
Phylogenetic Analysis and Protein Modelling of Isoflavonoid Synthase Highlights Key Catalytic Sites towards Realising New Bioengineering Endeavours



Abstract
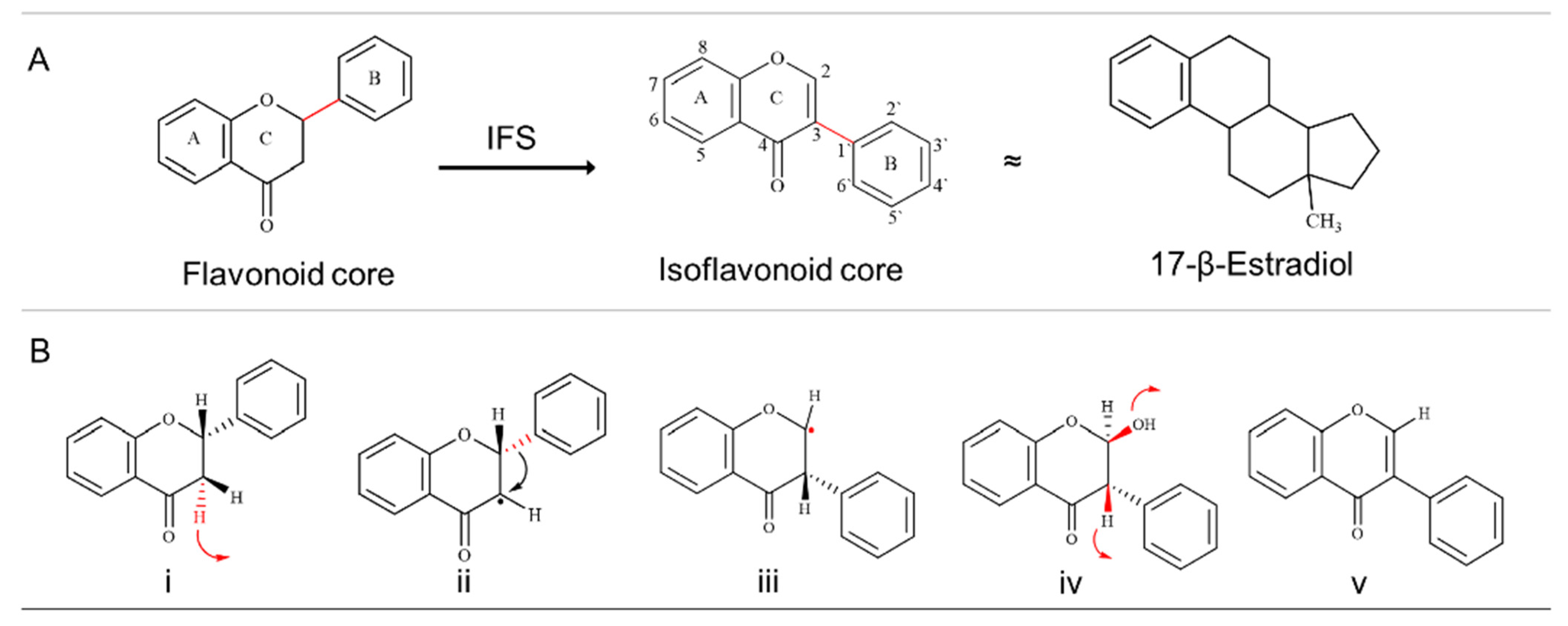
1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval
2.2. Sequence Alignment and Phylogenetic Analysis
2.3. Protein Modelling Analysis
2.4. Protein Docking Analysis
3. Results
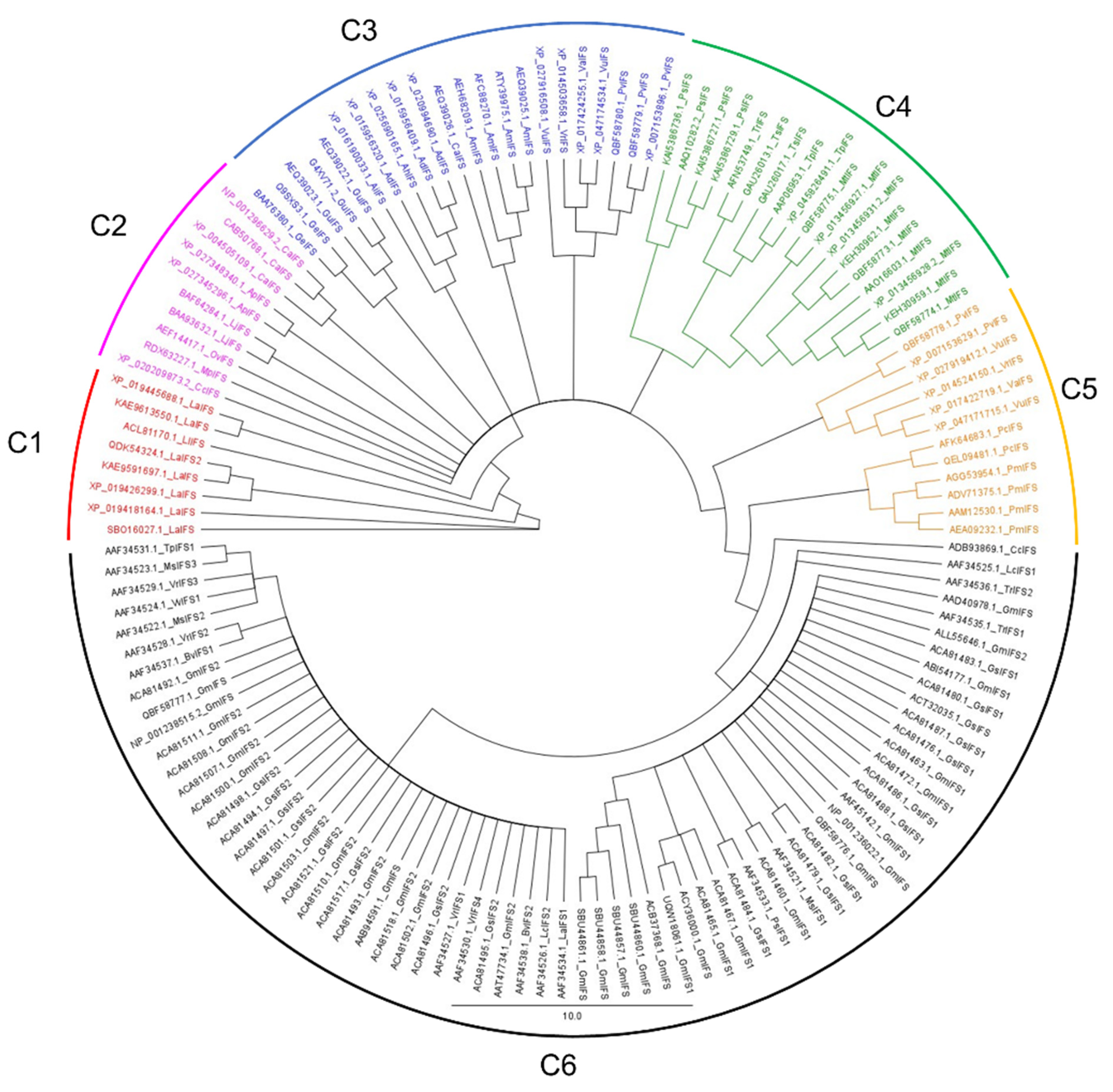
3.1. Phylogenetic Analysis of IFS Amino Acid Sequences
3.1.1. Clade 1
3.1.2. Clade 2
3.1.3. Clade 3
3.1.4. Clade 4
3.1.5. Clade 5
3.1.6. Clade 6
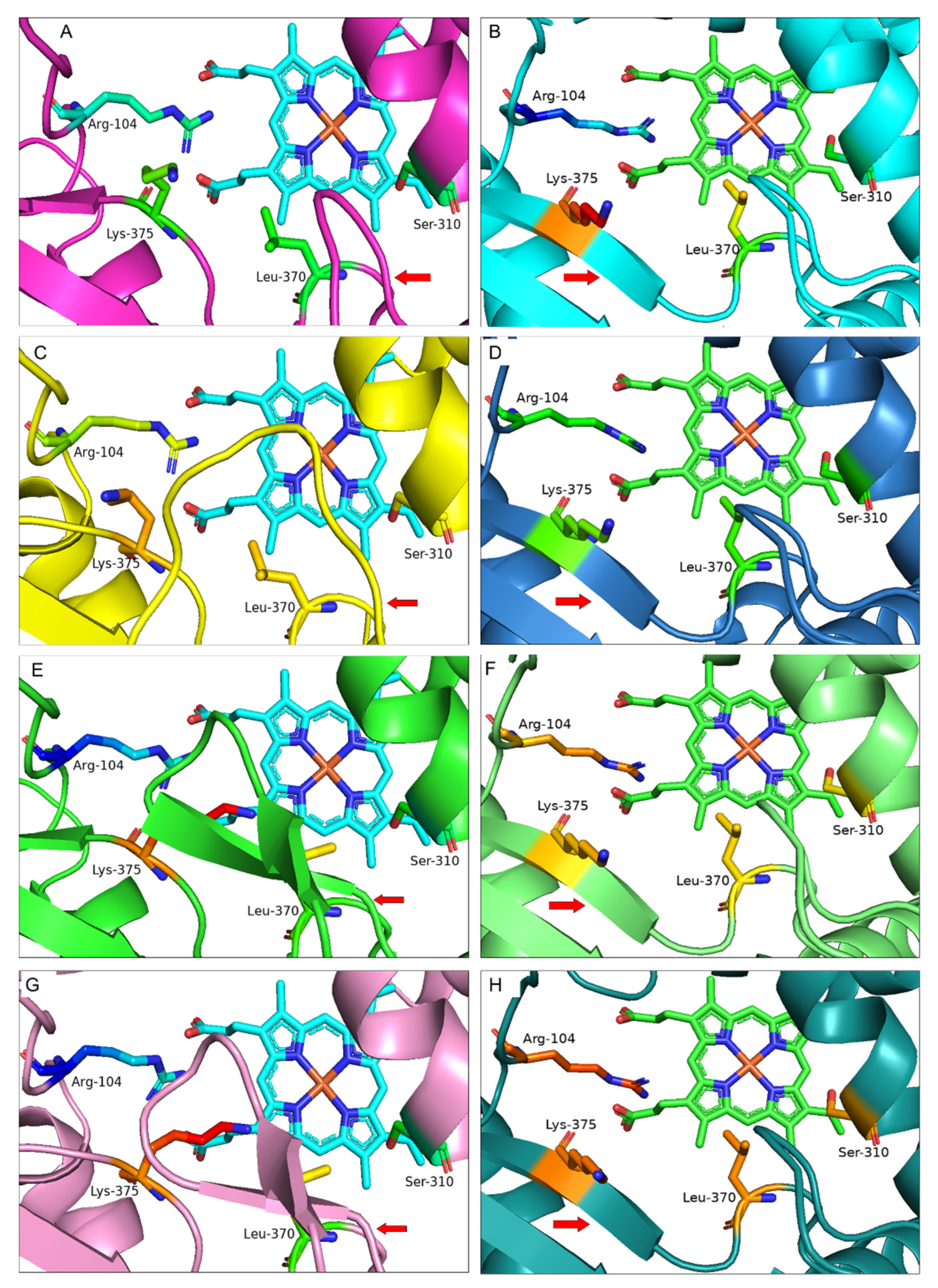
3.2. Protein Homology Modelling of Selected IFS Candidates
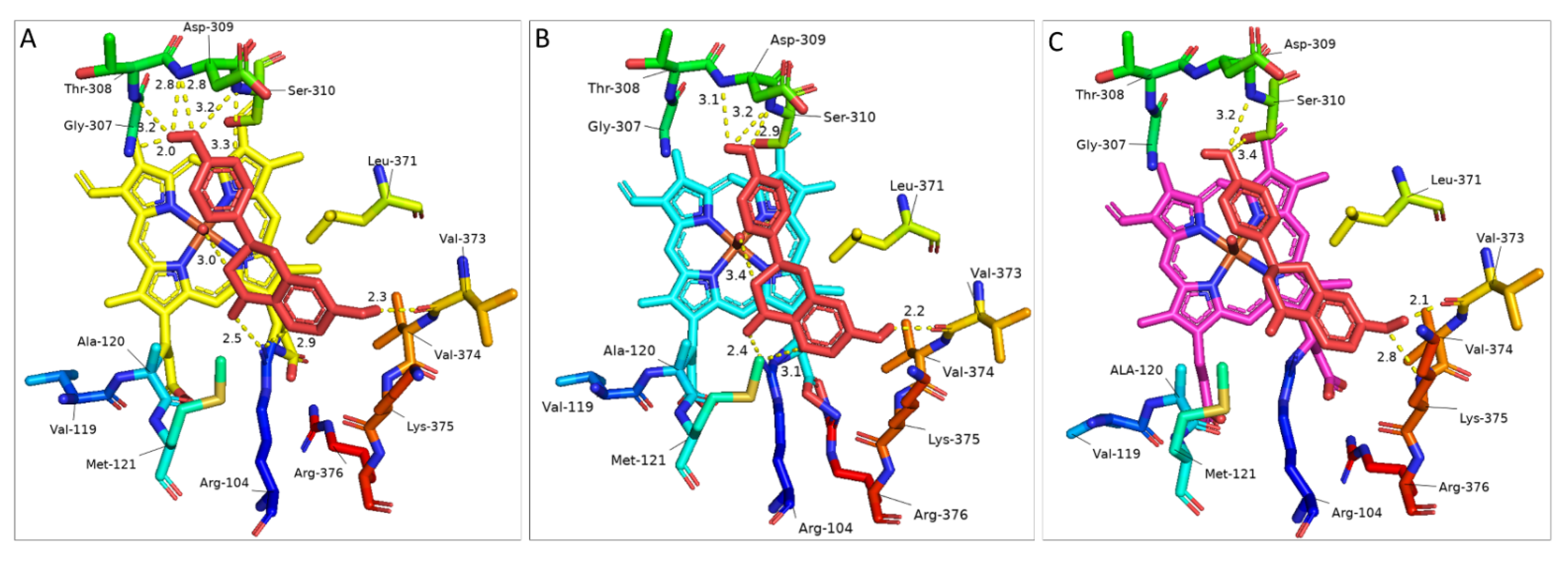
3.3. Protein Docking of Selected IFS Candidates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akashi, T.; Aoki, T.; Ayabe, S. Cloning and Functional Expression of a Cytochrome P450 CDNA Encoding 2-Hydroxyisoflavanone Synthase Involved in Biosynthesis of the Isoflavonoid Skeleton in Licorice. Plant Physiol. 1999, 121, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.L.; Gijzen, M.; Qutob, D.; Dixon, R.A. Molecular Characterization of the Enzyme Catalyzing the Aryl Migration Reaction of Isoflavonoid Biosynthesis in Soybean. Arch. Biochem. Biophys. 1999, 367, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Stone, S.R.; Kaur, P. Recent Advances in Heterologous Synthesis Paving Way for Future Green-Modular Bioindustries-A Review with Special Reference to Isoflavonoids. Front. Bioeng. Biotechnol. 2021, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Huang, Y.; Tang, Y. Genetic and Metabolic Engineering of Isoflavonoid Biosynthesis. Appl. Microbiol. Biotechnol. 2010, 86, 1293–1312. [Google Scholar] [CrossRef]
- Sajid, M.; Channakesavula, C.N.; Stone, S.R.; Kaur, P. Synthetic Biology towards Improved Flavonoid Pharmacokinetics. Biomolecules 2021, 11, 754. [Google Scholar] [CrossRef]
- Bustos-Salgado, P.; Andrade-Carrera, B.; Domínguez-Villegas, V.; Díaz-Garrido, N.; Rodríguez-Lagunas, M.J.; Badía, J.; Baldomà, L.; Mallandrich, M.; Calpena-Campmany, A.; Garduño-Ramírez, M.L. Screening Anti-Inflammatory Effects of Flavanones Solutions. Int. J. Mol. Sci. 2021, 22, 8878. [Google Scholar] [CrossRef]
- Kim, I.-S. Current Perspectives on the Beneficial Effects of Soybean Isoflavones and Their Metabolites for Humans. Antioxidants 2021, 10, 1064. [Google Scholar] [CrossRef]
- Gómez-Zorita, S.; González-Arceo, M.; Fernández-Quintela, A.; Eseberri, I.; Trepiana, J.; Portillo, M.P. Scientific Evidence Supporting the Beneficial Effects of Isoflavones on Human Health. Nutrients 2020, 12, 3853. [Google Scholar] [CrossRef]
- Lapčík, O. Isoflavonoids in Non-Leguminous Taxa: A Rarity or a Rule? Phytochemistry 2007, 68, 2909–2916. [Google Scholar] [CrossRef]
- Jung, W.; Yu, O.; Lau, S.-M.C.; O’Keefe, D.P.; Odell, J.; Fader, G.; McGonigle, B. Identification and Expression of Isoflavone Synthase, the Key Enzyme for Biosynthesis of Isoflavones in Legumes. Nat. Biotechnol. 2000, 18, 208–212. [Google Scholar] [CrossRef]
- Kim, B.G.; Kim, S.-Y.; Song, H.S.; Lee, C.; Hur, H.-G.; Kim, S.; Ahn, J.-H. Cloning and Expression of the Isoflavone Synthase Gene (IFS-Tp) from Trifolium Pratense. Mol. Cells (Springer Sci. Bus. Media BV) 2003, 15, 301–306. [Google Scholar]
- Shimada, N.; Akashi, T.; Aoki, T.; Ayabe, S. Induction of Isoflavonoid Pathway in the Model Legume Lotus Japonicus: Molecular Characterization of Enzymes Involved in Phytoalexin Biosynthesis. Plant Sci. 2000, 160, 37–47. [Google Scholar] [CrossRef]
- Tong, C.; Liu, X.; Wu, Y.; Zhao, Y.; Wang, J. Regulation of Endogenous Isoflavones on Alfalfa Nodulation and Nitrogen Fixation and Nitrogen Use Efficiency. Acta Prataculturae Sin. 2022, 31, 124. [Google Scholar]
- Sawada, Y.; Kinoshita, K.; Akashi, T.; Aoki, T.; Ayabe, S. Key Amino Acid Residues Required for Aryl Migration Catalysed by the Cytochrome P450 2-hydroxyisoflavanone Synthase. Plant J. 2002, 31, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Ran, F.; Dong, H.-L.; Wen, J.; Li, J.-N.; Liang, Z. Genome-Wide Analysis, Classification, Evolution, and Expression Analysis of the Cytochrome P450 93 Family in Land Plants. PLoS ONE 2016, 11, e0165020. [Google Scholar]
- Stafford, H.A.; Ibrahim, R.K. Recent Advances in Phytochemistry; Plenum Press: New York, NY, USA, 1992; Volume 26, ISBN 0306442310. [Google Scholar]
- Huccetogullari, D.; Luo, Z.W.; Lee, S.Y. Metabolic Engineering of Microorganisms for Production of Aromatic Compounds. Microb. Cell Factories 2019, 18, 1–29. [Google Scholar] [CrossRef]
- Cravens, A.; Payne, J.; Smolke, C.D. Synthetic Biology Strategies for Microbial Biosynthesis of Plant Natural Products. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Lv, Y.; Marsafari, M.; Koffas, M.; Zhou, J.; Xu, P. Optimizing Oleaginous Yeast Cell Factories for Flavonoids and Hydroxylated Flavonoids Biosynthesis. ACS Synth. Biol. 2019, 8, 2514–2523. [Google Scholar] [CrossRef]
- Zang, Y.; Zha, J.; Wu, X.; Zheng, Z.; Ouyang, J.; Koffas, M.A. In Vitro Naringenin Biosynthesis from P-Coumaric Acid Using Recombinant Enzymes. J. Agric. Food Chem. 2019, 67, 13430–13436. [Google Scholar] [CrossRef]
- Wang, Y.; Xue, P.; Cao, M.; Yu, T.; Lane, S.T.; Zhao, H. Directed Evolution: Methodologies and Applications. Chem. Rev. 2021, 121, 12384–12444. [Google Scholar] [CrossRef]
- Tian, L.; Dixon, R.A. Engineering Isoflavone Metabolism with an Artificial Bifunctional Enzyme. Planta 2006, 224, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Trantas, E.; Panopoulos, N.; Ververidis, F. Metabolic Engineering of the Complete Pathway Leading to Heterologous Biosynthesis of Various Flavonoids and Stilbenoids in Saccharomyces Cerevisiae. Metab. Eng. 2009, 11, 355–366. [Google Scholar] [CrossRef]
- Katsuyama, Y.; Miyahisa, I.; Funa, N.; Horinouchi, S. One-Pot Synthesis of Genistein from Tyrosine by Coincubation of Genetically Engineered Escherichia Coli and Saccharomyces Cerevisiae Cells. Appl. Microbiol. Biotechnol. 2007, 73, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Leonard, E.; Koffas, M.A. Engineering of Artificial Plant Cytochrome P450 Enzymes for Synthesis of Isoflavones by Escherichia Coli. Appl. Environ. Microbiol. 2007, 73, 7246–7251. [Google Scholar] [CrossRef] [PubMed]
- Trantas, E.A.; Koffas, M.A.G.; Xu, P.; Ververidis, F. When Plants Produce Not Enough or at All: Metabolic Engineering of Flavonoids in Microbial Hosts. Front. Plant Sci. 2015, 6, 1–16. [Google Scholar] [CrossRef]
- Kim, B.-G. Biological Synthesis of Genistein in Escherichia Coli. J. Microbiol. Biotechnol. 2020, 30, 770–776. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Li, G.; Savolainen, O.; Chen, Y.; Nielsen, J. De Novo Biosynthesis of Bioactive Isoflavonoids by Engineered Yeast Cell Factories. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Meng, Y.; Liu, X.; Zhang, L.; Zhao, G.-R. Modular Engineering of Saccharomyces Cerevisiae for De Novo Biosynthesis of Genistein. Microorganisms 2022, 10, 1402. [Google Scholar] [CrossRef]
- Ahuja, K.; Mamtani, K. Industry Analysis Report, Regional Outlook, Application Development Potential, Price Trends, Competitive Market Share & Forecast, 2019–2025. 2019. Available online: https://www.gminsights.com/industry-analysis/isoflavones-market (accessed on 10 February 2022).
- Sajid, M.; Stone, S.R.; Kaur, P. Transforming Traditional Nutrition Paradigms with Synthetic Biology Driven Microbial Production Platforms. Curr. Res. Biotechnol. 2021, 3, 260–268. [Google Scholar] [CrossRef]
- Sawada, Y.; Ayabe, S. Multiple Mutagenesis of P450 Isoflavonoid Synthase Reveals a Key Active-Site Residue. Biochem. Biophys. Res. Commun. 2005, 330, 907–913. [Google Scholar] [CrossRef]
- Gu, M.; Wang, M.; Guo, J.; Shi, C.; Deng, J.; Huang, L.; Huang, L.; Chang, Z. Crystal Structure of CYP76AH1 in 4-PI-Bound State from Salvia Miltiorrhiza. Biochem. Biophys. Res. Commun. 2019, 511, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Hameduh, T.; Haddad, Y.; Adam, V.; Heger, Z. Homology Modeling in the Time of Collective and Artificial Intelligence. Comput. Struct. Biotechnol. J. 2020, 18, 3494. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Wasukan, N.; Kuno, M.; Maniratanachote, R. Molecular Docking as a Promising Predictive Model for Silver Nanoparticle-Mediated Inhibition of Cytochrome P450 Enzymes. J. Chem. Inf. Model. 2019, 59, 5126–5134. [Google Scholar] [CrossRef]
- Fan, Y.; Tao, Y.; Liu, G.; Wang, M.; Wang, S.; Li, L. Interaction Study of Engeletin toward Cytochrome P450 3A4 and 2D6 by Multi-Spectroscopy and Molecular Docking. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 264, 120311. [Google Scholar] [CrossRef]
- Uchida, K.; Akashi, T.; Aoki, T. Functional Expression of Cytochrome P450 in Escherichia Coli: An Approach to Functional Analysis of Uncharacterized Enzymes for Flavonoid Biosynthesis. Plant Biotechnol. 2015, 32, 205–213. [Google Scholar] [CrossRef]
- Kaducová, M.; Eliašová, A.; Trush, K.; Bačovčinová, M.; Sklenková, K.; Pal’ove-Balang, P. Accumulation of Isoflavonoids in Lotus Corniculatus after UV-B Irradiation. Theor. Exp. Plant Physiol. 2022, 34, 53–62. [Google Scholar] [CrossRef]
- Chemler, J.A.; Lim, C.G.; Daiss, J.L.; Koffas, M.A. A Versatile Microbial System for Biosynthesis of Novel Polyphenols with Altered Estrogen Receptor Binding Activity. Chem. Biol. 2010, 17, 392–401. [Google Scholar] [CrossRef][Green Version]
- Fliegmann, J.; Furtwängler, K.; Malterer, G.; Cantarello, C.; Schüler, G.; Ebel, J.; Mithöfer, A. Flavone Synthase II (CYP93B16) from Soybean (Glycine max L.). Phytochemistry 2010, 71, 508–514. [Google Scholar] [CrossRef]
- Du, Y.; Chu, H.; Chu, I.K.; Lo, C. CYP93G2 Is a Flavanone 2-Hydroxylase Required for C-Glycosylflavone Biosynthesis in Rice. Plant Physiol. 2010, 154, 324–333. [Google Scholar] [CrossRef]
- Waki, T.; Yoo, D.; Fujino, N.; Mameda, R.; Denessiouk, K.; Yamashita, S.; Motohashi, R.; Akashi, T.; Aoki, T.; Ayabe, S. Identification of Protein–Protein Interactions of Isoflavonoid Biosynthetic Enzymes with 2-Hydroxyisoflavanone Synthase in Soybean (Glycine max (L.) Merr.). Biochem. Biophys. Res. Commun. 2016, 469, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Finnigan, J.D.; Young, C.; Cook, D.J.; Charnock, S.J.; Black, G.W. Cytochromes P450 (P450s): A Review of the Class System with a Focus on Prokaryotic P450s. Adv. Protein Chem. Struct. Biol. 2020, 122, 289–320. [Google Scholar] [PubMed]
- Anguraj Vadivel, A.K.; McDowell, T.; Renaud, J.B.; Dhaubhadel, S. A Combinatorial Action of GmMYB176 and GmbZIP5 Controls Isoflavonoid Biosynthesis in Soybean (Glycine max). Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mameda, R.; Waki, T.; Kawai, Y.; Takahashi, S.; Nakayama, T. Involvement of Chalcone Reductase in the Soybean Isoflavone Metabolon: Identification of GmCHR5, Which Interacts with 2-Hydroxyisoflavanone Synthase. Plant J. 2018, 96, 56–74. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, G.; Nandekar, P.P.; Wade, R.C. An Electron Transfer Competent Structural Ensemble of Membrane-Bound Cytochrome P450 1A1 and Cytochrome P450 Oxidoreductase. Commun. Biol. 2021, 4, 1–13. [Google Scholar] [CrossRef]
- Rani, D.; Vimolmangkang, S. Trends in the Biotechnological Production of Isoflavonoids in Plant Cell Suspension Cultures. Phytochem. Rev. 2022, 21, 1–20. [Google Scholar] [CrossRef]
- Rani, D.; Meelaph, T.; De-Eknamkul, W.; Vimolmangkang, S. Yeast Extract Elicited Isoflavonoid Accumulation and Biosynthetic Gene Expression in Pueraria Candollei Var. Mirifica Cell Cultures. Plant Cell Tissue Organ Cult. (PCTOC) 2020, 141, 661–667. [Google Scholar] [CrossRef]
- Sreevidya, V.S.; Srinivasa Rao, C.; Sullia, S.B.; Ladha, J.K.; Reddy, P.M. Metabolic Engineering of Rice with Soybean Isoflavone Synthase for Promoting Nodulation Gene Expression in Rhizobia. J. Exp. Bot. 2006, 57, 1957–1969. [Google Scholar] [CrossRef]
- Malla, A.; Shanmugaraj, B.; Srinivasan, B.; Sharma, A.; Ramalingam, S. Metabolic Engineering of Isoflavonoid Biosynthesis by Expressing Glycine Max Isoflavone Synthase in Allium Cepa L. for Genistein Production. Plants 2021, 10, 52. [Google Scholar] [CrossRef]
- Yang, D.; Park, S.Y.; Park, Y.S.; Eun, H.; Lee, S.Y. Metabolic Engineering of Escherichia Coli for Natural Product Biosynthesis. Trends Biotechnol. 2020, 38, 745–765. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, Y.; Guengerich, F.P.; Ma, L.; Li, S.; Zhang, W. Engineering Cytochrome P450 Enzyme Systems for Biomedical and Biotechnological Applications. J. Biol. Chem. 2020, 295, 833–849. [Google Scholar] [CrossRef]
- Siminszky, B.; Corbin, F.T.; Ward, E.R.; Fleischmann, T.J.; Dewey, R.E. Expression of a Soybean Cytochrome P450 Monooxygenase CDNA in Yeast and Tobacco Enhances the Metabolism of Phenylurea Herbicides. Proc. Natl. Acad. Sci. USA 1999, 96, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Wiriyaampaiwong, P.; Thanonkeo, S.; Thanonkeo, P. Molecular Characterization of Isoflavone Synthase Gene from Pueraria candollei var. mirifica. Afr. J. Agric. Res. 2012, 7, 4489–4498. [Google Scholar] [CrossRef]
- Pičmanová, M.; Reňák, D.; Feciková, J.; Růžička, P.; Mikšátková, P.; Lapčík, O.; Honys, D. Functional Expression and Subcellular Localization of Pea Polymorphic Isoflavone Synthase CYP93C18. Biol. Plant. 2013, 57, 635–645. [Google Scholar] [CrossRef]
- Misra, P.; Pandey, A.; Tewari, S.K.; Nath, P.; Trivedi, P.K. Characterization of Isoflavone Synthase Gene from Psoralea Corylifolia: A Medicinal Plant. Plant Cell Rep. 2010, 29, 747–755. [Google Scholar] [CrossRef]
- He, X.; Blount, J.W.; Ge, S.; Tang, Y.; Dixon, R.A. A Genomic Approach to Isoflavone Biosynthesis in Kudzu (Pueraria lobata). Planta 2011, 233, 843–855. [Google Scholar] [CrossRef]
- Kaur, N.; Murphy, J.B. Cloning, Characterization, and Functional Analysis of Cowpea Isoflavone Synthase (IFS) Homologs. J. Plant Mol. Biol. Biotechnol. PMBB 2010, 1. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Heterologous Host | Substrate (Input) | Isoflavonoid (Product) | Genetic Engineering Strategy | Ref. |
|---|---|---|---|---|---|
| Glycine max IFS Medicago sativa CHI | Yeast | IsoLQN/LQN | DEN | Construction of bi-functional enzyme by in-frame gene fusion for higher yield | [22] |
| G. max IFS Poplar hybrid CPR | Yeast | Tyrosine, p-coumaric acid | 0.1–7.7 mg/L GEN | Metabolic engineering of yeast using plasmid-based gene expression for heterologous biosynthesis | [23] |
| Glycyrrhiza echinata IFS | Yeast/ E. coli | 3 mM Tyrosine | 6 mg/L GEN | Co-culture approach for better expression and higher yield | [24] |
| G. max IFS Catharanthus roseus CPR | E. coli | 0.05 mM NGN | GEN | Engineered IFS architecture to complement a self-sufficient bacterial P450 enzyme | [25] |
| 0.05 mM LQN | DEN | ||||
| G. max IFS Putina hybrdia CPR | Yeast | 10 mM Phenylalanine | 0.1 mg/L GEN | Functional expression of plant enzyme and construction of pathway in yeast chassis | [26] |
| 1 mM p-Coumaric acid | 0.14 mg/L GEN | ||||
| 0.5 mM NGN | 7.7 mg/L GEN | ||||
| Trifolium pretense IFS Oryza sativa CPR | E. coli | 500 μM NGN | 35 mg/L GEN | Engineering of IFS for expression in prokaryotic system. Optimisation of culture system, medium, growth conditions and substrate concentration to increase overall yield | [27] |
| 300 μM p-Coumaric acid | 18.9 mg/L GEN | ||||
| G. echinata IFS C. roseus CPR | Yeast | - | 9.9 mg/L DEN | Metabolic engineering of a yeast strain for de-novo isoflavonoids biosynthesis | [28] |
| Lotus japonicas IFS L. japonicas CPR | Yeast | - | 19.32 mg/L GEN | Modular engineering of yeast and screening of IFS for de-novo biosynthesis of genistein | [29] |
| Species | ΔG for LQN | ΔG for NGN | ||
|---|---|---|---|---|
| Swiss-Model | Alphafold | Swiss-Model | Alphafold | |
| B. vulgaris | −8.03 | −7.68 | −7.80 | −7.18 |
| G. echinata | −7.37 | −7.98 | −6.97 | −7.49 |
| M. truncatula | −7.46 | −7.76 | −7.12 | −7.51 |
| T. pratense | −8.23 | −7.98 | −7.83 | −7.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sajid, M.; Stone, S.R.; Kaur, P. Phylogenetic Analysis and Protein Modelling of Isoflavonoid Synthase Highlights Key Catalytic Sites towards Realising New Bioengineering Endeavours. Bioengineering 2022, 9, 609. https://doi.org/10.3390/bioengineering9110609
Sajid M, Stone SR, Kaur P. Phylogenetic Analysis and Protein Modelling of Isoflavonoid Synthase Highlights Key Catalytic Sites towards Realising New Bioengineering Endeavours. Bioengineering. 2022; 9(11):609. https://doi.org/10.3390/bioengineering9110609
Chicago/Turabian StyleSajid, Moon, Shane R. Stone, and Parwinder Kaur. 2022. "Phylogenetic Analysis and Protein Modelling of Isoflavonoid Synthase Highlights Key Catalytic Sites towards Realising New Bioengineering Endeavours" Bioengineering 9, no. 11: 609. https://doi.org/10.3390/bioengineering9110609
APA StyleSajid, M., Stone, S. R., & Kaur, P. (2022). Phylogenetic Analysis and Protein Modelling of Isoflavonoid Synthase Highlights Key Catalytic Sites towards Realising New Bioengineering Endeavours. Bioengineering, 9(11), 609. https://doi.org/10.3390/bioengineering9110609