y.; Meng, X.; Li, D.; Xia, H.; Xu, L. miR-145 attenuates cardiac fibrosis through the AKT/GSK-3β/β-catenin signaling pathway by directly targeting SOX9 in fibroblasts. J. Cell. Biochem. 2021, 122, 209–221. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, L.; Song, J.; Wang, Z.; Huang, X.; Guo, Z.; Chen, F.; Zhao, X. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J. Cell. Physiol. 2019, 234, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, E.; Ikeda, G.; Tada, Y.; Wahlquist, C.; Mercola, M.; Yang, P.C. Sacubitril/valsartan improves cardiac function and decreases myocardial fibrosis via downregulation of exosomal mir-181a in a rodent chronic myocardial infarction model. J. Am. Heart Assoc. 2020, 9, e015640. [Google Scholar] [CrossRef]
- Chen, P.; Pan, J.; Zhang, X.; Shi, Z.; Yang, X. The role of microRNA-181a in myocardial fibrosis following myocardial infarction in a rat model. Med. Sci. Monit. 2018, 24, 4121–4127. [Google Scholar] [CrossRef] [PubMed]
- Bruen, R.; Fitzsimons, S.; Belton, O. MiR-155 in the resolution of atherosclerosis. Front. Pharmacol. 2019, 10, 463. [Google Scholar] [CrossRef]
- Schumacher, D.; Curaj, A.; Simsekyilmaz, S.; Schober, A.; Liehn, E.A.; Mause, S.F. Mir155 deficiency reduces myofibroblast density but fails to improve cardiac function after myocardial infarction in dyslipidemic mouse model. Int. J. Mol. Sci. 2021, 22, 5480. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, N.; Ma, Q.; Fan, F.; Ma, X. LncRNA TUG1 acts as a competing endogenous RNA to mediate CTGF expression by sponging miR-133b in myocardial fibrosis after myocardial infarction. Cell. Biol. Int. 2021, 45, 2534–2543. [Google Scholar] [CrossRef]
- Arango, D.; Diosa-toro, M.; Rojas-hernandez, L.S.; Jessica, L.; Schwartz, S.J.; Mo, X.; Jiang, J.; Schmittgen, T.D.; Doseff, A.I. Dietary apigenin reduces LPS-induced expression of miR-155 restoring immune balance during inflammation. Mol. Nutr. Food Res. 2015, 59, 763–772. [Google Scholar] [CrossRef]
- Eissa, M.G.; Artlett, C.M. The microRNA miR-155 is essential in fibrosis. Non-Coding RNA 2019, 5, 23. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, J.; Liu, F.; Zhang, J. Long non-coding RNA XIST promotes the proliferation of cardiac fibroblasts and the accumulation of extracellular matrix by sponging microRNA-155-5p. Exp. Ther. Med. 2021, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dakhlallah, D.; Zhang, J.; Yu, L.; Marsh, C.B.; Angelos, M.G.; Khan, M. MicroRNA-133a engineered mesenchymal stem cells augment cardiac function and cell survival in the infarct heart. J. Cardiovasc. Pharmacol. 2015, 65, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.T.; Yu, N.; Wang, Y.; Zhang, H.; Wan, K.; Sun, X.; Zhang, C.S. Role of MIR-133a in regulating TGF-β1 signaling pathway in myocardial fibrosis after acute myocardial infarction in rats. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8588–8597. [Google Scholar] [PubMed]
- Zhu, W.; Sun, L.; Zhao, P.; Liu, Y.; Zhang, J.; Zhang, Y.; Hong, Y.; Zhu, Y.; Lu, Y.; Zhao, W.; et al. Macrophage migration inhibitory factor facilitates the therapeutic efficacy of mesenchymal stem cells derived exosomes in acute myocardial infarction through upregulating miR-133a-3p. J. Nanobiotechnology 2021, 19, 1–16. [Google Scholar] [CrossRef]
- Hong, Y.; Cao, H.; Wang, Q.; Ye, J.; Sui, L.; Feng, J.; Cai, X.; Song, H.; Zhang, X.; Chen, X. MiR-22 may Suppress Fibrogenesis by Targeting TGFβR i in Cardiac Fibroblasts. Cell. Physiol. Biochem. 2016, 40, 1345–1353. [Google Scholar] [CrossRef]
- Zhang, L.; Yin, H.; Jiao, L.; Liu, T.; Gao, Y.; Shao, Y.; Zhang, Y.; Shan, H.; Zhang, Y.; Yang, B. Abnormal Downregulation of Caveolin-3 Mediates the Pro-Fibrotic Action of MicroRNA-22 in a Model of Myocardial Infarction. Cell. Physiol. Biochem. 2018, 45, 1641–1653. [Google Scholar] [CrossRef]
- Zhao, X.S.; Ren, Y.; Wu, Y.; Ren, H.K.; Chen, H. MiR-30b-5p and miR-22-3p restrain the fibrogenesis of post-myocardial infarction in mice via targeting PTAFR. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3993–4004. [Google Scholar]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis after Myocardial Infarction Via Targeting Smad7. Cell. Physiol. Biochem. 2017, 42, 2207–2219. [Google Scholar] [CrossRef]
- Cao, W.; Shi, P.; Ge, J.J. miR-21 enhances cardiac fibrotic remodeling and fibroblast proliferation via CADM1/STAT3 pathway. BMC Cardiovasc. Disord. 2017, 17, 1–11. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, S.; Chen, Y. Unraveling the biological functions of Smad7 with mouse models. Cell Biosci. 2011, 1, 44. [Google Scholar] [CrossRef]
- Dong, X.; Liu, S.; Zhang, L.; Yu, S.; Huo, L.; Qile, M.; Liu, L.; Yang, B.; Yu, J. Downregulation of miR-21 is Involved in Direct Actions of Ursolic Acid on the Heart: Implications for Cardiac Fibrosis and Hypertrophy. Cardiovasc. Ther. 2015, 33, 161–167. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, B.; Xu, Y.; Zhou, N.; Zhang, R.; Lu, L.; Feng, Z. MiR-208b/miR-21 Promotes the Progression of Cardiac Fibrosis Through the Activation of the TGF-β1/Smad-3 Signaling Pathway: An in vitro and in vivo Study. Front. Cardiovasc. Med. 2022, 9, 1–10. [Google Scholar] [CrossRef]
- Chiang, M.H.; Liang, C.J.; Lin, L.C.; Yang, Y.F.; Huang, C.C.; Chen, Y.H.; Kao, H.L.; Chen, Y.C.; Ke, S.R.; Lee, C.W.; et al. miR-26a attenuates cardiac apoptosis and fibrosis by targeting ataxia–telangiectasia mutated in myocardial infarction. J. Cell. Physiol. 2020, 235, 6085–6102. [Google Scholar] [CrossRef]
- Ge, Z.W.; Zhu, X.L.; Wang, B.C.; Hu, J.L.; Sun, J.J.; Wang, S.; Chen, X.J.; Meng, S.P.; Liu, L.; Cheng, Z.Y. MicroRNA-26b relieves inflammatory response and myocardial remodeling of mice with myocardial infarction by suppression of MAPK pathway through binding to PTGS2. Int. J. Cardiol. 2019, 280, 152–159. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Fan, X.; Yang, R.; Jiao, Y.; Li, Y. miR-29b-3p inhibits post-infarct cardiac fibrosis by targeting FOS. Biosci. Rep. 2020, 40, 1–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, X.R.; Wei, L.H.; Chung, A.C.; Yu, C.M.; Lan, H.Y. MiR-29b as a therapeutic agent for angiotensin ii-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol. Ther. 2014, 22, 974–985. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, B.J.; Chen, Q.; Yan, B.J.; Liu, Z.L. MicroRNA-29b upregulation improves myocardial fibrosis and cardiac function in myocardial infarction rats through targeting SH2B3. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10115–10122. [Google Scholar]
- Wang, R.; Peng, L.; Lv, D.; Shang, F.; Yan, J.; Li, G.; Li, D.; Ouyang, J.; Yang, J. Leonurine Attenuates Myocardial Fibrosis Through Upregulation of miR-29a-3p in Mice Post-myocardial Infarction. J. Cardiovasc. Pharmacol. 2021, 77, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, P.; Li, H.; Shi, Q.; Li, S.; Zhao, L. MicroRNA-29b mediates the antifibrotic effect of tanshinone IIA in postinfarct cardiac remodeling. J. Cardiovasc. Pharmacol. 2015, 65, 456–464. [Google Scholar] [CrossRef]
- Zhu, J.N.; Chen, R.; Fu, Y.H.; Lin, Q.X.; Huang, S.; Guo, L.L.; Zhang, M.Z.; Deng, C.Y.; Zou, X.; Zhong, S.L.; et al. Smad3 Inactivation and MiR-29b Upregulation Mediate the Effect of Carvedilol on Attenuating the Acute Myocardium Infarction-Induced Myocardial Fibrosis in Rat. PLoS ONE 2013, 8, 1–10. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, Y.; Wang, C.X. LncRNA-MIAT regulates fibrosis in hypertrophic cardiomyopathy (HCM) by mediating the expression of miR-29a-3p. J. Cell. Biochem. 2019, 120, 7265–7275. [Google Scholar] [CrossRef]
- Chen, L.; Ji, Q.; Zhu, H.; Ren, Y.; Fan, Z.; Tian, N. miR-30a attenuates cardiac fibrosis in rats with myocardial infarction by inhibiting CTGF. Exp. Ther. Med. 2018, 15, 4318–4324. [Google Scholar] [CrossRef]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; Van Der Made, I.; Herias, V.; Van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. MiR-133 and miR-30 Regulate connective tissue growth factor: Implications for a role of micrornas in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef]
- Li, J.; Salvador, A.M.; Li, G.; Valkov, N.; Ziegler, O.; Yeri, A.; Xiao, C.Y.; Meechoovet, B.; Alsop, E.; Rodosthenous, R.S.; et al. Mir-30d Regulates Cardiac Remodeling by Intracellular and Paracrine Signaling. Circ. Res. 2021, 128, E1–E23. [Google Scholar] [CrossRef]
- Wang, X.; Yong, C.; Yu, K.; Yu, R.; Zhang, R.; Yu, L.; Li, S.; Cai, S. Long noncoding RNA (lncRNA) n379519 promotes cardiac fibrosis in post-infarct myocardium by targeting mir-30. Med. Sci. Monit. 2018, 24, 3958–3965. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, W.; Xu, R.; Nie, Y.; Cao, X.; Meng, J.; Xu, X.; Hu, S.; Zheng, Z. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J. Cell. Mol. Med. 2012, 16, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Bei, Y.; Chen, P.; Lei, Z.; Fu, S.; Zhang, H.; Xu, J.; Che, L.; Chen, X.; Sluijter, J.P.G.; et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics 2016, 6, 2068–2083. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Liu, Q.; Ma, X.; Bai, S.; Chen, P.; Zhao, Y.; Bai, C.; Liu, Y.; Liu, K.; Xin, M.; et al. MicroRNA-146b-5p promotes atrial fibrosis in atrial fibrillation by repressing TIMP4. J. Cell. Mol. Med. 2021, 25, 10543–10553. [Google Scholar] [CrossRef] [PubMed]
- Shafei, S.; Khanmohammadi, M.; Ghanbari, H.; Nooshabadi, V.T.; Tafti, S.H.A.; Rabbani, S.; Kasaiyan, M.; Basiri, M.; Tavoosidana, G. Effectiveness of exosome mediated miR-126 and miR-146a delivery on cardiac tissue regeneration. Cell Tissue Res. 2022, 390, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lv, L.; Liang, R.; Guo, W.; Liao, Z.; Chen, Y.; Zhu, K.; Huang, R.; Zhao, H.; Pu, Q.; et al. miR-486 improves fibrotic activity in myocardial infarction by targeting SRSF3/p21-Mediated cardiac myofibroblast senescence. J. Cell. Mol. Med. 2022, 26, 5135–5149. [Google Scholar] [CrossRef]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles fromhuman cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function aftermyocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, R.; Ruan, Z.; Liu, L.; Li, Y.; Zhu, L. MicroRNA-132 attenuated cardiac fibrosis in myocardial infarction-induced heart failure rats. Biosci. Rep. 2020, 40, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Bao, Y.; Ding, J.; Li, H.; Liu, W.; Wang, X.; Guan, H.; Chen, Z. MicroRNA-130a attenuates cardiac fibrosis after myocardial infarction through TGF-β/Smad signaling by directly targeting TGF-β receptor 1. Bioengineered 2022, 13, 5779–5791. [Google Scholar] [CrossRef]
- Morelli, M.B.; Shu, J.; Sardu, C.; Matarese, A.; Santulli, G. Cardiosomal microRNAs are essential in post-infarction myofibroblast phenoconversion. Int. J. Mol. Sci. 2020, 21, 201. [Google Scholar] [CrossRef]
- Yuan, M.; Zhang, L.; You, F.; Zhou, J.; Ma, Y.; Yang, F.; Tao, L. MiR-145-5p regulates hypoxia-induced inflammatory response and apoptosis in cardiomyocytes by targeting CD40. Mol. Cell. Biochem. 2017, 431, 123–131. [Google Scholar] [CrossRef]
- Sun, N.; Meng, F.; Xue, N.; Pang, G.; Wang, Q.; Ma, H. Inducible miR-145 expression by HIF-1α protects cardiomyocytes against apoptosis via regulating SGK1 in simulated myocardial infarction hypoxic microenvironment. Cardiol. J. 2018, 25, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cao, H.; Zhu, G.; Liu, S.; Li, H. Overexpression of microRNA-145 protects against rat myocardial infarction through targeting PDCD4. Am. J. Transl. Res. 2017, 9, 5003–5011. [Google Scholar] [PubMed]
- Tan, L.; Liu, L.; Yao, J.; Piao, C. MiR-145-5p attenuates inflammatory response and apoptosis in myocardial ischemia-reperfusion injury by inhibiting (NADPH) oxidase homolog 1. Exp. Anim. 2021, 70, 311–321. [Google Scholar] [CrossRef]
- Liang, C.; Wang, S.; Zhao, L.; Han, Y.; Zhang, M. Effects of miR-145-5p on cardiomyocyte proliferation and apoptosis, GIGYF1 expression and oxidative stress response in rats with myocardial ischemia-reperfusion. Cell. Mol. Biol. 2022, 68, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheng, H.W.; Qiu, Y.; Dupee, D.; Noonan, M.; Lin, Y.D.; Fisch, S.; Unno, K.; Sereti, K.I.; Liao, R. MicroRNA-34a Plays a Key Role in Cardiac Repair and Regeneration Following Myocardial Infarction. Circ. Res. 2015, 117, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kang, W.S.; Hong, M.H.; Choe, N.; Kook, H.; Jeong, H.C.; Kang, J.; Hur, J.; Jeong, M.H.; Kim, Y.S.; et al. Involvement of miR-34c in high glucose-insulted mesenchymal stem cells leads to inefficient therapeutic effect on myocardial infarction. Cell. Signal. 2015, 27, 2241–2251. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Q.; Diao, J.; Lin, L.; Wei, J. MiR-23a Is Involved in Myocardial Ischemia/Reperfusion Injury by Directly Targeting CX43 and Regulating Mitophagy. Inflammation 2021, 44, 1581–1591. [Google Scholar] [CrossRef]
- Song, Y.S.; Joo, H.W.; Park, I.H.; Shen, G.Y.; Lee, Y.; Shin, J.H.; Kim, H.; Kim, K.S. Bone marrow mesenchymal stem cell-derived vascular endothelial growth factor attenuates cardiac apoptosis via regulation of cardiac miRNA-23a and miRNA-92a in a rat model of myocardial infarction. PLoS ONE 2017, 12, e0179972. [Google Scholar] [CrossRef]
- Luo, Q.; Guo, D.; Liu, G.; Chen, G.; Hang, M.; Jin, M. Exosomes from MiR-126-Overexpressing Adscs Are Therapeutic in Relieving Acute Myocardial Ischaemic Injury. Cell. Physiol. Biochem. 2018, 44, 2105–2116. [Google Scholar] [CrossRef]
- Xiao, L.; Gu, Y.; Ren, G.; Chen, L.; Liu, L.; Wang, X.; Gao, L. MiRNA-146a Mimic Inhibits NOX4/P38 Signalling to Ameliorate Mouse Myocardial Ischaemia Reperfusion (I/R) Injury. Oxidative Med. Cell. Longev. 2021, 2021, 6366254. [Google Scholar] [CrossRef]
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 628991. [Google Scholar] [CrossRef]
- Wang, S.; Ding, L.; Ji, H.; Xu, Z.; Liu, Q.; Zheng, Y. The role of p38 MAPK in the development of diabetic cardiomyopathy. Int. J. Mol. Sci. 2016, 17, 1037. [Google Scholar] [CrossRef]
- Yang, J.; Yang, J.; Chen, L.; Ding, J.; Li, S.; Wu, H.; Zhang, J.; Fan, Z.; Dong, W.; Li, X. MicroRNA-22 targeting CBP protects against myocardial ischemia-reperfusion injury through anti-apoptosis in rats. Mol. Biol. Rep. 2014, 41, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Cowan, D.B.; Wang1, D.Z. Non-coding RNAs in cardiac regeneration: Mechanism of action and therapeutic potential. Semin. Cell Dev. Biol. 2021, 118, 150–162. [Google Scholar] [CrossRef]
- Qiao, L.; Hu, S.; Liu, S.; Zhang, H.; Ma, H.; Huang, K.; Li, Z.; Su, T.; Vandergriff, A.; Tang, J.; et al. MicroRNA-21-5p dysregulation in exosomes derived from heart failure patients impairs regenerative potential. J. Clin. Investig. 2019, 129, 2237–2250. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Chen, Y.; Duan, C.; Zhu, K.; Huang, R.; Zhao, H.; Hintze, M.; Pu, Q.; Yuan, Z.; Lv, L.; et al. Cardiac telocytes inhibit cardiac microvascular endothelial cell apoptosis through exosomal miRNA-21-5p-targeted cdip1 silencing to improve angiogenesis following myocardial infarction. Theranostics 2020, 11, 268–291. [Google Scholar] [CrossRef]
- Asangani, I.A.; Rasheed, S.A.K.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef]
- Lu, Z.; Liu, M.; Stribinskis, V.; Klinge, C.M.; Ramos, K.S.; Colburn, N.H.; Li, Y. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008, 27, 4373–4379. [Google Scholar] [CrossRef]
- Zhou, X.H.; Chai, H.X.; Bai, M.; Zhang, Z. LncRNA-GAS5 regulates PDCD4 expression and mediates myocardial infarction-induced cardiomyocytes apoptosis via targeting MiR-21. Cell Cycle 2020, 19, 1363–1377. [Google Scholar] [CrossRef]
- Zhang, J.C.; Xia, L.; Jiang, Y.; Wu, D.Q.; Liu, S.C.; Zhou, X.N.; Zhang, F.X. Effect of lncRNA GAS5 on rats with acute myocardial infarction through regulating MIR-21. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8573–8579. [Google Scholar] [PubMed]
- Meloni, M.; Marchetti, M.; Garner, K.; Littlejohns, B.; Sala-Newby, G.; Xenophontos, N.; Floris, I.; Suleiman, M.S.; Madeddu, P.; Caporali, A.; et al. Local inhibition of microRNA-24 improves reparative angiogenesis and left ventricle remodeling and function in mice with myocardial infarction. Mol. Ther. 2013, 21, 1390–1402. [Google Scholar] [CrossRef]
- Minghua, W.; Zhijian, G.; Chahua, H.; Qiang, L.; Minxuan, X.; Luqiao, W.; Weifang, Z.; Peng, L.; Biming, Z.; Lingling, Y.; et al. Plasma exosomes induced by remote ischaemic preconditioning attenuate myocardial ischaemia/reperfusion injury by transferring MIR-24 article. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Mohammadi, A.; Karami, A.R.B.; Mashtani, V.D.; Sahraei, T.; Tarashoki, Z.B.; Khattavian, E.; Mobarak, S.; Kazerouni, H.M.; Radmanesh, E. Evaluation of Oxidative Stress, Apoptosis, and Expression of MicroRNA-208a and MicroRNA-1 in Cardiovascular Patients. Rep. Biochem. Mol. Biol. 2021, 10, 184–197. [Google Scholar] [CrossRef]
- Shyu, K.G.; Wang, B.W.; Cheng, W.P.; Lo, H.M. MicroRNA-208a increases myocardial endoglin expression and myocardial fibrosis in acute myocardial infarction. Can. J. Cardiol. 2015, 31, 679–690. [Google Scholar] [CrossRef]
- Lesizza, P.; Prosdocimo, G.; Martinelli, V.; Sinagra, G.; Zacchigna, S.; Giacca, M. Single-Dose Intracardiac Injection of Pro-Regenerative MicroRNAs Improves Cardiac Function after Myocardial Infarction. Circ. Res. 2017, 120, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Lock, M.C.; Tellam, R.L.; Botting, K.J.; Wang, K.C.W.; Selvanayagam, J.B.; Brooks, D.A.; Seed, M.; Morrison, J.L. The role of miRNA regulation in fetal cardiomyocytes, cardiac maturation and the risk of heart disease in adults. J. Physiol. 2018, 596, 5625–5640. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.-J.; Matkovich, S.J.; Dornall, G.W.; van Rooij, E.; Olson, E.N. miR-15 family regulates CM proliferation. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef]
- Mathiyalagan, P.; Liang, Y.; Kim, D.; Misener, S.; Thorne, T.; Kamide, C.E.; Klyachko, E.; Losordo, D.W.; Hajjar, R.J.; Sahoo, S. Angiogenic Mechanisms of Human CD34+ Stem Cell Exosomes in the Repair of Ischemic Hindlimb. Circ. Res. 2017, 120, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Mocharla, P.; Briand, S.; Giannotti, G.; Dörries, C.; Jakob, P.; Paneni, F.; Lüscher, T.; Landmesser, U. AngiomiR-126 expression and secretion from circulating CD34+ and CD14+ PBMCs: Role for proangiogenic effects and alterations in type 2 diabetics. Blood 2013, 121, 226–236. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, Q.; Liu, X.; Zhang, T.; Wang, S.; Zhou, L.; Zou, L.; Fan, F.; Chi, H.; Sun, J.; et al. Exosomal microRNA-98-5p from hypoxic bone marrow mesenchymal stem cells inhibits myocardial ischemia–reperfusion injury by reducing TLR4 and activating the PI3K/Akt signaling pathway. Int. Immunopharmacol. 2021, 101, 107592. [Google Scholar] [CrossRef]
- Gupta, S.K.; Foinquinos, A.; Thum, S.; Remke, J.; Zimmer, K.; Bauters, C.; de Groote, P.; Boon, R.A.; de Windt, L.J.; Preissl, S.; et al. Preclinical Development of a MicroRNA-Based Therapy for Elderly Patients With Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 68, 1557–1571. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, L.; Shan, H.; Zhang, Y.; Zhou, R.; Su, Z.; Du, Z. Microrna expression analysis: Clinical advantage of propranolol reveals key micrornas in myocardial infarction. PLoS ONE 2011, 6, e14736. [Google Scholar] [CrossRef]
- Zhou, J.; He, S.; Wang, B.; Yang, W.; Zheng, Y.; Jiang, S.; Li, D.; Lin, J. Construction and Bioinformatics Analysis of circRNA-miRNA-mRNA Network in Acute Myocardial Infarction. Front. Genet. 2022, 13, 1–17. [Google Scholar] [CrossRef]
- Ma, R.; Gao, L.; Liu, Y.; Du, P.; Chen, X.; Li, G. LncRNA TTTY15 knockdown alleviates H2O2-stimulated myocardial cell injury by regulating the miR-98-5p/CRP pathway. Mol. Cell. Biochem. 2021, 476, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Luo, L.; Wei, X.; Gong, D.; Li, Z.; Li, S.; Tang, W.; Jin, L. M1 Bone Marrow-Derived Macrophage-Derived Extracellular Vesicles Inhibit Angiogenesis and Myocardial Regeneration following Myocardial Infarction via the MALAT1/MicroRNA-25-3p/CDC42 Axis. Oxidative Med. Cell. Longev. 2021, 2021, 9959746. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.L.; Chen, Y.X.; Zhou, J.; Li, Y.; Gong, C.Y.; Wang, X.B. LncRNA HULC alleviates HUVEC inflammation and improves angiogenesis after myocardial infarction through down-regulating miR-29b. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6288–6298. [Google Scholar] [PubMed]
- Liang, H.; Li, F.; Li, H.; Wang, R.; Du, M. Overexpression of lncRNA HULC Attenuates Myocardial Ischemia/reperfusion Injury in Rat Models and Apoptosis of Hypoxia/reoxygenation Cardiomyocytes via Targeting miR-377-5p through NLRP3/Caspase-1/IL-1β Signaling Pathway Inhibition. Immunol. Investig. 2020, 50, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, J.; Shao, P. Long noncoding RNA MIAT2 alleviates lipopolysaccharide-induced inflammatory damage in WI-38 cells by sponging microRNA-15. J. Cell. Physiol. 2020, 235, 3690–3697. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Liang, H.; Wang, H.; Wen, D.; Wang, L.; Zhao, L.; Sun, M.; Wang, J. Mirt2 functions in synergy with miR-377 to participate in inflammatory pathophysiology of Sjögren’s syndrome. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2473–2480. [Google Scholar] [CrossRef]
- Li, T.; Tian, H.; Li, J.; Zuo, A.; Chen, J.; Xu, D.; Guo, Y.; Gao, H. Overexpression of lncRNA Gm2691 attenuates apoptosis and inflammatory response after myocardial infarction through PI3K/Akt signaling pathway. IUBMB Life 2019, 71, 1561–1570. [Google Scholar] [CrossRef]
- Li, X.; Zhou, J.; Huang, K. Erratum: Inhibition of the lnc RNA Mirt1 attenuates acute myocardial infarction by suppressing NF-κB activation. Cell. Physiol. Biochem. 2017, 42, 1153–1164. [Google Scholar] [CrossRef]
- Gast, M.; Rauch, B.H.; Haghikia, A.; Nakagawa, S.; Haas, J.; Stroux, A.; Schmidt, D.; Schumann, P.; Weiss, S.; Jensen, L.; et al. Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc. Res. 2019, 115, 1886–1906. [Google Scholar] [CrossRef]
- Lu, W.; Sheng, Z.; Zhang, Z.; Ma, G.; Chen, L.; Huang, J.; Ding, J.; Dai, Q. LncRNA-LUNAR1 Levels Are Closely Related to Coronary Collaterals in Patients with Chronic Total Coronary Occlusion. J. Cardiovasc. Transl. Res. 2020, 13, 171–180. [Google Scholar] [CrossRef]
- Zhang, Y.; Bian, Y. Long Non-Coding RNA SNHG8 Plays a Key Role in Myocardial Infarction Through Affecting Hypoxia-Induced Cardiomyocyte Injury. Med. Sci. Monit. 2020, 26, e924016. [Google Scholar] [CrossRef] [PubMed]
- Hao, K.; Lei, W.; Wu, H.; Wu, J.; Yang, Z.; Yan, S.; Lu, X.A.; Li, J.; Xia, X.; Han, X.; et al. LncRNA-Safe contributes to cardiac fibrosis through Safe-Sfrp2-HuR complex in mouse myocardial infarction. Theranostics 2019, 9, 7282–7297. [Google Scholar] [CrossRef] [PubMed]
- Micheletti, R.; Plaisance, I.; Abraham, B.J.; Sarre, A.; Ting, C.-C.; Alexanian, M.; Maric, D.; Maison, D.; Nemir, M.; Young, R.A.; et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci. Transl. Med. 2017, 9, eaai9118. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhang, M.; Wu, H.; Ding, X.; Li, D.; Dong, X.; Hu, X.; Su, S.; Shang, W.; Wu, J.; et al. SAIL: A new conserved anti-fibrotic lncRNA in the heart. Basic Res. Cardiol. 2021, 116, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fu, X.; Kataoka, M.; Liu, N.; Wang, Y.; Gao, F.; Liang, T.; Dong, X.; Pei, J.; Hu, X.; et al. Long noncoding RNA Cfast regulates cardiac fibrosis. Mol. Ther. Nucleic Acids 2021, 23, 377–392. [Google Scholar] [CrossRef]
- Sun, F.; Zhuang, Y.; Zhu, H.; Wu, H.; Li, D.; Zhan, L.; Yang, W.; Yuan, Y.; Xie, Y.; Yang, S.; et al. LncRNA PCFL promotes cardiac fibrosis via miR-378/GRB2 pathway following myocardial infarction. J. Mol. Cell. Cardiol. 2019, 133, 188–198. [Google Scholar] [CrossRef]
- Xiong, X.; Liu, J.; He, Q.; Dai, R.; Zhang, H.; Cao, Z.; Liao, Y.; Liu, B.; Zhou, Y.; Chen, J.; et al. Long non-coding RNA NORAD aggravates acute myocardial infarction by promoting fibrosis and apoptosis via miR-577/COBLL1 axis. Environ. Toxicol. 2021, 36, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Pan, Z.; Zhao, X.; Liu, L.; Sun, J.; Su, X.; Xu, C.; Zhou, Y.; Zhao, D.; Xu, B.; et al. Erratum: LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics 2018, 8, 1180–1194. [Google Scholar] [CrossRef]
- Lang, M.; Ou, D.; Liu, Z.; Li, Y.; Zhang, X.; Zhang, F. LncRNA MHRT promotes cardiac fibrosis via miR-3185 pathway following myocardial infarction. Int. Heart J. 2021, 62, 891–899. [Google Scholar] [CrossRef]
- Luo, B.; He, Z.; Huang, S.; Wang, J.; Han, D.; Xue, H.; Liu, P.; Zeng, X.; Lu, D. Long Non-Coding RNA 554 Promotes Cardiac Fibrosis via TGF-β1 Pathway in Mice Following Myocardial Infarction. Front. Pharmacol. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Chen, G.; Huang, S.; Song, F.; Zhou, Y.; He, X. Lnc-Ang362 is a pro-fibrotic long non-coding RNA promoting cardiac fibrosis after myocardial infarction by suppressing Smad7. Arch. Biochem. Biophys. 2020, 685, 108354. [Google Scholar] [CrossRef]
- Zhuang, Y.; Li, T.; Zhuang, Y.; Li, Z.; Yang, W.; Huang, Q.; Li, D.; Wu, H.; Zhang, G.; Yang, T.; et al. Involvement of lncR-30245 in Myocardial Infarction–Induced Cardiac Fibrosis Through Peroxisome Proliferator-Activated Receptor-γ–Mediated Connective Tissue Growth Factor Signalling Pathway. Can. J. Cardiol. 2019, 35, 480–489. [Google Scholar] [CrossRef]
- Du, L.; Chen, J.; Wu, Y.; Xia, G.; Chen, M.; Zhao, P.; Wang, Y.; Yao, D.; Liu, F.; Zhang, L.; et al. Long Non-coding RNA N1LR Protects Against Myocardial Ischemic/Reperfusion Injury Through Regulating the TGF-β Signaling Pathway. Front. Cardiovasc. Med. 2021, 8, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, T.; Zhang, M.; Chen, P.; Yu, Y. Down-regulation of myocardial infarction associated transcript 1 improves myocardial ischemia-reperfusion injury in aged diabetic rats by inhibition of activation of NF-κB signaling pathway. Chem. Interactions 2019, 300, 111–122. [Google Scholar] [CrossRef]
- Yan, M.; Liu, Q.; Jiang, Y.; Wang, B.; Ji, Y.; Liu, H.; Xie, Y. Long Noncoding RNA LNC_000898 Alleviates Cardiomyocyte Apoptosis and Promotes Cardiac Repair after Myocardial Infarction through Modulating the miR-375/PDK1 Axis. J. Cardiovasc. Pharmacol. 2020, 76, 77–85. [Google Scholar]
- Chen, Y.; Li, S.; Zhang, Y.; Wang, M.; Li, X.; Liu, S.; Xu, D.; Bao, Y.; Jia, P.; Wu, N.; et al. The lncRNA Malat1 regulates microvascular function after myocardial infarction in mice via miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics. Redox Biol. 2021, 41, 101910. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Yuan, Z.; Chen, A.; Liu, Z.; Wang, Z.; Li, H. LncRNA-MALAT1 promotes CPC proliferation and migration in hypoxia by up-regulation of JMJD6 via sponging miR-125. Biochem. Biophys. Res. Commun. 2018, 499, 711–718. [Google Scholar] [CrossRef]
- Zhang, B.F.; Jiang, H.; Chen, J.; Hu, Q.; Yang, S.; Liu, X.P.; Liu, G. LncRNA H19 ameliorates myocardial infarction-induced myocardial injury and maladaptive cardiac remodelling by regulating KDM3A. J. Cell. Mol. Med. 2020, 24, 1099–1115. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Yuan, Z.; Chen, A.; Liu, Z.; Li, H.; Wang, Z. Long non-coding RNA H19 contributes to hypoxia-induced CPC injury by suppressing Sirt1 through miR-200a-3p. Acta Biochim. Biophys. Sin. 2018, 50, 950–959. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Liu, D.; Zang, S.; Ma, N.; Zhao, L.; Zhang, L.; Zhang, X.; Qiao, C. The lncRNA H19/miR-675 axis regulates myocardial ischemic and reperfusion injury by targeting PPARα. Mol. Immunol. 2018, 105, 46–54. [Google Scholar] [CrossRef]
- Choong, O.K.; Chen, C.Y.; Zhang, J.; Lin, J.H.; Lin, P.J.; Ruan, S.C.; Kamp, T.J.; Hsieh, P.C.H. Hypoxia-induced H19/YB-1 cascade modulates cardiac remodeling after infarction. Theranostics 2019, 9, 6550–6567. [Google Scholar] [CrossRef]
- Zhao, X.; Ren, Y.; Ren, H.; Wu, Y.; Liu, X.; Chen, H.; Ying, C. The mechanism of myocardial fibrosis is ameliorated by myocardial infarction-associated transcript through the PI3K/Akt signaling pathway to relieve heart failure. J. Int. Med. Res. 2021, 49, 3000605211031433. [Google Scholar] [CrossRef]
- Dong, Q.; Wang, Q.; Yan, X.; Wang, X.; Li, Z.; Zhang, L. Long noncoding RNA MIAT inhibits the progression of diabetic nephropathy and the activation of NF-κB pathway in high glucose-treated renal tubular epithelial cells by the miR-182-5p/GPRC5A axis. Open Med. 2021, 16, 1336–1349. [Google Scholar] [CrossRef]
- Sun, Q.; Luo, M.; Gao, Z.; Han, X.; Yan, Z.; Xie, S.; Zhao, H.; Sun, H. TUG1 knockdown suppresses cardiac fibrosis after myocardial infarction. Mamm. Genome 2021, 32, 435–442. [Google Scholar] [CrossRef]
- Li, M.; Zheng, H.; Han, Y.; Chen, Y.; Li, B.; Chen, G.; Chen, X.; Huang, S.; He, X.; Wei, G.; et al. LncRNA Snhg1-driven self-reinforcing regulatory network promoted cardiac regeneration and repair after myocardial infarction. Theranostics 2021, 11, 9397–9414. [Google Scholar] [CrossRef]
- Cai, B.; Ma, W.; Wang, X.; Sukhareva, N.; Hua, B.; Zhang, L.; Xu, J.; Li, X.; Li, S.; Liu, S.; et al. Targeting LncDACH1 promotes cardiac repair and regeneration after myocardium infarction. Cell Death Differ. 2020, 27, 2158–2175. [Google Scholar] [CrossRef]
- Fu, W.; Ren, H.; Shou, J.; Liao, Q.; Li, L.; Shi, Y.; Jose, P.A.; Zeng, C.; Wang, W.E. Loss of NPPA-AS1 promotes heart regeneration by stabilizing SFPQ–NONO heteromer-induced DNA repair. Basic Res. Cardiol. 2022, 117, 1–19. [Google Scholar] [CrossRef]
- Ponnusamy, M.; Liu, F.; Zhang, Y.H.; Li, R.B.; Zhai, M.; Liu, F.; Zhou, L.Y.; Liu, C.Y.; Yan, K.W.; Dong, Y.H.; et al. Long Noncoding RNA CPR (Cardiomyocyte Proliferation Regulator) Regulates Cardiomyocyte Proliferation and Cardiac Repair. Circulation 2019, 139, 2668–2684. [Google Scholar] [CrossRef]
- Trembinski, D.J.; Bink, D.I.; Theodorou, K.; Sommer, J.; Fischer, A.; van Bergen, A.; Kuo, C.C.; Costa, I.G.; Schürmann, C.; Leisegang, M.S.; et al. Aging-regulated anti-apoptotic long non-coding RNA Sarrah augments recovery from acute myocardial infarction. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Hosen, M.R.; Militello, G.; Weirick, T.; Ponomareva, Y.; Dassanayaka, S.; MooreIV, J.B.; Döring, C.; Wysoczynski, M.; Jones, S.P.; Dimmeler, S.; et al. Airn Regulates Igf2bp2 Translation in Cardiomyocytes. Circ. Res. 2018, 122, 1347–1353. [Google Scholar] [CrossRef]
- Cai, B.; Ma, W.; Ding, F.; Zhang, L.; Huang, Q.; Wang, X.; Hua, B.; Xu, J.; Li, J.; Bi, C.; et al. The Long Noncoding RNA CAREL Controls Cardiac Regeneration. J. Am. Coll. Cardiol. 2018, 72, 534–550. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Huang, S.; Chen, Y.; Chen, X.; Li, M.; Si, X.; He, X.; Zheng, H.; Zhong, L.; et al. Inhibition of AZIN2-sv induces neovascularization and improves prognosis after myocardial infarction by blocking ubiquitin-dependent talin1 degradation and activating the Akt pathway. EBioMedicine 2019, 39, 69–82. [Google Scholar] [CrossRef]
- Li, X.; He, X.; Wang, H.; Li, M.; Huang, S.; Chen, G.; Jing, Y.; Wang, S.; Chen, Y.; Liao, W.; et al. Loss of AZIN2 splice variant facilitates endogenous cardiac regeneration. Cardiovasc. Res. 2018, 114, 1642–1655. [Google Scholar] [CrossRef]
- Safaei, S.; Tahmasebi-Birgani, M.; Bijanzadeh, M.; Seyedian, S.M. Increased expression level of long noncoding RNA H19 in plasma of patients with myocardial infarction. Int. J. Mol. Cell. Med. 2020, 9, 122–129. [Google Scholar]
- Yang, L.; Deng, J.; Ma, W.; Qiao, A.; Xu, S.; Yu, Y.; Boriboun, C.; Kang, X.; Han, D.; Ernst, P.; et al. Ablation of lncrna miat attenuates pathological hypertrophy and heart failure. Theranostics 2021, 11, 7995–8007. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Z.; Peng, X.; Zheng, Z.; Le, A.; Guo, J.; Ma, L.; Shi, H.; Yao, K.; Zhang, S.; et al. Extracellular Vesicle-Derived circITGB1 Regulates Dendritic Cell Maturation and Cardiac Inflammation via miR-342-3p/NFAM1. Oxidative Med. Cell. Longev. 2022, 2022, 1–23. [Google Scholar] [CrossRef]
- Ren, K.; Li, B.; Jiang, L.; Liu, Z.; Wu, F.; Zhang, Y.; Liu, J.; Duan, W. Circ_0023461 Silencing Protects Cardiomyocytes from Hypoxia-Induced Dysfunction through Targeting miR-370-3p/PDE4D Signaling. Oxidative Med. Cell. Longev. 2021, 2021, 1–18. [Google Scholar] [CrossRef]
- Chaorui, X.; Jia, Z.; Cao, X.; Wang, S.; Wang, J.; An, L. Hsa_circ_0007059 promotes apoptosis and inflammation in cardiomyocytes during ischemia by targeting microRNA-378 and microRNA-383. Cell Cycle 2022, 21, 1003–1019. [Google Scholar]
- Zhang, Y.; Li, Z.; Wang, J.; Chen, H.; He, R.; Wu, H. CircTRRAP Knockdown Has Cardioprotective Function in Cardiomyocytes via the Signal Regulation of miR-370-3p/PAWR Axis. Cardiovasc. Ther. 2022, 2022, 1–12. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, Z.; Chen, X.; Lu, G.; Zhu, X.; Xu, G. CircUBXN7 mitigates H/R-induced cell apoptosis and inflammatory response through the miR-622-MCL1 axis. Am. J. Transl. Res. 2021, 13, 8711–8727. [Google Scholar]
- Zhou, D.; Dai, Z.; Ren, M.; Yang, M. Adipose-Derived Stem Cells-Derived Exosomes with High Amounts of Circ_0001747 Alleviate Hypoxia/Reoxygenation-Induced Injury in Myocardial Cells by Targeting MiR-199b-3p/MCL1 Axis. Int. Heart J. 2022, 63, 356–366. [Google Scholar] [CrossRef]
- Li, F.; Long, T.Y.; Bi, S.S.; Sheikh, S.A.; Zhang, C.L. circPAN3 exerts a profibrotic role via sponging miR-221 through FoxO3/ATG7-activated autophagy in a rat model of myocardial infarction. Life Sci. 2020, 257, 118015. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.Y.; Zhao, J.C.; Ge, X.M.; Zhang, H.; Wang, C.M.; Bie, Z.D. Circ_LAS1L regulates cardiac fibroblast activation, growth, and migration through miR-125b/SFRP5 pathway. Cell Biochem. Funct. 2020, 38, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pan, W.; Yang, T.; Meng, X.; Jiang, Z.; Tao, L.; Wang, L. Upregulation of circular RNA CircNFIB attenuates cardiac fibrosis by sponging miR-433. Front. Genet. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Li, C.; Xu, B.; Xiang, Y.; Jia, X.; Yuan, Z.; Wu, L.; Zhong, L.; Li, Y. Circular RNA mmu_circ_0005019 inhibits fibrosis of cardiac fibroblasts and reverses electrical remodeling of cardiomyocytes. BMC Cardiovasc. Disord. 2021, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Mu, B.; Li, X.; Bie, Z.D. circCELF1 Inhibits Myocardial Fibrosis by Regulating the Expression of DKK2 Through FTO/m6A and miR-636. J. Cardiovasc. Transl. Res. 2022, 15, 998–1009. [Google Scholar] [CrossRef]
- Wang, Y.; Li, C.; Zhao, R.; Qiu, Z.; Shen, C.; Wang, Z.; Liu, W.; Zhang, W.; Ge, J.; Shi, B. CircUbe3a from M2 macrophage-derived small extracellular vesicles mediates myocardial fibrosis after acute myocardial infarction. Theranostics 2021, 11, 6315–6333. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Shen, J.F.; Wei, X.F.; Qi, G.X. Circular RNA foxo3 relieves myocardial ischemia/reperfusion injury by suppressing autophagy via inhibiting HMGB1 by repressing KAT7 in myocardial infarction. J. Inflamm. Res. 2021, 14, 6397–6407. [Google Scholar] [CrossRef]
- Lan, Z.; Wang, T.; Zhang, L.; Jiang, Z.; Zou, X. CircSLC8A1 Exacerbates Hypoxia-Induced Myocardial Injury via Interacting with MiR-214-5p to Upregulate TEAD1 Expression. Int. Heart J. 2022, 63, 591–601. [Google Scholar] [CrossRef]
- Huang, S.; Li, X.; Zheng, H.; Si, X.; Li, B.; Wei, G.; Li, C.; Chen, Y.; Chen, Y.; Liao, W.; et al. Loss of Super-Enhancer-Regulated circRNA Nfix Induces Cardiac Regeneration after Myocardial Infarction in Adult Mice. Circulation 2019, 139, 2857–2876. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Z.; Cheng, Q.; Wang, Z.; Lv, X.; Wang, Z.; Li, N. Circular RNA (circRNA) CDYL induces myocardial regeneration by ceRNA after myocardial infarction. Med. Sci. Monit. 2020, 26, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wang, X.; Sun, H.; Xu, B.; Song, R.; Tian, Y.; Zhao, L.; Xu, Y.; Zhao, Y.; Yang, F.; et al. Oxidant stress-sensitive circRNA Mdc1 controls cardiomyocyte chromosome stability and cell cycle re-entry during heart regeneration. Pharmacol. Res. 2022, 184, 106422. [Google Scholar] [CrossRef]
- Zheng, H.; Huang, S.; Wei, G.; Sun, Y.; Li, C.; Si, X.; Chen, Y.; Tang, Z.; Li, X.; Chen, Y.; et al. CircRNA Samd4 induces cardiac repair after myocardial infarction by blocking mitochondria-derived ROS output. Mol. Ther. 2022, 30, 3477–3498. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Ma, R.; Cao, J.; Du, X.; Cai, X.; Fan, Y. CircSAMD4A aggravates H/R-induced cardiomyocyte apoptosis and inflammatory response by sponging miR-138-5p. J. Cell. Mol. Med. 2022, 26, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Si, X.; Zheng, H.; Wei, G.; Li, M.; Li, W.; Wang, H.; Guo, H.; Sun, J.; Li, C.; Zhong, S.; et al. CircRNA Hipk3 Induces Cardiac Regeneration after Myocardial Infarction in Mice by Binding to Notch1 and miR-133a. Mol. Ther. Nucleic Acids 2020, 21, 636–655. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, J.; Xie, G.; Zeng, X.; Li, H. Circ-hipk3 strengthens the effects of adrenaline in heart failure by mir-17-3p-adcy6 axis. Int. J. Biol. Sci. 2019, 15, 2484–2496. [Google Scholar] [CrossRef]
- Bai, M.; Pan, C.L.; Jiang, G.X.; Zhang, Y.M. CircRNA 010567 improves myocardial infarction rats through inhibiting TGF-β1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 369–375. [Google Scholar]
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef]
- Truby, L.K.; Rogers, J.G. Advanced Heart Failure: Epidemiology, Diagnosis, and Therapeutic Approaches. JACC Heart Fail. 2020, 8, 523–536. [Google Scholar] [CrossRef]
- Dalen, J.E.; Alpert, J.S.; Goldberg, R.J.; Weinstein, R.S. The epidemic of the 20th century: Coronary heart disease. Am. J. Med. 2014, 127, 807–812. [Google Scholar] [CrossRef]
- Duggan, J.P.; Peters, A.S.; Trachiotis, G.D.; Antevil, J.L. Epidemiology of Coronary Artery Disease. Surg. Clin. North Am. 2022, 102, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef] [PubMed]
- Trajano, L.F.; Smart, N. Immunomodulation for optimal cardiac regeneration: Insights from comparative analyses. NPJ Regen. Med. 2021, 6, 1–11. [Google Scholar]




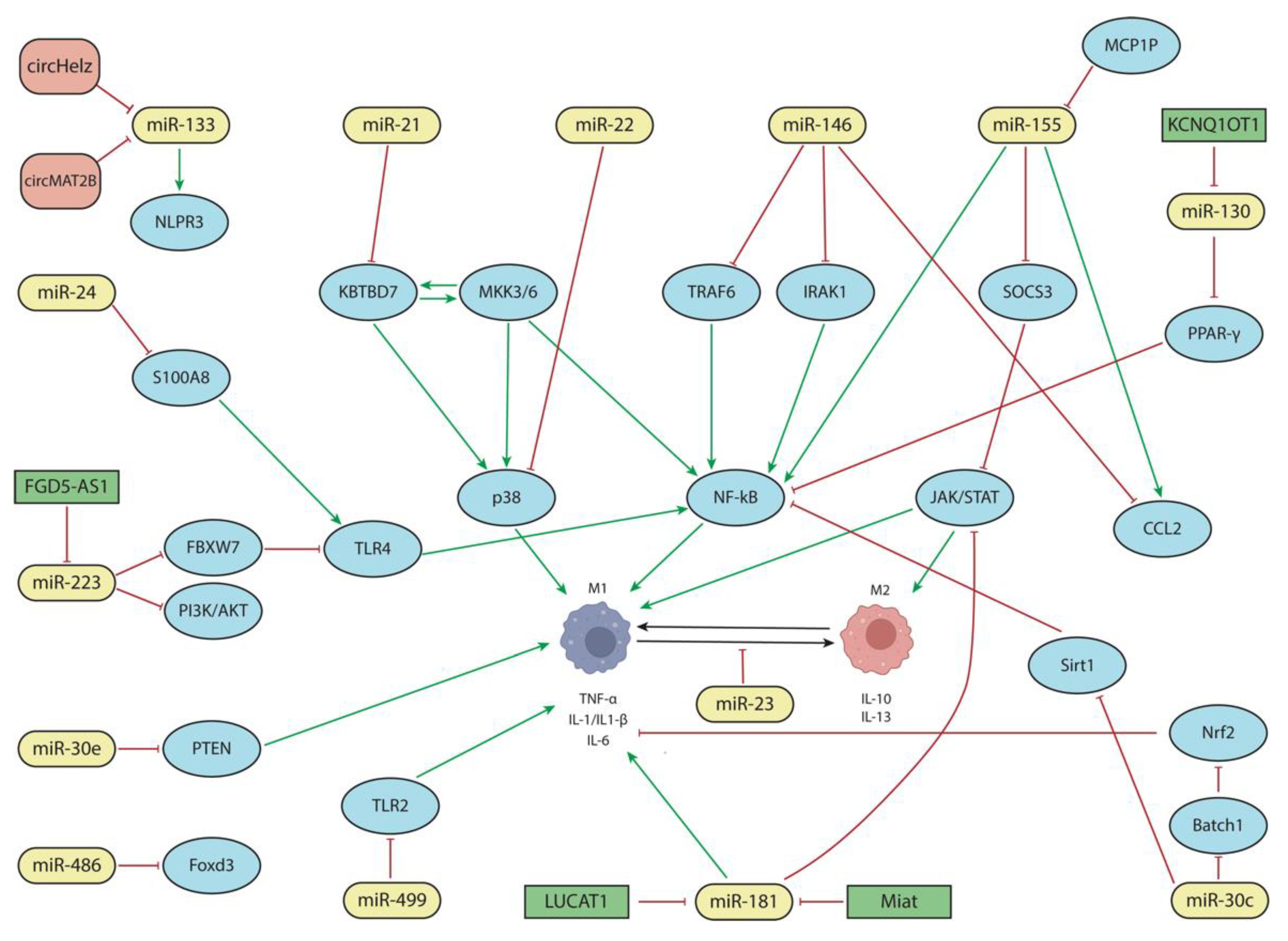{kind=link}
{kind=link}
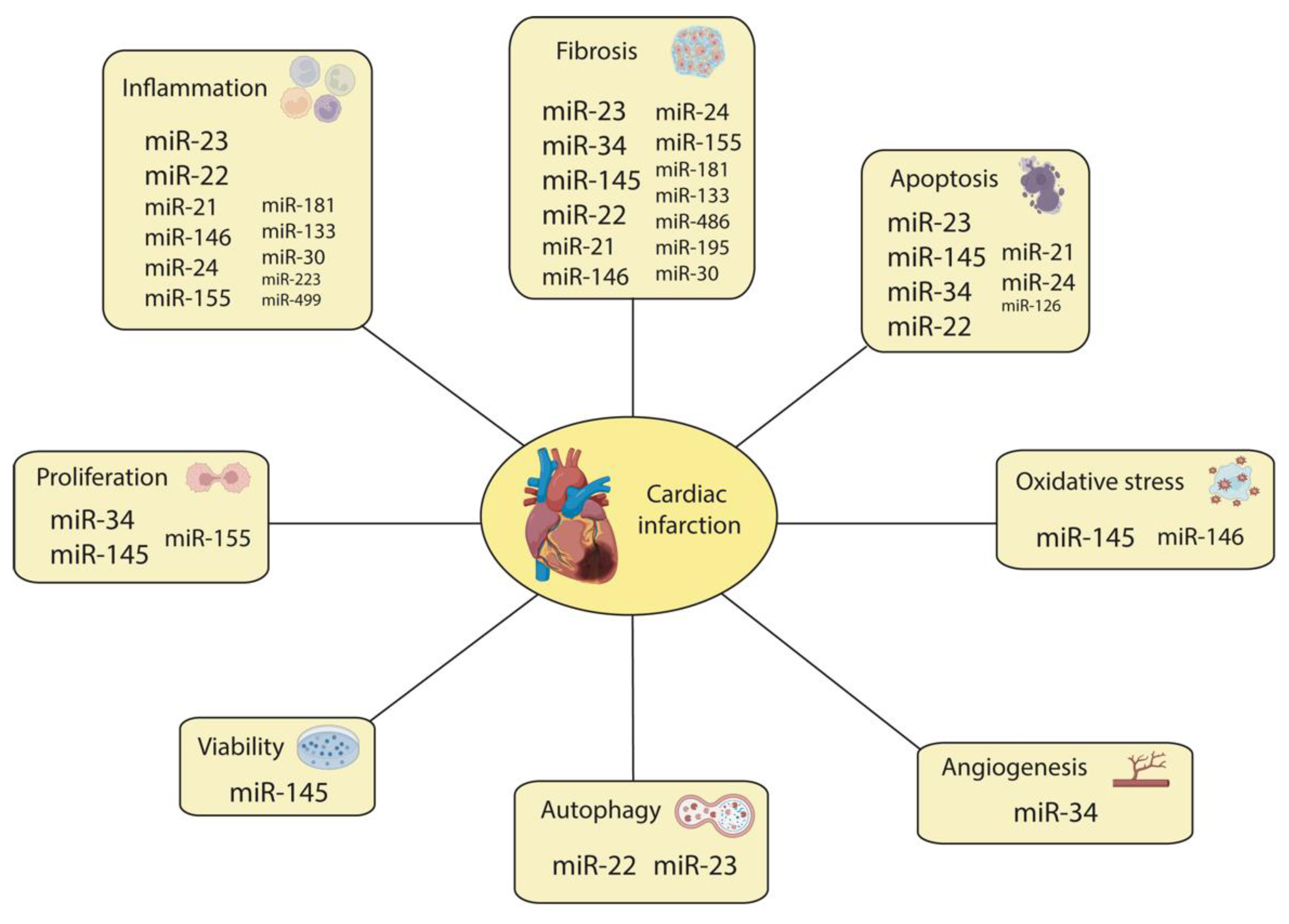{kind=link}
| Cardiac Inflammation | |||||
|---|---|---|---|---|---|
| lncRNA | miRNA | Protein | Signaling | Functional Role | Ref. |
| KCNQ1OT1 | miR-130 | ZNF91 | - | [83] | |
| LUCAT1 | miR-181 | - | - | ROS, inflammation, apoptosis | [40] |
| HULC | miR-29 | - | - | Inflammation, angiogenesis | [194] |
| HULC | miR-377 | NLRP3/Caspase-1/IL-1β | - | Inflammation, apoptosis | [195] |
| TTTY15 | miR-98 | CRP | - | Myocardial injury | [192] |
| MIAT2 | miR-15 | - | - | Inflammation | [196] |
| MIRT2 | miR-377 | - | - | [197] | |
| GM2691 | - | - | Akt | Inflammation | [198] |
| MIRT1 | - | - | NF-kB | Cardiomyocytes, inflammation | [199] |
| NEAT1 | - | - | - | Myocardial inflammation | [200] |
| LUNAR1 | - | - | Inflammation | [201] | |
| SNHG8 | - | - | - | Inflammation | [202] |
| Cardiac Fibrosis | |||||
| lncRNA | miRNA | Protein | Signaling | Functional Role | Ref. |
| SAFE | - | Sfrp2, HuR | - | Fibroblasts, fibrosis | [203] |
| WISPER | - | Tia1-related protein | Collagen | Proliferation, fibrosis | [204] |
| SAIL | - | Safb | Collagen | Fibrosis | [205] |
| CFAST | - | Colt1, Trap1 | - | Fibrosis | [206] |
| PCFL | miR-378 | Grb2 | Collagen | Fibrosis | [207] |
| NORAD | miR-577 | Cobll1 | Collagen | Fibrosis | [208] |
| PFL | Let-7d | Ptafr | - | Fibrosis | [209] |
| MHRT | miR-3185 | - | Collagen | Fibrosis | [210] |
| n379519 | miR-30 | - | Collagen | Fibrosis | [147] |
| XIST | miR-155 | - | - | Fibrosis | [121] |
| lncRNA554 | - | - | TGF-β1 | Fibrosis | [211] |
| lnc-ANG362 | - | Smad7 | - | Fibrosis | [212] |
| lncR-30245 | - | - | TGF-β1, Pparg | Fibrosis | [213] |
| N1LR | - | TGF-β1, Smads | - | Fibrosis, Inflammation | [214] |
| FGD5-AS1 | miR-223 | - | Akt | Cardiac fibrosis, inflammation, apoptosis | [91] |
| Other Biological Processes Related to Cardiac Infarction | |||||
| lncRNA | miRNA | Protein | Signaling | Functional Role | Ref. |
| MIRT1 | - | - | NF-kβ | Fibroblasts, apoptosis, ROS, inflammation | [215] |
| GAS5 | miR-21 | - | - | Myocardial apoptosis | [178] |
| LNC_000898 | miR-375 | Pdk1 | - | Cardiac apoptosis | [216] |
| Cardiac regeneration | |||||
| lncRNA | miRNA | Protein | Signaling | Functional Role | Ref. |
| MALAT1 | miR-26 | Mfn1 | - | Mitochondrial activity | [217] |
| MALAT1 | miR-25-3p | CDC42 | - | - | [193] |
| MALAT1 | miR-125 | Jmj6b | - | Endothelial cell viability | [218] |
| MALAT1 | miR-145 | TGF-β1 | - | Fibroblast proliferation | [113] |
| H19 | miR-22-3p | Kdm3a | - | Fibrosis | [219] |
| H19 | miR-200a-3p | Sirt1 | - | Cardiac progenitor proliferation | [220] |
| H19 | miR-675 | PPar | - | Cell viability, apoptosis, ROS, inflammation | [221] |
| H19 | - | YB-1 | - | Fibrosis | [222] |
| MIAT | - | - | PI3K/AKT | - | [223] |
| MIAT | miR-181a-5p | JAK2 | - | Cell proliferation, apoptosis, inflammation | [41] |
| MIAT | miR-182-5p | GPRC5A | - | - | [224] |
| TUG1 | miR-133b | - | - | - | [118] |
| TUG1 | miR-590 | Fgf1 | TGF-β | Fibrosis | [225] |
| SNHG1 | - | PTEN | PI3K/AKT | - | [226] |
| lncDACH1 | - | PPQA | YAP1 | - | [227] |
| NPPA-AS1 | - | SFPQ | - | - | [228] |
| CPR | - | Mcm3 | - | Cardiomyocyte proliferation, scar formation | [229] |
| SARRAH | - | Nrf2 | - | - | [230] |
| AIM | - | Igf2bp2, Rap1 | - | - | [231] |
| CAREL | miR-296 | Trp53inp1, Itm2a | - | Cardiomyocyte replication | [232] |
| AZIN2-sv | miR-214 | PTEN | PI3K/AKT | Cardiomyocyte proliferation | [233] |
| AZIN2-sv | - | Tln1, ITGB1 | - | Angiogenesis | [234] |
| Cardiac Inflammation | ||||
|---|---|---|---|---|
| circRNA | miRNA | Protein | Signaling | Ref. |
| circITGB1 | miR-342-3p | NFAM1 | - | [237] |
| circ_0023461 | miR-370-3p | PDE4D | - | [238] |
| circHelz | miR-133a-3p | NLRP3 | - | [81] |
| circ_0007059 | miR-378, miR-383 | - | - | [239] |
| circTRRAP | miR-370-3p | PAWR | - | [240] |
| circMAT2B | miR-133 | - | PI3K/AKT, Raf/MEK/ERK | [80] |
| circUBXN7 | miR-622 | MCL1 | - | [241] |
| circ_0001747 | miR-199-3p | MCL1 | - | [242] |
| Cardiac Fibrosis | ||||
| lncRNA | miRNA | Protein | Signaling | Ref. |
| circPAN | miR-221 | Foxo3 | TGF-β | [243] |
| circ_LAS1L | miR-125b | SFRP5 | - | [244] |
| circNFIB | miR-433 | - | TGF-β | [245] |
| circ_0005019 | miR-499-5p | Kcnn3 | - | [246] |
| circCELF1 | miR-636 | Dkk2 | TGF-β | [247] |
| circUbe3a | miR-138-5p | - | - | [248] |
| Other Biological Processes Related to Cardiac Infarction | ||||
| lncRNA | miRNA | Protein | Signaling | Ref. |
| circFoxo3 | - | KAT7 | - | [249] |
| circSLC8A1 | miR-214 | TEAD4 | - | [250] |
| Cardiac Regeneration | ||||
| lncRNA | miRNA | Protein | Signaling | Ref. |
| circNfix | - | Neddl4, Ybx1 | - | [251] |
| circCDYL | miR-4793-5p | APP | - | [252] |
| circMdc1 | - | PABP/MDC1 | - | [253] |
| circSmad4 | - | - | - | [254] |
| circSmad4 | miR-138-5p | - | - | [255] |
| circHipk3 | miR-133 | - | - | [256] |
| cirHipk3 | miR-17-3p | - | - | [257] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caño-Carrillo, S.; Lozano-Velasco, E.; Castillo-Casas, J.M.; Sánchez-Fernández, C.; Franco, D. The Role of ncRNAs in Cardiac Infarction and Regeneration. J. Cardiovasc. Dev. Dis. 2023, 10, 123. https://doi.org/10.3390/jcdd10030123
Caño-Carrillo S, Lozano-Velasco E, Castillo-Casas JM, Sánchez-Fernández C, Franco D. The Role of ncRNAs in Cardiac Infarction and Regeneration. Journal of Cardiovascular Development and Disease. 2023; 10(3):123. https://doi.org/10.3390/jcdd10030123
Chicago/Turabian StyleCaño-Carrillo, Sheila, Estefanía Lozano-Velasco, Juan Manuel Castillo-Casas, Cristina Sánchez-Fernández, and Diego Franco. 2023. "The Role of ncRNAs in Cardiac Infarction and Regeneration" Journal of Cardiovascular Development and Disease 10, no. 3: 123. https://doi.org/10.3390/jcdd10030123
APA StyleCaño-Carrillo, S., Lozano-Velasco, E., Castillo-Casas, J. M., Sánchez-Fernández, C., & Franco, D. (2023). The Role of ncRNAs in Cardiac Infarction and Regeneration. Journal of Cardiovascular Development and Disease, 10(3), 123. https://doi.org/10.3390/jcdd10030123