Right Ventricular Adaptation in Congenital Heart Diseases


Abstract
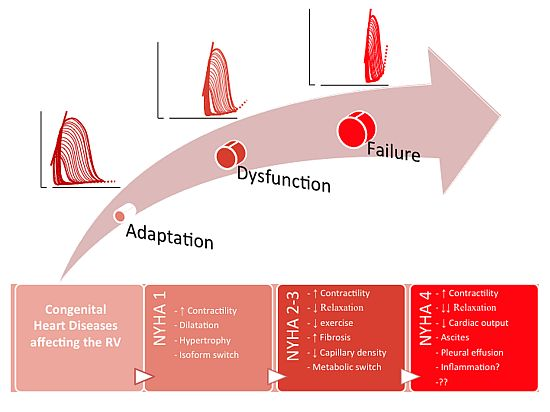
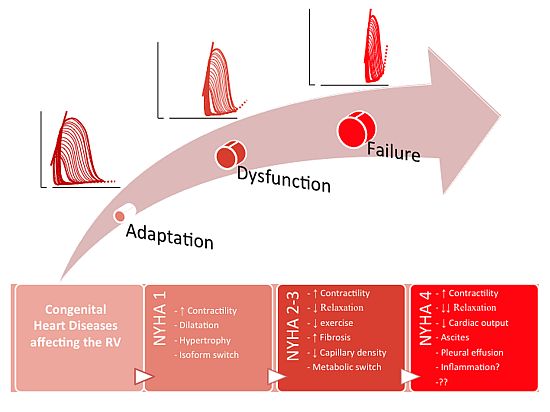
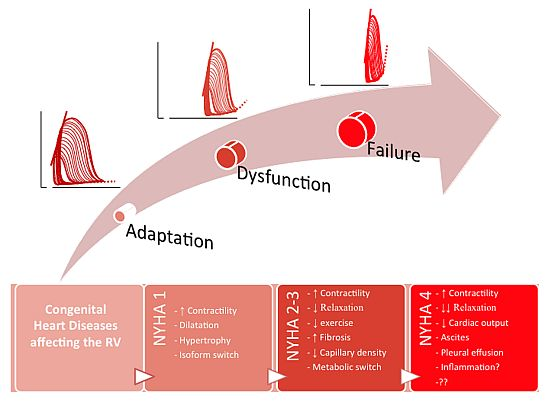
:
1. Introduction
2. RV Response to Stress
2.1. Building Blocks of RV Adaptation
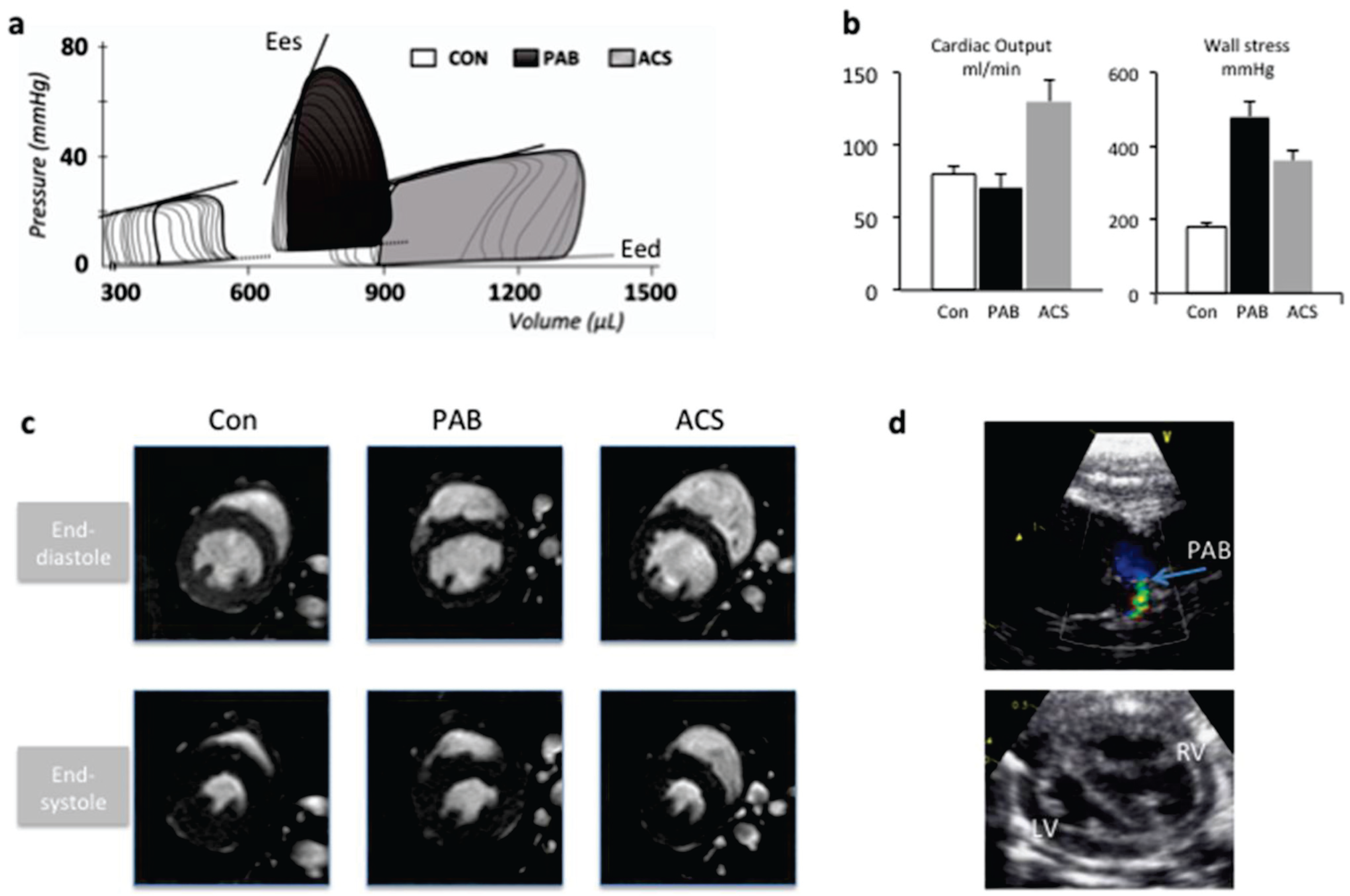
2.2. Modeling Abnormal Loading Conditions of the RV
2.3. RV Adaptation to Pressure Load
2.4. RV Adaptation to Volume Load
2.5. Treatment of RV Failure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RV dysfunction described? | ref | |
|---|---|---|
| Increased volume load | ||
| Atrial septal defect | >25% mild RV dysfunction 30 years after correction (CMR) | [62] |
| Pulmonary insufficiency | 30% symptomatic after 40 years | [52] |
| Tricuspid insufficiency | Reduces survival independent of other defects | [63] |
| Increased pressure load | ||
| Pulmonary stenosis | Reduced exercise capacity | [64] |
| Pulmonary Hypertension | RV failure predicts outcome | [7] |
| Tetralogy of Fallot | Fatal if uncorrected | |
| Congenitally corrected transposition of the great arteries (ccTGA) | >25% RV failure after 45 y when there are no associated lesions | [65] |
| Transposition of the great arteries after atrial switch procedure (TGA-as) | Reduced RV function similar to ccTGA | [66] |
| Mixed Lesions | ||
| Atrial Septal Defect + Pulmonary stenosis/Pulmonary Hypertension | Reduced survival in patients with ASD-PH | [67,68] |
| Corrected Fallot + pulmonary insufficiency | Risk for Sudden Death, arrhythmias | [69] |
| ccTGA + tricuspid insufficiency | Risk of RV failure increases from 25 → 60% | [65] |
| TGA-as + tricuspid insufficiency | Idem ccTGA |
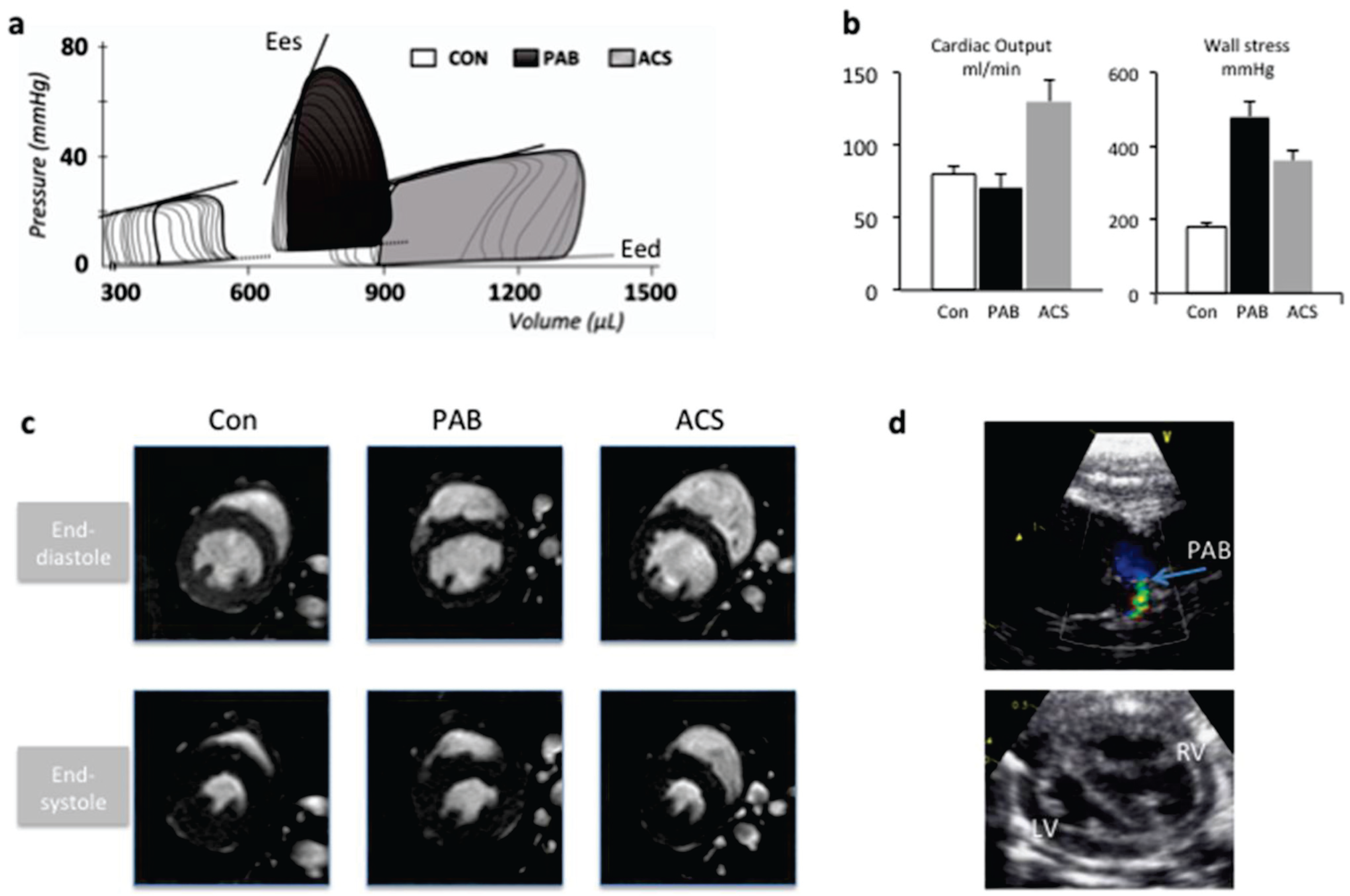
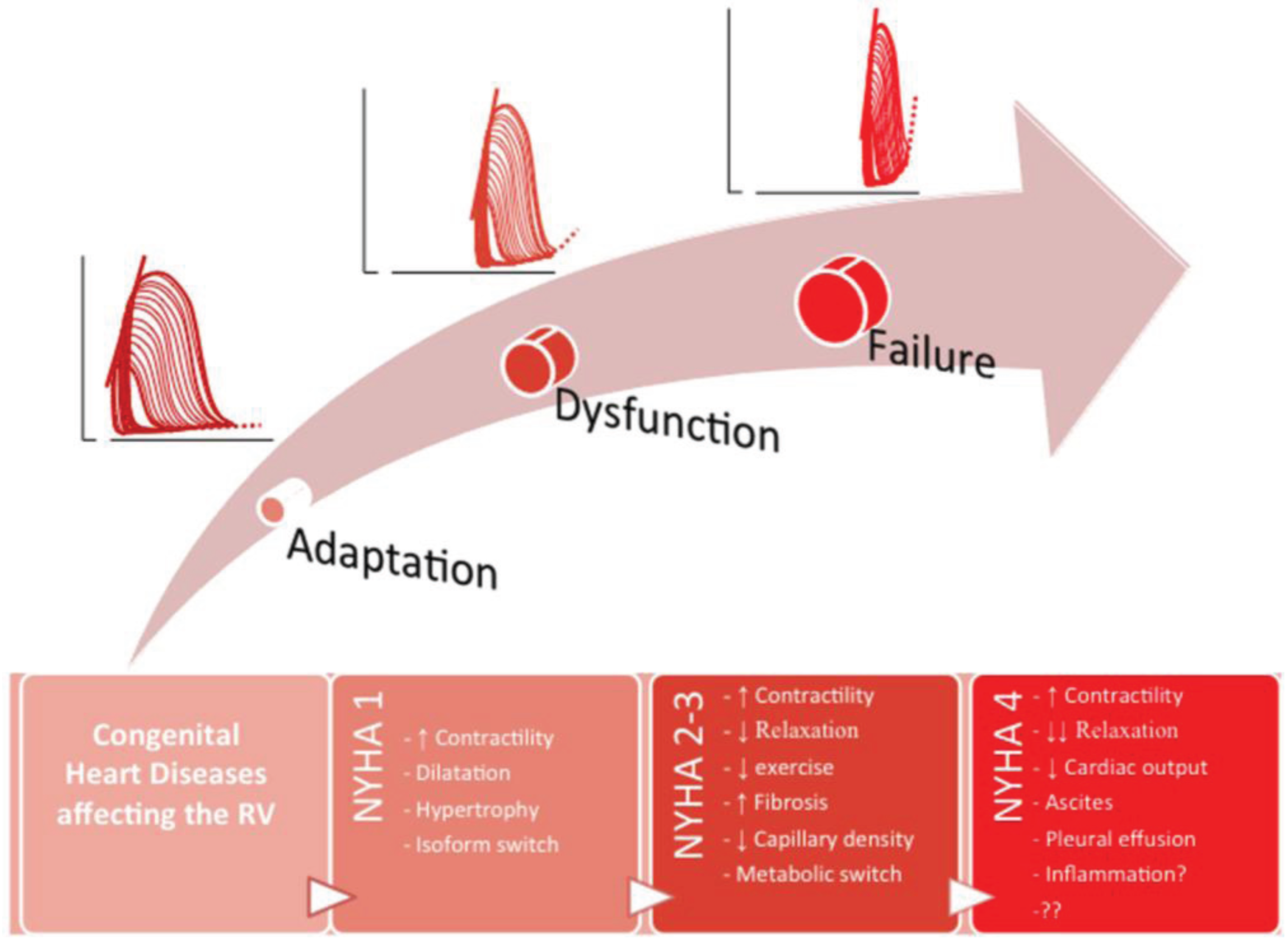
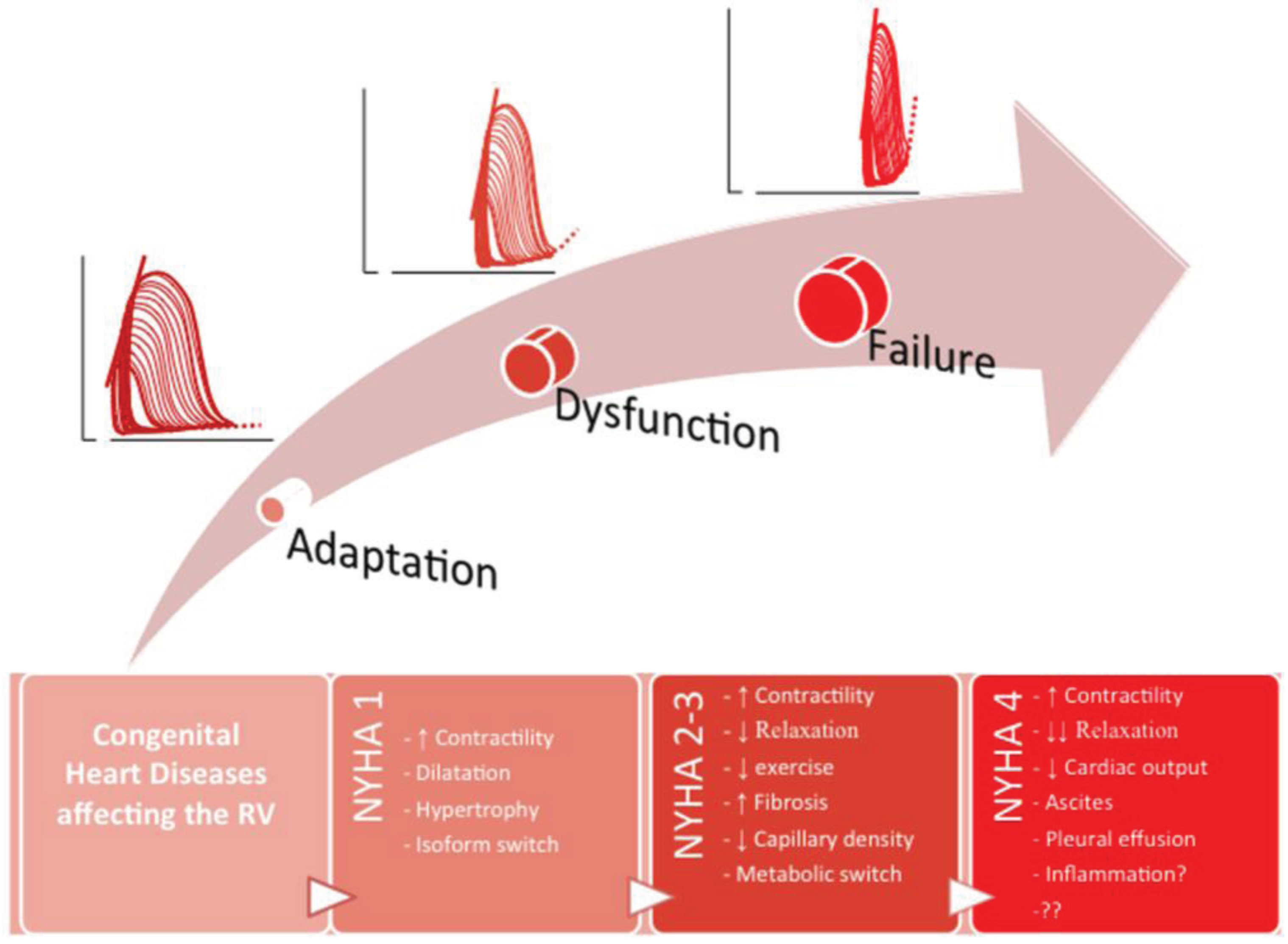
3. Summary and Future Perspectives
Author Contributions
Conflicts of Interest
References
- Zomer, A.C.; Vaartjes, I.; Uiterwaal, C.S.; van der Velde, E.T.; van den Merkhof, L.F.; Baur, L.H.; Ansink, T.J.; Cozijnsen, L.; Pieper, P.G.; Meijboom, F.J.; et al. Circumstances of death in adult congenital heart disease. Int. J. Cardiol. 2012, 154, 168–172. [Google Scholar] [CrossRef]
- Norozi, K.; Wessel, A.; Alpers, V.; Arnhold, J.O.; Geyer, S.; Zoege, M.; Buchhorn, R. Incidence and risk distribution of heart failure in adolescents and adults with congenital heart disease after cardiac surgery. Am. J. Cardiol. 2006, 97, 1238–1243. [Google Scholar] [CrossRef]
- Kelly, R.G.; Brown, N.A.; Buckingham, M.E. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev. Cell 2001, 1, 435–440. [Google Scholar] [CrossRef]
- Ho, S.Y.; Nihoyannopoulos, P. Anatomy, echocardiography, and normal right ventricular dimensions. Heart 2006, 92 (Suppl. 1), i2–i13. [Google Scholar] [CrossRef]
- Bartelds, B.; Berger, R.M.F. The right ventricle in congenital heart diseases. In The Right Ventricle; Gaine, S., Naeije, R., Paecock, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Haddad, F.; Doyle, R.; Murphy, D.J.; Hunt, S.A. Right ventricular function in cardiovascular disease, part II: Pathophysiology, clinical importance, and management of right ventricular failure. Circulation 2008, 117, 1717–1731. [Google Scholar] [CrossRef]
- Van Wolferen, S.A.; Marcus, J.T.; Boonstra, A.; Marques, K.M.; Bronzwaer, J.G.; Spreeuwenberg, M.D.; Postmus, P.E.; Vonk-Noordegraaf, A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur. Heart J. 2007, 28, 1250–1257. [Google Scholar] [CrossRef]
- Ghio, S.; Gavazzi, A.; Campana, C.; Inserra, C.; Klersy, C.; Sebastiani, R.; Arbustini, E.; Recusani, F.; Tavazzi, L. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J. Am. Coll. Cardiol. 2001, 37, 183–188. [Google Scholar] [CrossRef]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef]
- Freling, H.G.; Pieper, P.G.; Vermeulen, K.M.; van Swieten, J.M.; Sijens, P.E.; van Veldhuisen, D.J.; Willems, T.P. Improved cardiac MRI volume measurements in patients with tetralogy of Fallot by independent end-systolic and end-diastolic phase selection. PLoS ONE 2013, 8, e55462. [Google Scholar] [CrossRef]
- Bartelds, B.; Borgdorff, M.A.; Smit-van Oosten, A.; Takens, J.; Boersma, B.; Nederhoff, M.G.; Elzenga, N.J.; van Gilst, W.H.; de Windt, L.J.; Berger, R.M. Differential responses of the right ventricle to abnormal loading conditions in mice: pressure vs. volume load. Eur. J. Heart Fail. 2011, 13, 1275–1282. [Google Scholar] [CrossRef]
- Bouzas, B.; Kilner, P.J.; Gatzoulis, M.A. Pulmonary regurgitation: not a benign lesion. Eur. Heart J. 2005, 26, 433–439. [Google Scholar]
- Apitz, C.; Honjo, O.; Humpl, T.; Li, J.; Assad, R.S.; Cho, M.Y.; Hong, J.; Friedberg, M.K.; Redington, A.N. Biventricular structural and functional responses to aortic constriction in a rabbit model of chronic right ventricular pressure overload. J. Thorac. Cardiovasc. Surg. 2012, 144, 1494–1501. [Google Scholar] [CrossRef] [Green Version]
- Borgdorff, M.A.; Bartelds, B.; Dickinson, M.G.; Steendijk, P.; de Vroomen, M.; Berger, R.M. Distinct loading conditions reveal various patterns of right ventricular adaptation. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H354–H364. [Google Scholar] [CrossRef]
- Leeuwenburgh, B.P.; Helbing, W.A.; Steendijk, P.; Schoof, P.H.; Baan, J. Biventricular systolic function in young lambs subject to chronic systemic right ventricular pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2697–H2704. [Google Scholar]
- Gaynor, S.L.; Maniar, H.S.; Bloch, J.B.; Steendijk, P.; Moon, M.R. Right atrial and ventricular adaptation to chronic right ventricular pressure overload. Circulation 2005, 112, I212–I218. [Google Scholar]
- Hessel, M.H.; Steendijk, P.; den Adel, B.; Schutte, C.I.; van der Laarse, A. Characterization of right ventricular function after monocrotaline-induced pulmonary hypertension in the intact rat. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2424–H2430. [Google Scholar] [CrossRef]
- Dickinson, M.G.; Bartelds, B.; Borgdorff, M.A.; Berger, R.M. The role of disturbed blood flow in the development of pulmonary arterial hypertension: lessons from preclinical animal models. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2013, 305, L1–L14. [Google Scholar] [CrossRef]
- Piao, L.; Marsboom, G.; Archer, S.L. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J. Mol. Med. 2010, 88, 1011–1020. [Google Scholar] [CrossRef]
- Kreymborg, K.; Uchida, S.; Gellert, P.; Schneider, A.; Boettger, T.; Voswinckel, R.; Wietelmann, A.; Szibor, M.; Weissmann, N.; Ghofrani, A.H.; et al. Identification of right heart-enriched genes in a murine model of chronic outflow tract obstruction. J. Mol. Cell. Cardiol. 2010, 49, 598–605. [Google Scholar] [CrossRef]
- Urashima, T.; Zhao, M.; Wagner, R.; Fajardo, G.; Farahani, S.; Quertermous, T.; Bernstein, D. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1351–H1368. [Google Scholar] [CrossRef]
- Borgdorff, M.A.; Bartelds, B.; Dickinson, M.G.; Boersma, B.; Weij, M.; Zandvoort, A.; Sillje, H.H.; Steendijk, P.; de Vroomen, M.; Berger, R.M. Sildenafil enhances systolic adaptation, but does not prevent diastolic dysfunction, in the pressure-loaded right ventricle. Eur. J. Heart Fail. 2012, 14, 1067–1074. [Google Scholar] [CrossRef]
- Faber, M.J.; Dalinghaus, M.; Lankhuizen, I.M.; Steendijk, P.; Hop, W.C.; Schoemaker, R.G.; Duncker, D.J.; Lamers, J.M.; Helbing, W.A. Right and left ventricular function after chronic pulmonary artery banding in rats assessed with biventricular pressure-volume loops. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1580–H1586. [Google Scholar] [CrossRef]
- Rondelet, B.; Dewachter, L.; Kerbaul, F.; Dewachter, C.; Hubloue, I.; Fesler, P.; Franck, S.; Remmelink, M.; Brimioulle, S.; Naeije, R. Sildenafil added to sitaxsentan in overcirculation-induced pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1118–H1123. [Google Scholar] [CrossRef]
- Reddy, S.; Zhao, M.; Hu, D.Q.; Fajardo, G.; Katznelson, E.; Punn, R.; Spin, J.M.; Chan, F.P.; Bernstein, D. Physiologic and molecular characterization of a murine model of right ventricular volume overload. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1314–H1327. [Google Scholar] [CrossRef]
- Bove, T.; Bouchez, S.; de Hert, S.; Wouters, P.; de Somer, F.; Devos, D.; Somers, P.; van Nooten, G. Acute and chronic effects of dysfunction of right ventricular outflow tract components on right ventricular performance in a porcine model: Implications for primary repair of tetralogy of fallot. J. Am. Coll. Cardiol. 2012, 60, 64–71. [Google Scholar] [CrossRef]
- Borgdorff, M.A.; Bartelds, B.; Dickinson, M.G.; Steendijk, P.; Berger, R.M. A cornerstone of heart failure treatment is not effective in experimental right ventricular failure. Int. J. Cardiol. 2013, 169, 183–189. [Google Scholar] [CrossRef]
- Borgdorff, M.A.; Bartelds, B.; Dickinson, M.G.; Steendijk, P.; Koops, A.C.; Berger, R.M. Characterization of right ventricular failure in chronic experimental pressure load. J. Mol. Cell. Cardiol. 2014. submitted. [Google Scholar]
- Rain, S.; Handoko, M.L.; Trip, P.; Gan, C.T.; Westerhof, N.; Stienen, G.J.; Paulus, W.J.; Ottenheijm, C.A.; Marcus, J.T.; Dorfmuller, P.; et al. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation 2013, 128, 2016–2025. [Google Scholar] [CrossRef]
- Van Berlo, J.H.; Maillet, M.; Molkentin, J.D. Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Invest. 2013, 123, 37–45. [Google Scholar] [CrossRef]
- Bartelds, B.; Knoester, H.; Smid, G.B.; Takens, J.; Visser, G.H.; Penninga, L.; van der Leij, F.R.; Beaufort-Krol, G.C.; Zijlstra, W.G.; Heymans, H.S.; et al. Perinatal changes in myocardial metabolism in lambs. Circulation 2000, 102, 926–931. [Google Scholar] [CrossRef]
- Bartelds, B.; Borgdorff, M.A.; Boersma, B.; Takens, J.; Smit-van Oosten, A.; de WIndt, L.J.; Berger, R.M.F. Right ventricular adaptation to pressure load in mice is improved after blockade of calcineurin activation. Eur. Heart J. 2011, 31, 305. [Google Scholar]
- Reddy, S.; Zhao, M.; Hu, D.Q.; Fajardo, G.; Hu, S.; Ghosh, Z.; Rajagopalan, V.; Wu, J.C.; Bernstein, D. Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol. Genomics 2012, 44, 562–575. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Bonnet, S.; Haromy, A.; McMurtry, M.S.; Bleackley, R.C.; Michelakis, E.D. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFalpha contributes to the pathogenesis of pulmonary arterial hypertension. J. Mol. Med. 2011, 89, 771–783. [Google Scholar] [CrossRef]
- De Man, F.S.; Handoko, M.L.; van Ballegoij, J.J.; Schalij, I.; Bogaards, S.J.; Postmus, P.E.; van der Velden, J.; Westerhof, N.; Paulus, W.J.; Vonk-Noordegraaf, A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ. Heart Fail. 2012, 5, 97–105. [Google Scholar]
- Bogaard, H.J.; Abe, K.; Vonk Noordegraaf, A.; Voelkel, N.F. The right ventricle under pressure: Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009, 135, 794–804. [Google Scholar] [CrossRef]
- Van Albada, M.E.; Berger, R.M.; Niggebrugge, M.; van Veghel, R.; Cromme-Dijkhuis, A.H.; Schoemaker, R.G. Prostacyclin therapy increases right ventricular capillarisation in a model for flow-associated pulmonary hypertension. Eur. J. Pharmacol. 2006, 549, 107–116. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Paulin, R.; Zervopoulos, S.; Haromy, A.; Nagendran, J.; Michelakis, E.D. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J. Mol. Med. 2013, 91, 1315–1327. [Google Scholar] [CrossRef]
- Fang, Y.H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. 2012, 90, 31–43. [Google Scholar] [CrossRef]
- Nagendran, J.; Michelakis, E.D. Mitochondrial NOS is upregulated in the hypoxic heart: Implications for the function of the hypertrophied right ventricle. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1723–H1726. [Google Scholar] [CrossRef]
- Piao, L.; Fang, Y.H.; Cadete, V.J.; Wietholt, C.; Urboniene, D.; Toth, P.T.; Marsboom, G.; Zhang, H.J.; Haber, I.; Rehman, J.; et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J. Mol. Med. 2010, 88, 47–60. [Google Scholar] [CrossRef]
- Dromparis, P.; Michelakis, E.D. Mitochondria in vascular health and disease. Annu. Rev. Physiol. 2013, 75, 95–126. [Google Scholar] [CrossRef]
- Redout, E.M.; Wagner, M.J.; Zuidwijk, M.J.; Boer, C.; Musters, R.J.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res. 2007, 75, 770–781. [Google Scholar] [CrossRef]
- Redout, E.M.; van der Toorn, A.; Zuidwijk, M.J.; van de Kolk, C.W.; van Echteld, C.J.; Musters, R.J.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1038–H1047. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef]
- Hassoun, P.M.; Mouthon, L.; Barbera, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; Jones, P.L.; Maitland, M.L.; Michelakis, E.D.; Morrell, N.W.; et al. Inflammation, growth factors, and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 2009, 54, S10–S19. [Google Scholar] [CrossRef]
- Bartelds, B.; van Loon, R.L.; Mohaupt, S.; Wijnberg, H.; Dickinson, M.G.; Boersma, B.; Takens, J.; van Albada, M.; Berger, R.M. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest 2012, 141, 651–660. [Google Scholar] [CrossRef]
- Van Albada, M.E.; Bartelds, B.; Wijnberg, H.; Mohaupt, S.; Dickinson, M.G.; Schoemaker, R.G.; Kooi, K.; Gerbens, F.; Berger, R.M. Gene expression profile in flow-associated pulmonary arterial hypertension with neointimal lesions. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L483–L491. [Google Scholar] [CrossRef]
- Rajagopalan, V.; Zhao, M.; Reddy, S.; Fajardo, G.A.; Wang, X.; Dewey, S.; Gomes, A.V.; Bernstein, D. Altered Ubiquitin-Proteasome Signaling in Right Ventricular Hypertrophy and Failure. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H551–H562. [Google Scholar] [CrossRef]
- Dewachter, C.; Dewachter, L.; Rondelet, B.; Fesler, P.; Brimioulle, S.; Kerbaul, F.; Naeije, R. Activation of apoptotic pathways in experimental acute afterload-induced right ventricular failure. Crit. Care 2010, 38, 1405–1413. [Google Scholar] [CrossRef]
- Rondelet, B.; Dewachter, C.; Kerbaul, F.; Kang, X.; Fesler, P.; Brimioulle, S.; Naeije, R.; Dewachter, L. Prolonged overcirculation-induced pulmonary arterial hypertension as a cause of right ventricular failure. Eur. Heart J. 2012, 33, 1017–1026. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Blackstone, E.H.; Kirklin, J.W. The natural history of isolated congenital pulmonaryvalve incompetence: surgical implications. Thorac. Cardiovasc. Surg. 1984, 32, 257–259. [Google Scholar] [CrossRef]
- Toischer, K.; Rokita, A.G.; Unsold, B.; Zhu, W.; Kararigas, G.; Sossalla, S.; Reuter, S.P.; Becker, A.; Teucher, N.; Seidler, T.; et al. Differential cardiac remodeling in preload versus afterload. Circulation 2010, 122, 993–1003. [Google Scholar] [CrossRef]
- Kehat, I.; Davis, J.; Tiburcy, M.; Accornero, F.; Saba-El-Leil, M.K.; Maillet, M.; York, A.J.; Lorenz, J.N.; Zimmermann, W.H.; Meloche, S.; et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ. Res. 2010, 108, 176–183. [Google Scholar]
- Van der Bom, T.; Winter, M.M.; Bouma, B.J.; Groenink, M.; Vliegen, H.W.; Pieper, P.G.; van Dijk, A.P.; Sieswerda, G.T.; Roos-Hesselink, J.W.; Zwinderman, A.H. Effect of valsartan on systemic right ventricular function: A double-blind, randomized, placebo-controlled pilot trial. Circulation 2013, 127, 322–330. [Google Scholar] [CrossRef]
- Archer, S.L.; Michelakis, E.D. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N. Engl. J. Med. 2009, 361, 1864–1871. [Google Scholar] [CrossRef]
- Borgdorff, M.A.; Bartelds, B.; Dickinson, M.G.; Steendijk, P.; de Vroomen, M.; Berger, R.M. Sildenafil treatement in established right ventricular dysfunction improves diastolic function and attenuates interstitial fibrosis independent from afterload. Am. J. Physiol.-Heart Circ. Physiol. 2013. submitted. [Google Scholar]
- Lang, M.; Kojonazarov, B.; Tian, X.; Kalymbetov, A.; Weissmann, N.; Grimminger, F.; Kretschmer, A.; Stasch, J.P.; Seeger, W.; Ghofrani, H.A.; et al. The soluble guanylate cyclase stimulator riociguat ameliorates pulmonary hypertension induced by hypoxia and SU5416 in rats. PLoS ONE 2012, 7, e43433. [Google Scholar] [CrossRef]
- Choudhary, G.; Troncales, F.; Martin, D.; Harrington, E.O.; Klinger, J.R. Bosentan attenuates right ventricular hypertrophy and fibrosis in normobaric hypoxia model of pulmonary hypertension. J. Heart Lung Transplant. 2011, 30, 827–833. [Google Scholar] [CrossRef]
- Nagendran, J.; Sutendra, G.; Paterson, I.; Champion, H.C.; Webster, L.; Chiu, B.; Haromy, A.; Rebeyka, I.M.; Ross, D.B.; Michelakis, E.D. Endothelin axis is upregulated in human and rat right ventricular hypertrophy. Circ. Res. 2013, 112, 347–354. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Mizuno, S.; Hussaini, A.A.; Toldo, S.; Abbate, A.; Kraskauskas, D.; Kasper, M.; Natarajan, R.; Voelkel, N.F. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am. J. Respir. Crit. Care Med. 2011, 183, 1402–1410. [Google Scholar] [CrossRef]
- Cuypers, J.A.; Opic, P.; Menting, M.E.; Utens, E.M.; Witsenburg, M.; Helbing, W.A.; van den Bosch, A.E.; Ouhlous, M.; van Domburg, R.T.; Meijboom, F.J.; et al. The unnatural history of an atrial septal defect: Longitudinal 35 year follow up after surgical closure at young age. Heart 2013, 99, 1346–1352. [Google Scholar] [CrossRef]
- Nath, J.; Foster, E.; Heidenreich, P.A. Impact of tricuspid regurgitation on long-term survival. J. Am. Coll. Cardiol. 2004, 43, 405–409. [Google Scholar] [CrossRef]
- Luijnenburg, S.E.; de Koning, W.B.; Romeih, S.; van den Berg, J.; Vliegen, H.W.; Mulder, B.J.; Helbing, W.A. Exercise capacity and ventricular function in patients treated for isolated pulmonary valve stenosis or tetralogy of Fallot. Int. J. Cardiol. 2012, 158, 359–363. [Google Scholar] [CrossRef]
- Graham, T.P., Jr.; Bernard, Y.D.; Mellen, B.G.; Celermajer, D.; Baumgartner, H.; Cetta, F.; Connolly, H.M.; Davidson, W.R.; Dellborg, M.; Foster, E.; et al. Long-term outcome in congenitally corrected transposition of the great arteries: A multi-institutional study. J. Am. Coll. Cardiol. 2000, 36, 255–261. [Google Scholar] [CrossRef]
- Grothoff, M.; Fleischer, A.; Abdul-Khaliq, H.; Hoffmann, J.; Lehmkuhl, L.; Luecke, C.; Gutberlet, M. The systemic right ventricle in congenitally corrected transposition of the great arteries is different from the right ventricle in dextro-transposition after atrial switch: a cardiac magnetic resonance study. Cardiol. Young 2013, 23, 239–247. [Google Scholar] [CrossRef]
- Diller, G.P.; Dimopoulos, K.; Broberg, C.S.; Kaya, M.G.; Naghotra, U.S.; Uebing, A.; Harries, C.; Goktekin, O.; Gibbs, J.S.; Gatzoulis, M.A. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: A combined retrospective and case-control study. Eur. Heart J. 2006, 27, 1737–1742. [Google Scholar] [CrossRef]
- Van Loon, R.L.; Roofthooft, M.T.; Hillege, H.L.; Ten Harkel, A.D.; van Osch-Gevers, M.; Delhaas, T.; Kapusta, L.; Strengers, J.L.; Rammeloo, L.; Clur, S.A.; et al. Pediatric pulmonary hypertension in the Netherlands: Epidemiology and characterization during the period 1991 to 2005. Circulation 2011, 124, 1755–1764. [Google Scholar] [CrossRef]
- Gatzoulis, M.A.; Balaji, S.; Webber, S.A.; Siu, S.C.; Hokanson, J.S.; Poile, C.; Rosenthal, M.; Nakazawa, M.; Moller, J.H.; Gillette, P.C.; et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: A multicentre study. Lancet 2000, 356, 975–981. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bartelds, B.; Borgdorff, M.A.J.; Berger, R.M.F. Right Ventricular Adaptation in Congenital Heart Diseases. J. Cardiovasc. Dev. Dis. 2014, 1, 83-97. https://doi.org/10.3390/jcdd1010083
Bartelds B, Borgdorff MAJ, Berger RMF. Right Ventricular Adaptation in Congenital Heart Diseases. Journal of Cardiovascular Development and Disease. 2014; 1(1):83-97. https://doi.org/10.3390/jcdd1010083
Chicago/Turabian StyleBartelds, Beatrijs, Marinus A. J. Borgdorff, and Rolf M. F. Berger. 2014. "Right Ventricular Adaptation in Congenital Heart Diseases" Journal of Cardiovascular Development and Disease 1, no. 1: 83-97. https://doi.org/10.3390/jcdd1010083