
Loss of Sec-1 Family Domain-Containing 1 (scfd1) Causes Severe Cardiac Defects and Endoplasmic Reticulum Stress in Zebrafish

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Zebrafish Husbandry
2.2. Generation of hahvcc43 Mutant
2.3. Mapping of hahvcc43 Mutation
2.4. Morpholino-Mediated Knockdown of scfd1
2.5. CRISPR—CRISPR-Associated Protein 9 (CAS9)-Mediated Generation of scfd1vcc44
2.6. Video-Microscopy
2.7. Echocardiography
2.8. qPCR
2.9. mRNA Injection/Rescue
2.10. Western Blot
2.11. Electron Microscopy
2.12. Statistical Analysis
3. Results
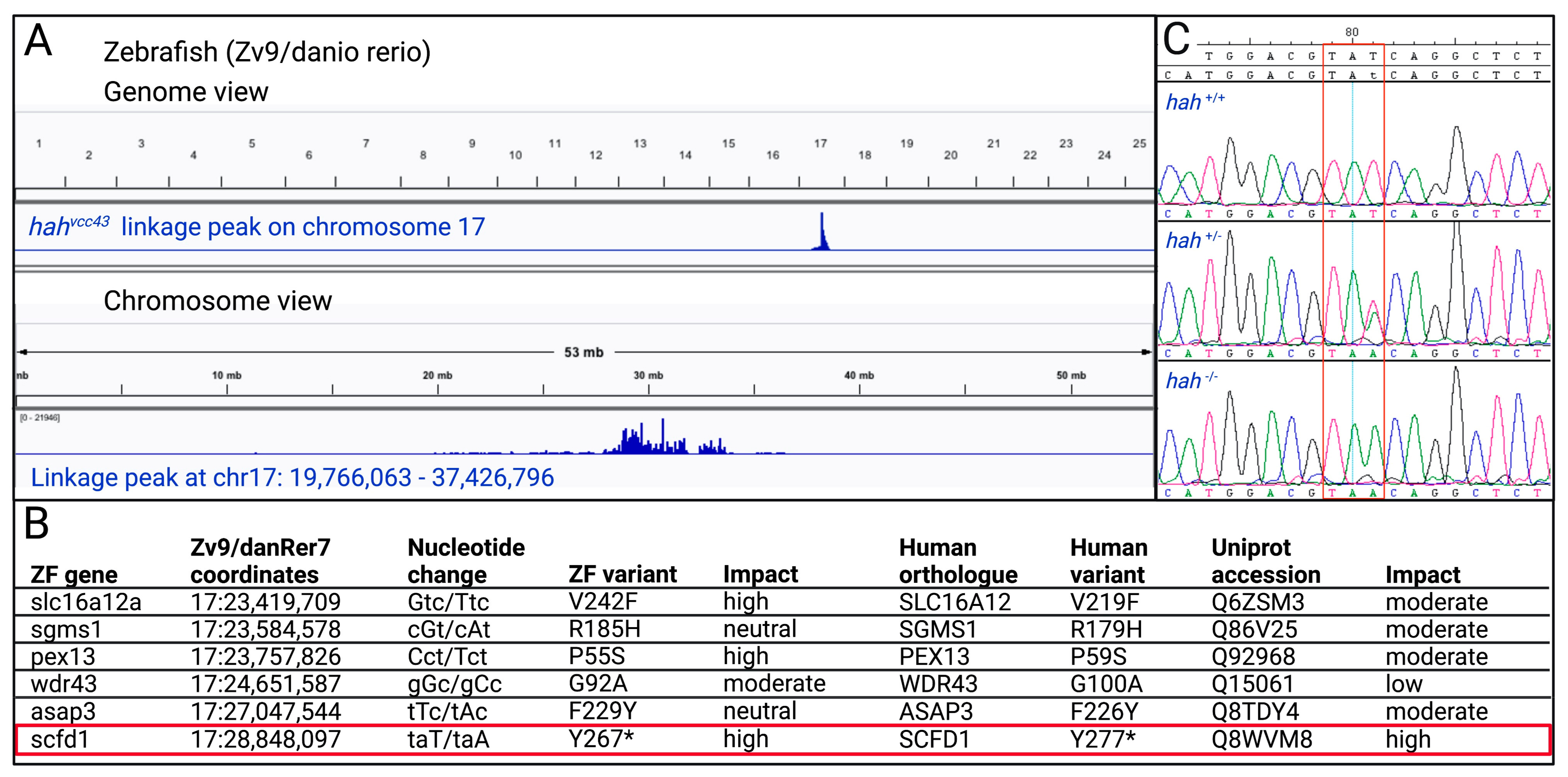
3.1. Identification of scfd1 as a Candidate Gene at the hahvcc43 Locus
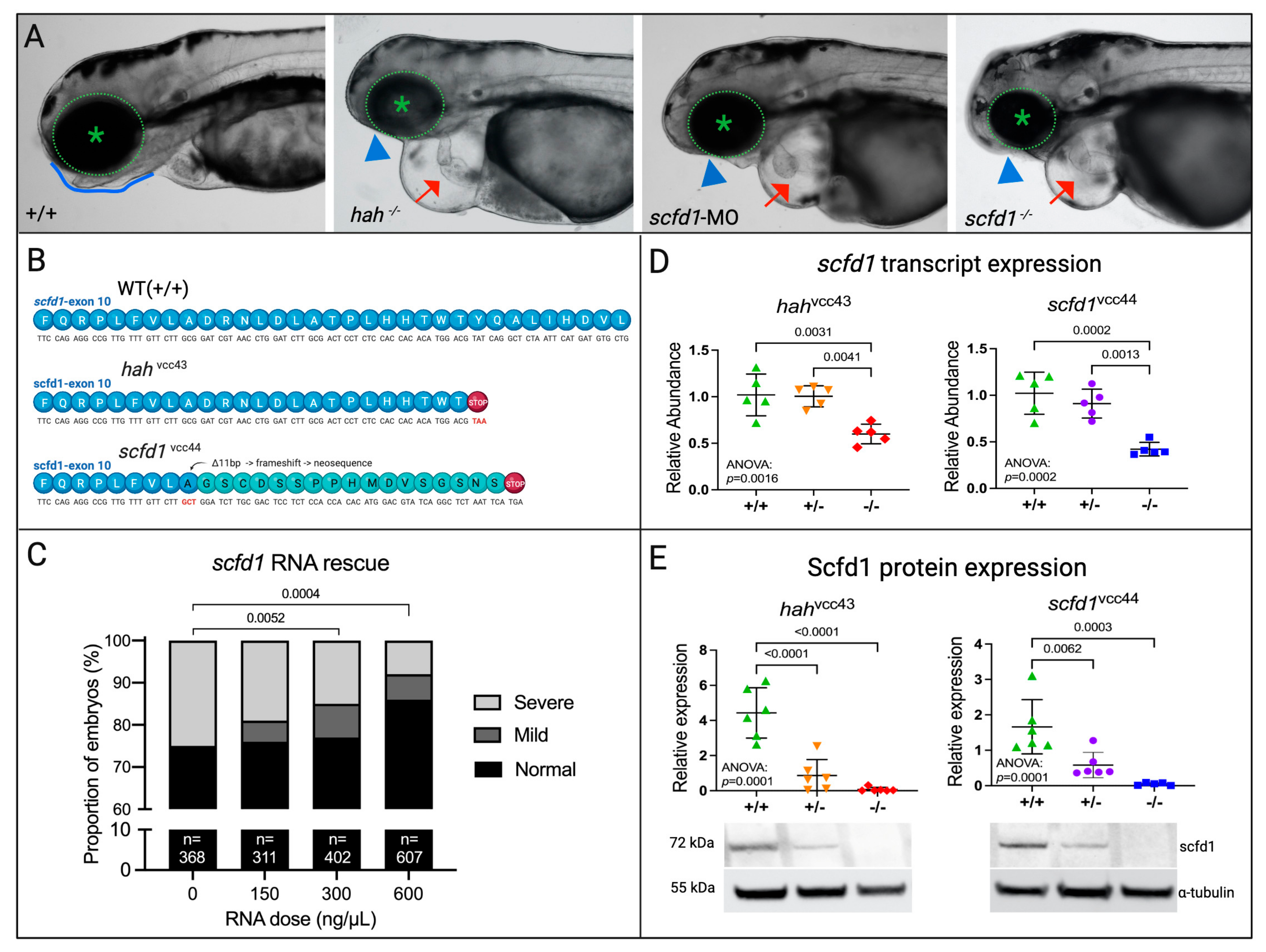
3.2. A CRISPR-Cas9-Engineered scfd1 Null Mutant Phenocopies hahvcc43
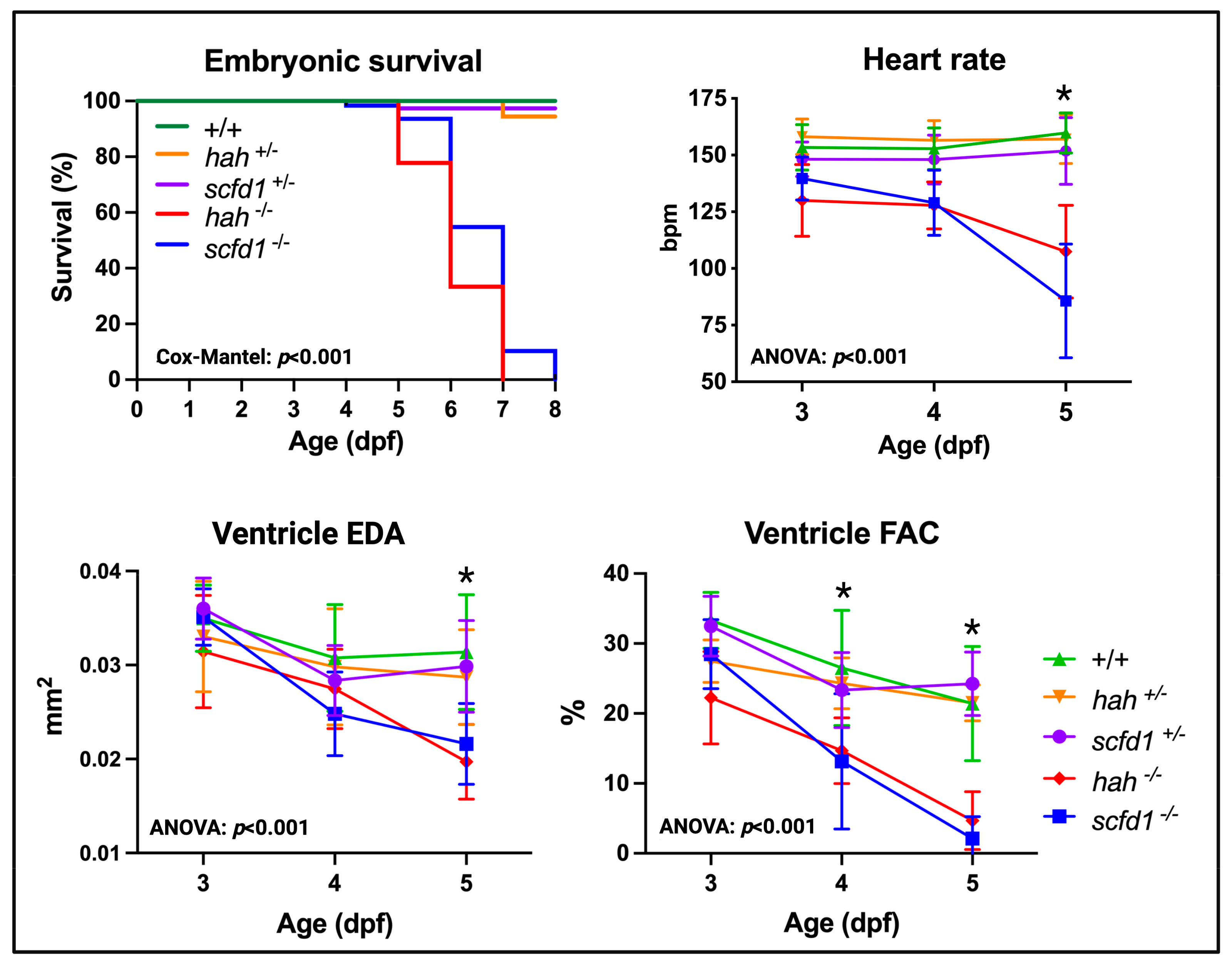
3.3. Scfd1 Loss Leads to Cardiac Dysfunction in Embryonic Zebrafish
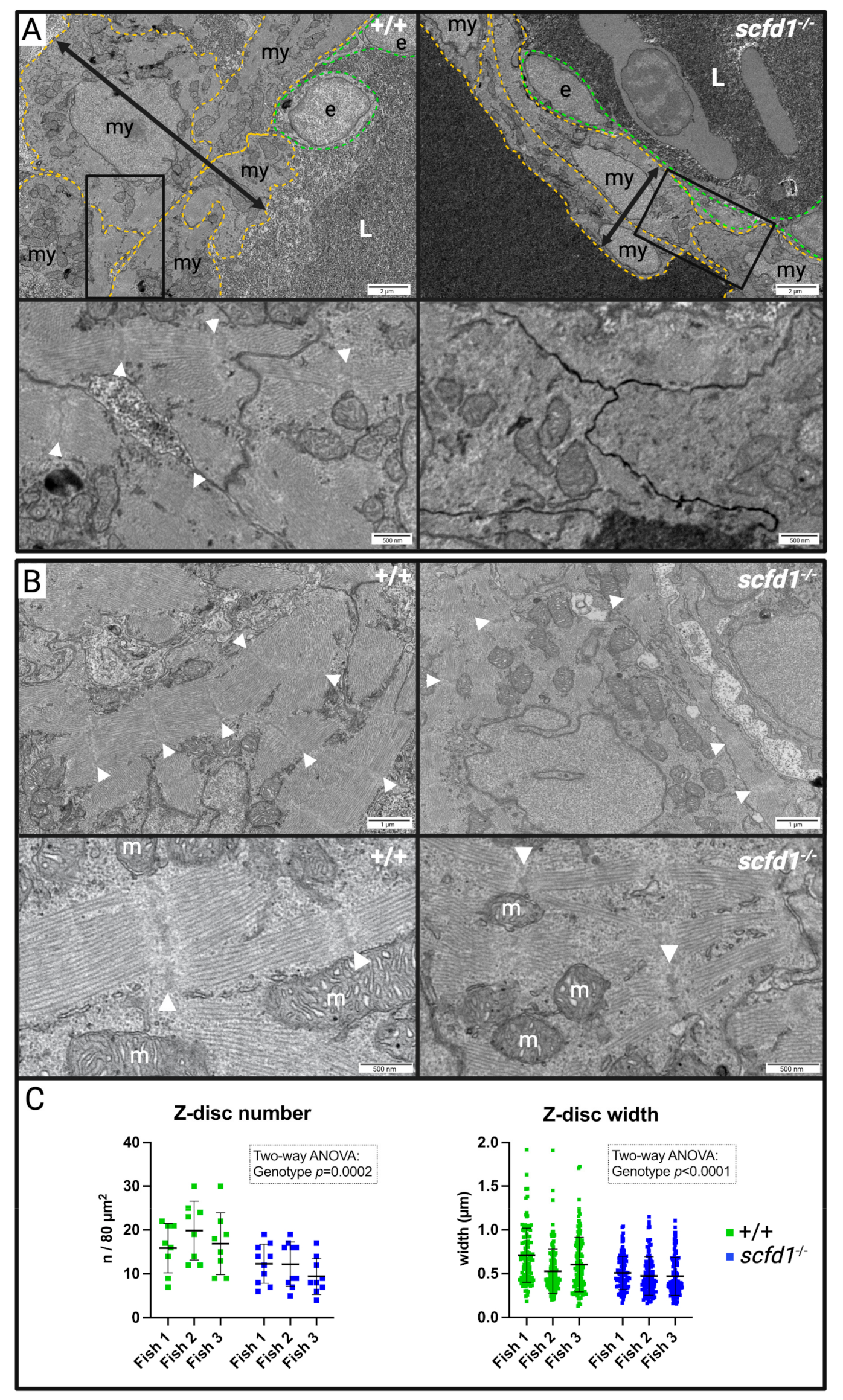
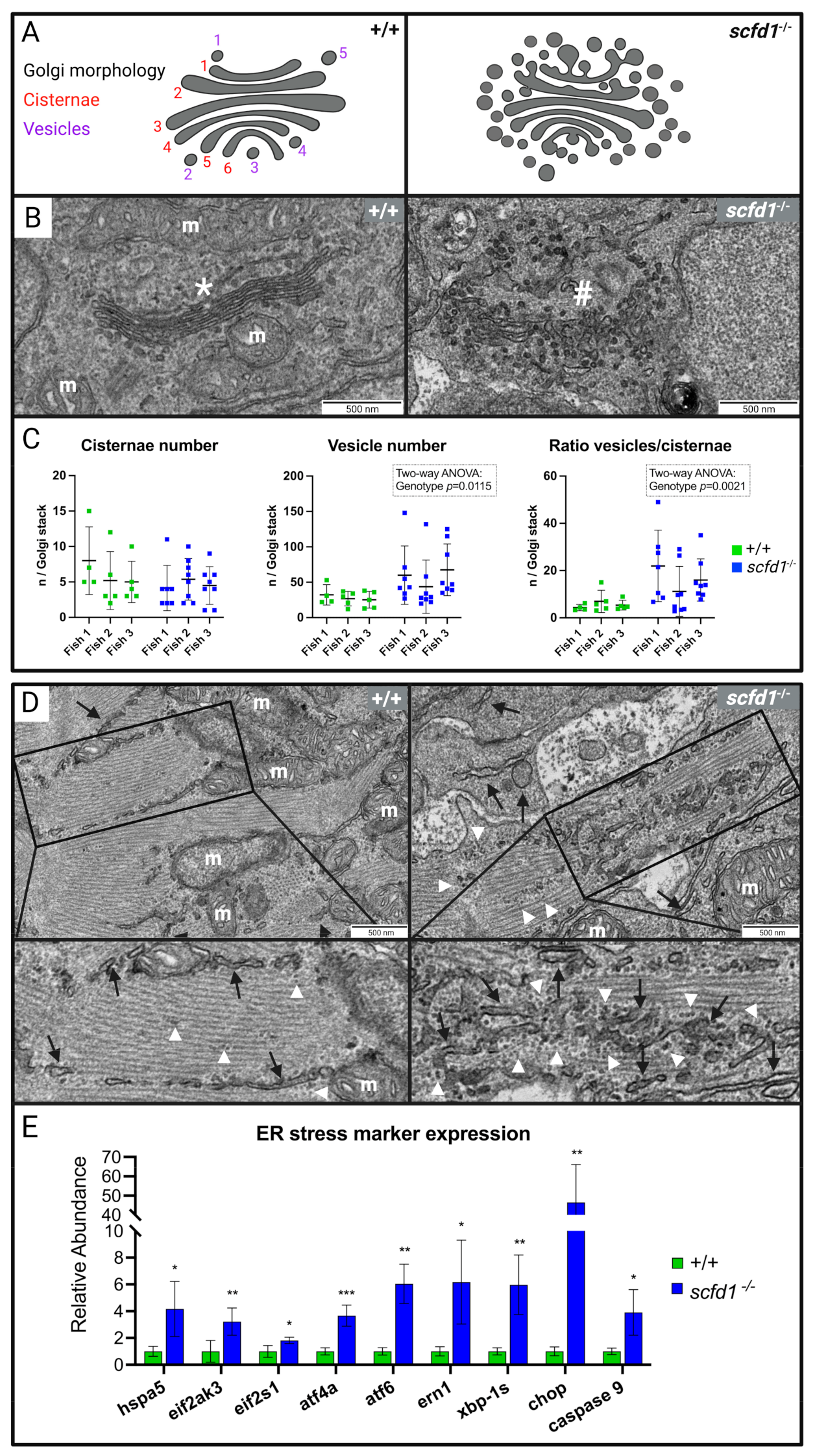
3.4. Electron Microscopy Showed Reduced Myofibril Width and Sarcomere Density, as well as Abnormal Golgi Apparatus and Reticular Network Morphology in scfd1vcc44−/− Mutant Embryos
3.5. Increased ER Stress Responses in scfd1vcc44−/− Embryos
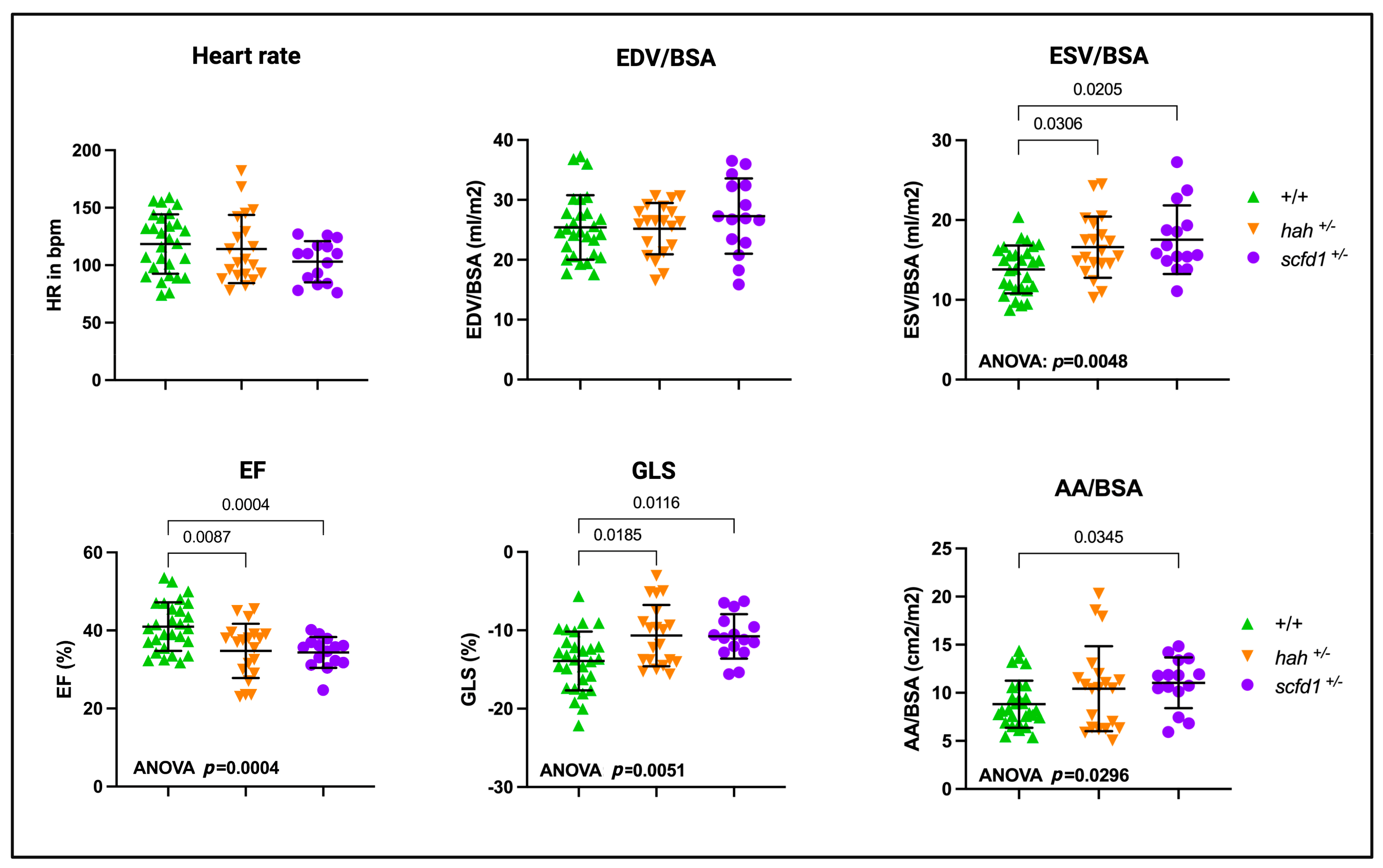
3.6. Partial Scfd1 Deficiency Leads to Mild Cardiac Dysfunction in Adult Zebrafish
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fatkin, D.; Huttner, I.G.; Kovacic, J.C.; Seidman, J.G.; Seidman, C.E. Precision Medicine in the Management of Dilated Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 2921–2938. [Google Scholar] [CrossRef]
- Burke, M.A.; Cook, S.A.; Seidman, J.G.; Seidman, C.E. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2871–2886. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.G.; Myers, V.D.; Dubey, P.; Dubey, S.; Perez, E.; Moravec, C.S.; Willis, M.S.; Feldman, A.M.; Kirk, J.A. Cardiomyocyte contractile impairment in heart failure results from reduced BAG3-mediated sarcomeric protein turnover. Nat. Commun. 2021, 12, 2942. [Google Scholar] [CrossRef]
- Agarwal, R.; Paulo, J.A.; Toepfer, C.N.; Ewoldt, J.K.; Sundaram, S.; Chopra, A.; Zhang, Q.; Gorham, J.; DePalma, S.R.; Chen, C.S.; et al. Filamin C Cardiomyopathy Variants Cause Protein and Lysosome Accumulation. Circ. Res. 2021, 129, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yussman, M.G.; Barrett, T.J.; Hahn, H.S.; Osinska, H.; Hilliard, G.M.; Wang, X.; Toyokawa, T.; Yatani, A.; Lynch, R.A.; et al. Increased myocardial Rab GTPase expression: A consequence and cause of cardiomyopathy. Circ. Res. 2001, 89, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Shih, Y.H.; Zhang, Y.; Ding, Y.; Ross, C.A.; Li, H.; Olson, T.M.; Xu, X. Cardiac transcriptome and dilated cardiomyopathy genes in zebrafish. Circ. Cardiovasc. Genet. 2015, 8, 261–269. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Xu, X.; Meiler, S.E.; Zhong, T.P.; Mohideen, M.; Crossley, D.A.; Burggren, W.W.; Fishman, M.C. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat. Genet. 2002, 30, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Sehnert, A.J.; Huq, A.; Weinstein, B.M.; Walker, C.; Fishman, M.C.; Stainier, D.Y. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Genet. 2002, 31, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Rottbauer, W.; Wessels, G.; Dahme, T.; Just, S.; Trano, N.; Hassel, D.; Burns, C.G.; Katus, H.A.; Fishman, M.C. Cardiac myosin light chain-2: A novel essential component of thick-myofilament assembly and contractility of the heart. Circ. Res. 2006, 99, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Huttner, I.G.; Wang, L.W.; Santiago, C.F.; Horvat, C.; Johnson, R.; Cheng, D.; von Frieling-Salewsky, M.; Hillcoat, K.; Bemand, T.J.; Trivedi, G.; et al. Titin Truncation in Zebrafish Causes Dilated Cardiomyopathy and Hemodynamic Stress Intolerance. Circ. Genom. Precis. Med. 2018, 11, e002135. [Google Scholar] [CrossRef]
- Carr, C.M.; Rizo, J. At the junction of SNARE and SM protein function. Curr. Opin. Cell Biol. 2010, 22, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Dascher, C.; Ossig, R.; Gallwitz, D.; Schmitt, H.D. Identification and structure of four yeast genes (SLY) that are able to suppress the functional loss of YPT1, a member of the RAS superfamily. Mol. Cell. Biol. 1991, 11, 872–885. [Google Scholar] [CrossRef]
- Lobingier, B.T.; Nickerson, D.P.; Lo, S.-Y.; Merz, A.J. SM proteins Sly1 and Vps33 co-assemble with Sec17 and SNARE complexes to oppose SNARE disassembly by Sec18. eLife 2014, 3, e02272. [Google Scholar] [CrossRef]
- Nishiwaki, Y.; Yoshizawa, A.; Kojima, Y.; Oguri, E.; Nakamura, S.; Suzuki, S.; Yuasa-Kawada, J.; Kinoshita-Kawada, M.; Mochizuki, T.; Masai, I. The BH3-only SNARE BNip1 mediates photoreceptor apoptosis in response to vesicular fusion defects. Dev. Cell 2013, 25, 374–387. [Google Scholar] [CrossRef]
- Hou, N.; Yang, Y.; Scott, I.C.; Lou, X. The Sec domain protein Scfd1 facilitates trafficking of ECM components during chondrogenesis. Dev. Biol. 2017, 421, 8–15. [Google Scholar] [CrossRef]
- Nechiporuk, A.; Poss, K.D.; Johnson, S.L.; Keating, M.T. Positional cloning of a temperature-sensitive mutant emmental reveals a role for sly1 during cell proliferation in zebrafish fin regeneration. Dev. Biol. 2003, 258, 291–306. [Google Scholar] [CrossRef]
- Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio Rerio), 4th ed.; University of Oregon Press: Eugene, OR, USA, 2000. [Google Scholar]
- Koltowska, K.; Paterson, S.; Bower, N.I.; Baillie, G.J.; Lagendijk, A.K.; Astin, J.W.; Chen, H.; Francois, M.; Crosier, P.S.; Taft, R.J.; et al. mafba is a downstream transcriptional effector of Vegfc signaling essential for embryonic lymphangiogenesis in zebrafish. Genes Dev. 2015, 29, 1618–1630. [Google Scholar] [CrossRef]
- Baek, S.; Oh, T.G.; Secker, G.; Sutton, D.L.; Okuda, K.S.; Paterson, S.; Bower, N.I.; Toubia, J.; Koltowska, K.; Capon, S.J.; et al. The Alternative Splicing Regulator Nova2 Constrains Vascular Erk Signaling to Limit Specification of the Lymphatic Lineage. Dev. Cell 2019, 49, 279–292.e5. [Google Scholar] [CrossRef] [PubMed]
- Leshchiner, I.; Alexa, K.; Kelsey, P.; Adzhubei, I.; Austin-Tse, C.A.; Cooney, J.D.; Anderson, H.; King, M.J.; Stottmann, R.W.; Garnaas, M.K.; et al. Mutation mapping and identification by whole-genome sequencing. Genome Res. 2012, 22, 1541–1548. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- López-Ferrando, V.; Gazzo, A.; de la Cruz, X.; Modesto Orozco, M.; Gelpí, J.L. PMut: A web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res. 2017, 45, W222–W228. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef]
- Wang, L.W.; Huttner, I.G.; Santiago, C.F.; Kesteven, S.H.; Yu, Z.Y.; Feneley, M.P.; Fatkin, D. Standardized echocardiographic assessment of cardiac function in normal adult zebrafish and heart disease models. Dis. Models Mech. 2017, 10, 63–76. [Google Scholar] [CrossRef]
- Lan, C.C.; Tang, R.; Leong, I.U.S.; Love, D.R. Quantitative real-time RT-PCR (qRT-PCR) of zebrafish transcripts: Optimization of RNA extraction, quality control considerations, and data analysis. Cold Spring Harb. Protoc. 2009, 4, pdb.prot5314. [Google Scholar] [CrossRef]
- Deerinck, T.J.; Bushong, E.A.; Ellisman, M.H.; Thor, A. Preparation of Biological Tissues for Serial Block Face Scanning Electron Microscopy (SBEM) V.2; University of California: San Diego, CA, USA, 2022. [Google Scholar] [CrossRef]
- Tapia, J.C.; Kasthuri, N.; Hayworth, K.J.; Schalek, R.; Lichtman, J.W.; Smith, S.J.; Buchanan, J. High-contrast en bloc staining of neuronal tissue for field emission scanning electron microscopy. Nat. Protoc. 2012, 7, 193–206. [Google Scholar] [CrossRef]
- Berger, J.; Berger, S.; Jacoby, A.S.; Wilton, S.D.; Currie, P.D. Evaluation of exon-skipping strategies for Duchenne muscular dystrophy utilizing dystrophin-deficient zebrafish. J. Cell. Mol. Med. 2011, 15, 2643–2651. [Google Scholar] [CrossRef]
- Michalak, M.; Opas, M. ER and SR in the heart. Trends Cell Biol. 2009, 19, 253–259. [Google Scholar] [CrossRef]
- Doroudgar, S.; Glembotski, C.C. New concepts of endoplasmic reticulum function in the heart: Programmed to conserve. J. Mol. Cell. Cardiol. 2013, 55, 85–91. [Google Scholar] [CrossRef]
- Prola, A.; Nichtova, Z.; Da Silva, J.P.; Piquereau, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Ventura-Clapier, R.; Garnier, A.; Zahradnik, I.; et al. Endoplasmic reticulum stress induces cardiac dysfunction through architectural modifications and alteration of mitochondrial function in cardiomyocytes. Cardiovasc. Res. 2019, 115, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Glembotski, C.C. Endoplasmic reticulum stress in the heart. Circ. Res. 2007, 101, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L. Cleaning Up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Ossig, R.; Dascher, C.; Trepte, H.H.; Schmitt, H.D.; Gallwitz, D. The yeast SLY gene products, suppressors of defects in the essential GTP-binding Ypt1 protein, may act in endoplasmic reticulum-to-Golgi transport. Mol. Cell. Biol. 1991, 11, 2980–2993. [Google Scholar] [CrossRef]
- Mizuta, K.; Warner, J.R. Continued functioning of the secretory pathway is essential for ribosome synthesis. Mol. Cell. Biol. 1994, 14, 2493–2502. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Dulubova, I.; Min, S.W.; Chen, X.; Rizo, J.; Südhof, T.C. Sly1 binds to Golgi and ER syntaxins via a conserved N-terminal peptide motif. Dev. Cell 2002, 2, 295–305. [Google Scholar] [CrossRef]
- Linders, P.T.; Horst, C.V.; Beest, M.T.; van den Bogaart, G. Stx5-Mediated ER-Golgi Transport in Mammals and Yeast. Cells 2019, 8, 780. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.; Erlmann, P.; Villeneuve, J.; Santos, A.J.M.; Martinez-Alonso, E.; Martinez-Menarguez, J.A.; Malhotra, V. SLY1 and Syntaxin 18 specify a distinct pathway for procollagen VII export from the endoplasmic reticulum. eLife 2014, 3, e02784. [Google Scholar] [CrossRef] [PubMed]
- Courchaine, K.; Rykiel, G.; Rugonyi, S. Influence of blood flow on cardiac development. Prog. Biophys. Mol. Biol. 2018, 137, 95–110. [Google Scholar] [CrossRef]
- Renna, M.; Schaffner, C.; Winslow, A.R.; Menzies, F.M.; Peden, A.A.; Floto, R.A.; Rubinsztein, D.C. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J. Cell Sci. 2011, 124 Pt 3, 469–482. [Google Scholar] [CrossRef]
- Clark, E.M.; Link, B.A. Complementary and divergent functions of zebrafish Tango1 and Ctage5 in tissue development and homeostasis. Mol. Biol. Cell 2021, 32, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef]
- Chen, K.; Li, X.; Song, G.; Zhou, T.; Long, Y.; Li, Q.; Zhong, S.; Cui, Z. Deficiency in the membrane protein Tmbim3a/Grinaa initiates cold-induced ER stress and cell death by activating an intrinsic apoptotic pathway in zebrafish. J. Biol. Chem. 2019, 294, 11445–11457. [Google Scholar] [CrossRef]
- Wang, D.Y.; Abbasi, C.; El-Rass, S.; Li, J.Y.; Dawood, F.; Naito, K.; Sharma, P.; Bousette, N.; Singh, S.; Backx, P.H.; et al. Endoplasmic reticulum resident protein 44 (ERp44) deficiency in mice and zebrafish leads to cardiac developmental and functional defects. J. Am. Heart Assoc. 2014, 3, e001018. [Google Scholar] [CrossRef]
- Lee, S.H.; Hadipour-Lakmehsari, S.; Murthy, H.R.; Gibb, N.; Miyake, T.; Teng, A.C.T.; Cosme, J.; Yu, J.C.; Moon, M.; Lim, S.; et al. REEP5 depletion causes sarco-endoplasmic reticulum vacuolization and cardiac functional defects. Nat. Commun. 2020, 11, 965. [Google Scholar] [CrossRef]
- Stephens, S.B.; Dodd, R.D.; Brewer, J.W.; Lager, P.J.; Keene, J.D.; Nicchitta, C.V. Stable ribosome binding to the endoplasmic reticulum enables compartment-specific regulation of mRNA translation. Mol. Biol. Cell 2005, 16, 5819–5831. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, J.; Jiang, Y.; Chen, T. Fine particulate matter induces heart defects via AHR/ROS-mediated endoplasmic reticulum stress. Chemosphere 2022, 307 Pt 2, 135962. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Dang, Y.; Xu, Q.; Yu, L.; Liu, C.; Yuan, Y.; Wang, J. Microcystin-LR induced developmental toxicity and apoptosis in zebrafish (Danio rerio) larvae by activation of ER stress response. Chemosphere 2016, 157, 166–173. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huttner, I.G.; Santiago, C.F.; Jacoby, A.; Cheng, D.; Trivedi, G.; Cull, S.; Cvetkovska, J.; Chand, R.; Berger, J.; Currie, P.D.; et al. Loss of Sec-1 Family Domain-Containing 1 (scfd1) Causes Severe Cardiac Defects and Endoplasmic Reticulum Stress in Zebrafish. J. Cardiovasc. Dev. Dis. 2023, 10, 408. https://doi.org/10.3390/jcdd10100408
Huttner IG, Santiago CF, Jacoby A, Cheng D, Trivedi G, Cull S, Cvetkovska J, Chand R, Berger J, Currie PD, et al. Loss of Sec-1 Family Domain-Containing 1 (scfd1) Causes Severe Cardiac Defects and Endoplasmic Reticulum Stress in Zebrafish. Journal of Cardiovascular Development and Disease. 2023; 10(10):408. https://doi.org/10.3390/jcdd10100408
Chicago/Turabian StyleHuttner, Inken G., Celine F. Santiago, Arie Jacoby, Delfine Cheng, Gunjan Trivedi, Stephen Cull, Jasmina Cvetkovska, Renee Chand, Joachim Berger, Peter D. Currie, and et al. 2023. "Loss of Sec-1 Family Domain-Containing 1 (scfd1) Causes Severe Cardiac Defects and Endoplasmic Reticulum Stress in Zebrafish" Journal of Cardiovascular Development and Disease 10, no. 10: 408. https://doi.org/10.3390/jcdd10100408
APA StyleHuttner, I. G., Santiago, C. F., Jacoby, A., Cheng, D., Trivedi, G., Cull, S., Cvetkovska, J., Chand, R., Berger, J., Currie, P. D., Smith, K. A., & Fatkin, D. (2023). Loss of Sec-1 Family Domain-Containing 1 (scfd1) Causes Severe Cardiac Defects and Endoplasmic Reticulum Stress in Zebrafish. Journal of Cardiovascular Development and Disease, 10(10), 408. https://doi.org/10.3390/jcdd10100408