
Hematopoietic Stem Cells: Nature and Niche Nurture


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
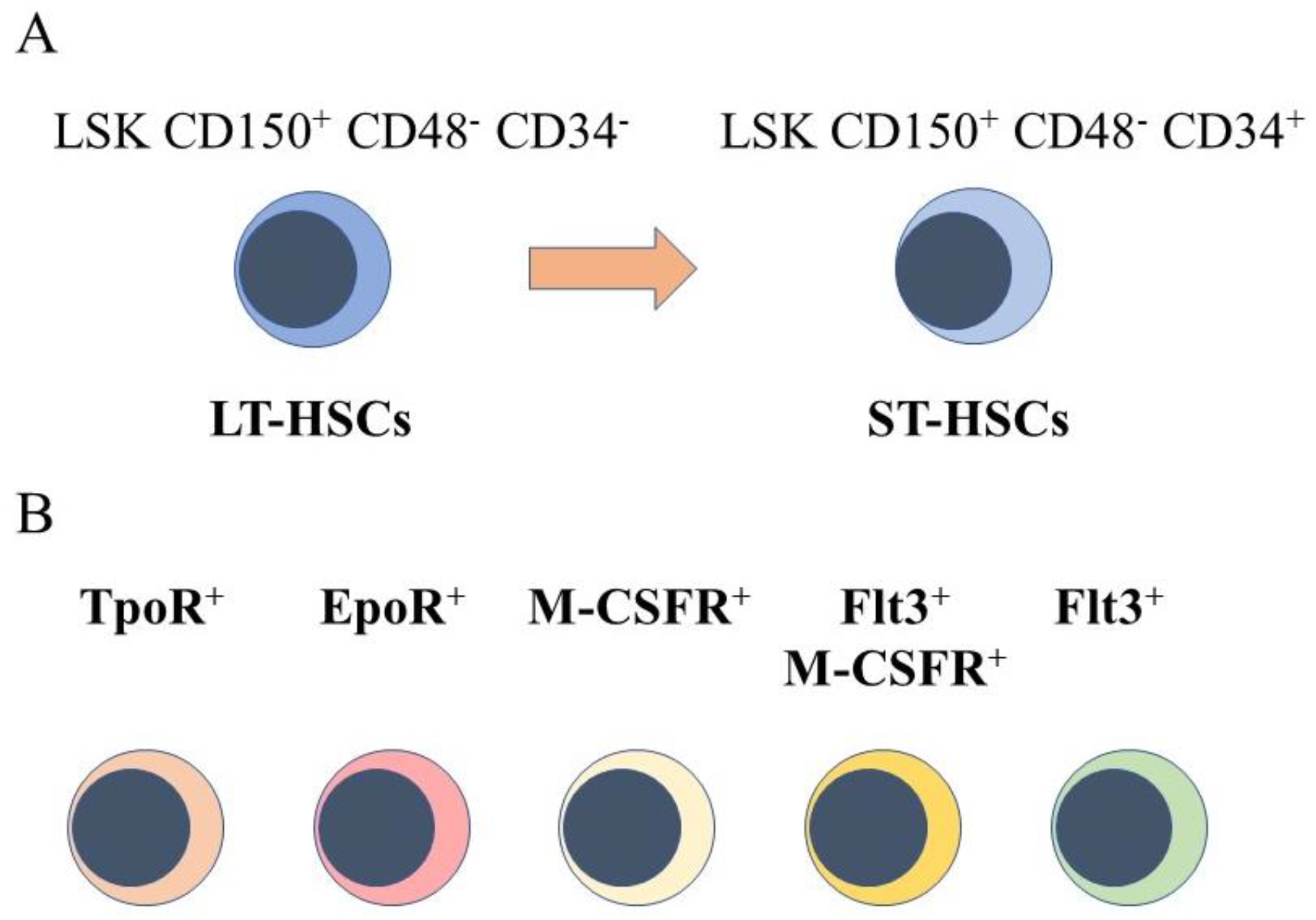
2. HSCs Are a Heterogeneous Population of Cells
3. Bone Marrow Niche Cells Are Heterogeneous
4. Mapping HSC Developmental Progression
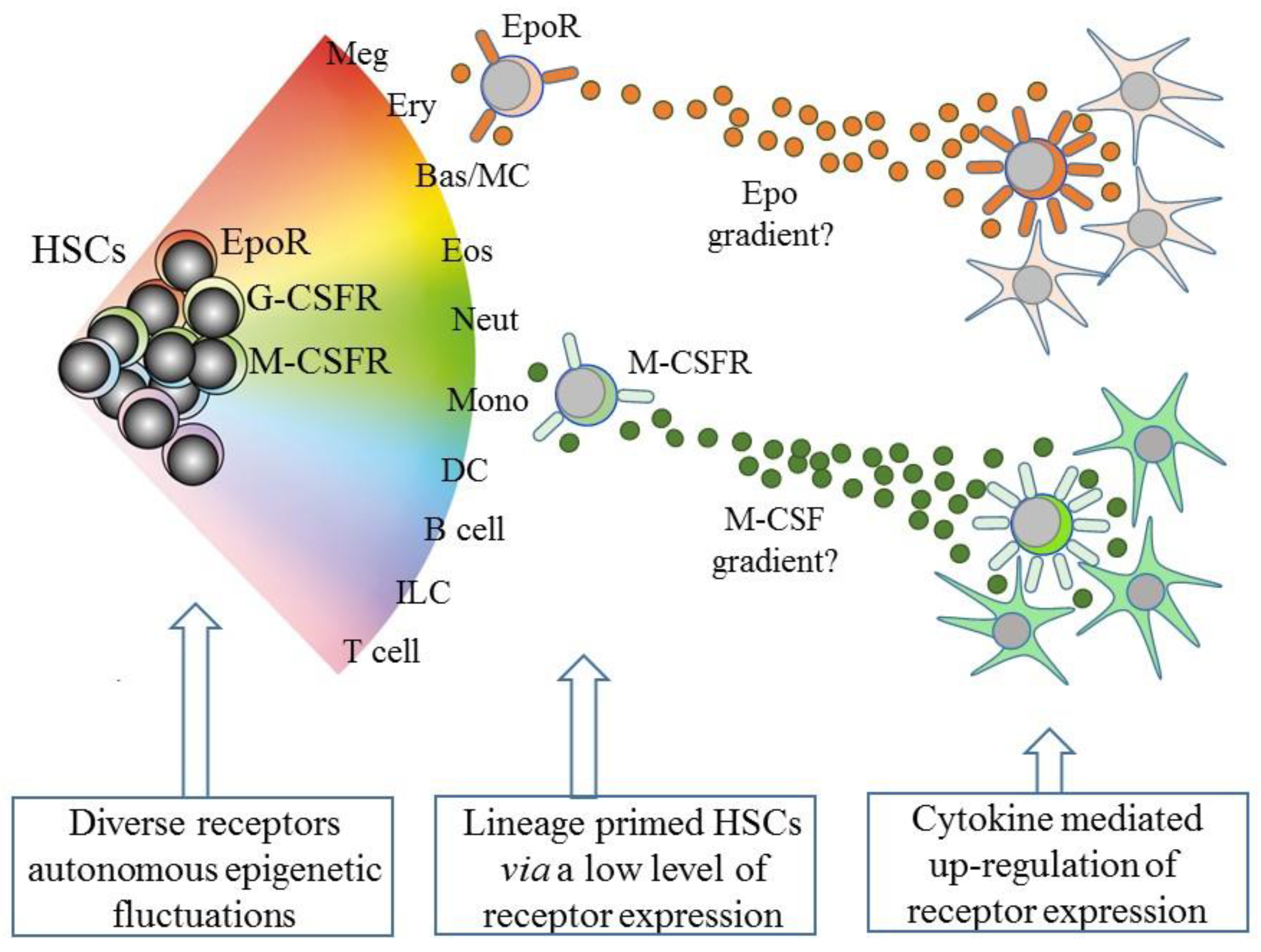
5. Intrinsic Events Influence HSCs Lineage “Choice”
6. Some Hematopoietic Cytokines Instruct Lineage Fate
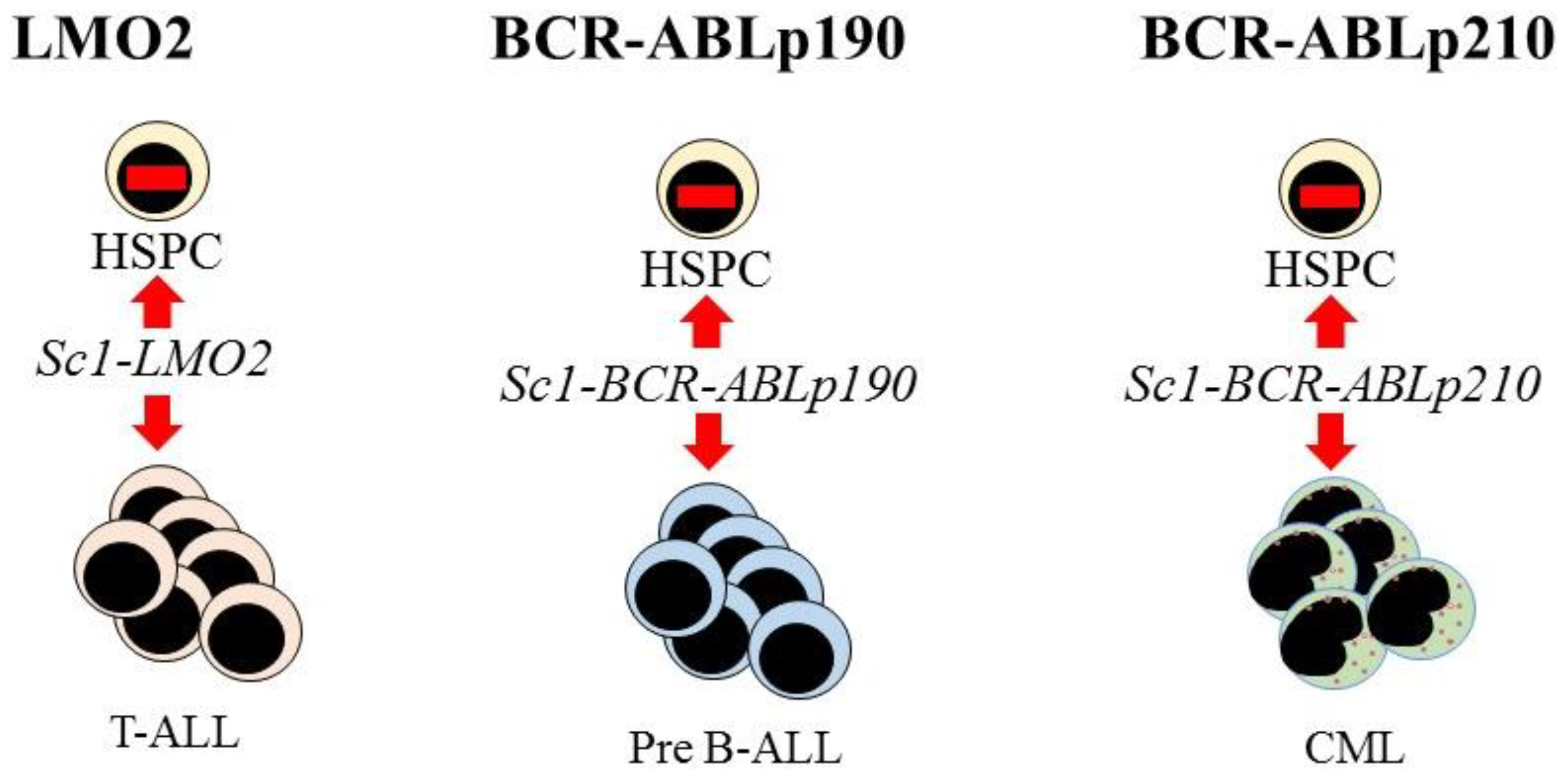
7. Hematopoietic Malignancies
8. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raff, M.C. Social controls on cell survival and cell death. Nature 1992, 356, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2629–2671. [Google Scholar] [CrossRef]
- Robb, L. Cytokine receptors and hematopoietic differentiation. Oncogene 2007, 26, 6715–6723. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Takizawa, H.; Strowig, T.; Willinger, T.; Eynon, E.E.; Flavell, R.A.; Manz, M.G. Human hemato-lymphoid system mice: Current use and future potential for medicine. Annu. Rev. Immunol. 2013, 31, 635–674. [Google Scholar] [CrossRef]
- Gbyli, R.; Song, Y.; Halene, S. Humanized mice as preclinical models for myeloid malignancies. Biochem. Pharmacol. 2020, 174, 113794. [Google Scholar] [CrossRef]
- Osawa, M.; Hanada, K.; Hamada, H.; Nakauchi, H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 1996, 273, 242–245. [Google Scholar] [CrossRef]
- Sanjuan-Pla, A.; Macauley, I.C.; Jensen, C.T.; Wall, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carellla, C.; Matsuoka, S.; Bouriez-Jones, T.; et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 2013, 154, 1112–1126. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Hu, W.; Naramura, M.; Park, C.Y. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J. Exp. Med. 2014, 211, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Mooney, C.J.; Cunningham, A.; Tsapogas, P.; Toellner, K.-M.; Brown, G. Selective expression of Flt3 within the mouse hematopoietic stem cell compartment. Int. J. Mol. Sci. 2017, 18, 1037. [Google Scholar] [CrossRef]
- Schuettpelz, L.G.; Borderding, J.N.; Christopher, M.D.; Gopalan, P.K.; Romine, M.P.; Herman, A.C.; Woloszynek, J.R.; Greenbaum, A.M.; Link, D.C. G-CSF regulates hematopoietic stem cell activity, in part, through activation of toll-like receptor signaling. Leukaemia 2014, 28, 1851–1860. [Google Scholar] [CrossRef]
- Kondo, M.; Scherer, D.C.; Miyamoto, T.; King, A.G.; Akashi, K.; Sugamura, K.; Weismann, I.L. Cell-fate conversion of lymphoid-committed progenitors by instructive cytokines. Nature 2000, 407, 383–386. [Google Scholar] [CrossRef]
- Gekas, C.; Graf, T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 2013, 121, 4463–4472. [Google Scholar] [CrossRef]
- Shimazu, T.; Iida, R.; Zhang, Q.; Weiner, R.S.; Medina, K.L.; Alberola-Ila, J.; Kincade, P.W. CD86 is expressed on murine hematopoietic stem cells and denotes lymphopoietic potential. Blood 2012, 119, 4889–4897. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Bhattacharaya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Doulatove, S.; Laurenti, E.; Poeppl, A.; Jurisica, I.; Dick, J.E. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 2011, 333, 218–221. [Google Scholar] [CrossRef]
- Robin, C.; Bollerot, K.; Mendes, S.; Hoak, E.; Crisan, M.; Cerisoli, F.; Lauw, I.; Kaimakis, P.; Jorna, R.; Vermeulen, M.; et al. Human placenta is a potent hematopoietic niche containing hematopoietic stem and progenitor cells throughout development. Cell Stem Cell. 2009, 5, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ema, H.; Karlsson, G.; Yamaguchi, T.; Miyoshi, H.; Shioda, S.; Taketo, M.M.; Karlson, S.; Iwama, A.; Nakauchi, H. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 2011, 147, 1146–1158. [Google Scholar] [CrossRef]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olsen, D.P.; Knight, M.C.; Martin, R.P.; Shipani, M.E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.J.C.; Montei, N.M.; Dumortier, A.; Sutur, U.; MacDonald, H.R.; Radtke, F. Jagged-dependent Notch signalling is dispensible for hematopoietic stem cell self-renewal and differentiation. Blood 2005, 105, 2340–2342. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef]
- Bromberg, O.; Frisch, B.J.; Weber, J.M.; Porter, R.L.; Civitelli, R.; Calvi, L.M. Osteoblastic N-cadherin is not required for microenvironmental support and regulation of hematopoietic stem and progenitor cells. Blood 2012, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Stier, S.; Ko, Y.; Forkert, R. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J. Exp. Med. 2005, 201, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 2006, 12, 657–664. [Google Scholar] [CrossRef]
- Shook, B.; Gonzablez, S.E.; Grisotti, G.; Zwick, R.; Horsley, V. The role of adipocytes in tissue regeneration and stem cell niches. Ann. Rev. Cell Dev. Biol. 2016, 32, 609–631. [Google Scholar] [CrossRef]
- Spangrude, G.J.; Heimfeld, S.; Weissman, I.L. Purification and characterization of mouse hematopoietic stem cells. Science 1988, 241, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Ceredig, R.; Rolink, A.G.; Brown, G. Models of haematopoiesis: Seeing the wood for the trees. Nat. Rev. Immunol. 2009, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Nestorowa, S.; Hamey, F.K.; Pijuane Sala, B.; Diamanti, E.; Sheperd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Gottens, B. A single cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Mead, A.J. Sinlge cell aproaches reveal novel pathways for megakaryocyte and erythroid differnetiation. Blood 2019, 133, 1427–1435. [Google Scholar] [CrossRef]
- Balciunaite, E.; Ceredig, R.; Rolink, A.G. The earliest subpopulations of mouse thymocytes contains potent T, significant macrophage and natural killer cell but no B-lymphocyte potential. Blood 2005, 105, 1930–1936. [Google Scholar] [CrossRef]
- Glatman Zaretsky, A.; Taylor, J.J.; King, I.L.; Marshall, F.A.; Mohrs, M.; Pearce, E.J. T follicular helper cells differentiate from th2 cells in response to helminth antigens. J. Exp. Med. 2009, 206, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Gatzka, M.; Peters, T.; Borkner, L.; Hainzl, A.; Wang, H.; Sindrilaru, A.; Scharffetter-Kochanek, K. Reduced cd18 levels drive regulatory t cell conversion into th17 cells in the cd18hypo pl/j mouse model of psoriasis. J. Immunol. 2013, 190, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Kim, I.K.; Park, Y.J.; Kim, Y.S.; Kim, Y.J.; Chang, W.S.; Lee, Y.S.; Kweon, M.N.; Chung, Y.; Kang, C.Y. Conversion of th2 memory cells into foxp3+ regulatory T cells suppressing th2-mediated allergic asthma. Proc. Natl. Acad. Sci. USA 2010, 107, 8742–8747. [Google Scholar] [CrossRef]
- Eizenberg-Magar, I.; Rimer, J.; Zaretsky, I.; Lara-Astiaso, D.; Reich-Zeliger, S.; Friedman, N. Diverse continuum of cd4(+) t-cell states is determined by hierarchical additive integration of cytokine signals. Proc. Natl. Acad. Sci. USA 2017, 114, E6447–E6456. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Hemberg, M.; Barahana, M.; Ingber, D.E.; Huang, S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 2008, 453, 544–547. [Google Scholar] [CrossRef]
- Teles, J.; Pina, C.; Eden, P.; Ohlsson, M.; Enver, T.; Peterson, C. Transcriptional regulation of lineage commitment—A stochastic model of cell fate decisions. PLoS Comput. Biol. 2013, 98, e1003197. [Google Scholar] [CrossRef]
- Seshasayee, D.; Gaines, P.; Wojehowski, D.M. GATA-1 dominantly activates a program of erythroid gene expression in factor-dependent myeloid FDCW2 cells. Mol. Cell. Biol. 1998, 18, 3278–3288. [Google Scholar] [CrossRef]
- Gaine, M.E.; Sharpe, D.J.; Smith, J.S.; Colyer, H.A.A.; Hodges, V.M.; Lappin, T.R.; Mills, K.I. GATA2 regulates the erythropoietin receptor in t(12;21) ALL. Oncotarget 2017, 8, 66061–66074. [Google Scholar] [CrossRef][Green Version]
- Vicente, C.; Conchillo, A.; Garcia-Sanchez, M.A.; Odero, M.D. The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit. Rev. Oncol. Hematol. 2012, 82, 1–17. [Google Scholar] [CrossRef]
- Barroso, G.V.; Puzovic, N.; Dutheil, J.Y. The evolution of gene-specific transcriptional noise is driven by selection at the pathway level. Genetics 2018, 208, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Yokota, Y.; Satoh, Y.; Okuzaki, D.; Tokunaga, M.; Ishibashi, T.; Sudo, T.; Ueda, T.; Shingai, Y.; Ichi, M. Variable SATB1 levels regulate hematopoietic stem cell heterogeneity with distinct lineage fates. Cell Rep. 2018, 23, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Lund, R.J.; Narva, E.; Lahesmaa, R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 2012, 13, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Krause, D.; Greaves, M.; Sharkis, S.; Dexter, M.; Heyworth, C.; Enver, T. Multilineage gene expression precedes commitment in the hematopoietic system. Genes Dev. 1997, 11, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Mossadegh-Keller, N.; Sarrazin, S.; Kandalla, P.K.; Espinosa, L.; Stanley, E.R.; Nutt, S.L.; Moore, J.; Sieweke, M.H. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 2013, 497, 239–243. [Google Scholar] [CrossRef]
- Metcalf, D.; Burgess, A.W. Clonal analysis of progenitor cell commitment of granulocyte or macrophage production. J. Cell. Physiol. 1982, 111, 275–283. [Google Scholar] [CrossRef]
- Rieger, M.A.; Hoppe, P.S.; Smejkal, B.M.; Eitelhuber, A.C.; Schroeder, T. Hematopoietic cytokines can instruct lineage choice. Science 2009, 325, 217–218. [Google Scholar] [CrossRef]
- Grover, A.; Mancini, E.; Moore, S.; Mead, A.J.; Atkinson, D.; Rasmussen, K.D.; O’Carroll, D.; Jacobsen, S.E.; Nerlove, C. Erythropoietin guides multipotent hematopoietic progenitor cells towards an erythroid fate. J. Exp. Med. 2014, 211, 181–188. [Google Scholar] [CrossRef]
- Tsapogas, P.; Swee, L.K.; Nusser, A.; Nuber, N.; Kreuzaler, M.; Capoferri, G.; Rolink, H.; Ceredig, R.; Rolink, A. In vivo evidence for an instructive role of fms-like tyrosine kinase-3 (FLT3) ligand in hematopoietic development. Haematologica 2014, 99, 638–646. [Google Scholar] [CrossRef]
- Luis, T.; Barkas, N.; Giustacchini, A.; Guerrero, J.; Wu, B.; Bouriez-Jones, T.; Macaulay, I.; Mead, A.; Nerlove, C.; Ghevaert, C.; et al. Perivascular niche cells sense thrombocytopenia and activate platelet-biased HSCs in an IL-1 dependent manner. Exp. Hematol. 2019, 76, S77. [Google Scholar] [CrossRef]
- Singh, R.P.; Grinenko, T.; Ramasz, B.; Franke, K.; Lesche, M.; Dahl, A.; Gassman, M.; Chavakis, T.; Henry, I.; Wielockx, B. Hematopoietic stem cells but not multipotent progenitors drive erythropoiesis during chronic erythroid stress in Epo transgenic mice. Stem Cell Rep. 2018, 10, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Colmore, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukaemia cells create bone marrow niches that disrupt the behaviour of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef]
- Zwezdaryk, K.J.; Coffelt, S.B.; Figueroa, Y.G.; Liu, J.; Phinney, D.G.; LaMarca, H.L.; Florez, L.; Morris, C.B.; Hoyle, G.W.; Scandurro, A.B. Erythropoietin, a hypoxia-regulated factor, elicits a pro-angiogenic program in human mesenchymal stem cells. Exp. Hematol. 2007, 35, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Anagnostou, A.; Lee, E.S.; Keissiman, N.; Levinson, R.; Steiner, M. Erytrhopoietin has a mitogenic and postive chemotactic effect on endothelial cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5082–5972. [Google Scholar] [CrossRef]
- Poniewierska-Baran, A.; Rajewska, J.R.; Ratajczak, M.Z. Erythropoietin enhances migration of human nuroblastoma cells: In vitro studies and potential therapeutic implication. J. Cancer Stem Cell Res. 2017, 5, e1003. [Google Scholar]
- Fuller, K.; Owens, J.M.; Jagger, A.; Wilson, A.; Moss, R.; Chambers, T.J. Macrophage colony-stimulating factors stimulates survival and chemotactic behaviour in isolated osteoclasts. J. Exp. Med. 1993, 178, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Griffin, J.D.; Rambaldi, A.; Chen, Z.D.; Mantovani, A. Induction of monocyte migration by recombinant macrophage colony-stimulating factor. J. Immunol. 1988, 141, 575–579. [Google Scholar] [PubMed]
- Vedham, V.; Phee, H.; Coggeshall, K.M. Vav activation and function as a Rac guanine nucleotide exchange factor in macrophage colony-stimulating factor-induced macrophage chemotaxis. Mol. Cell. Biol. 2005, 25, 4211–4220. [Google Scholar] [CrossRef][Green Version]
- Pierce, J.H.; Di Marco, E.; Cox, G.W.; Lombardi, D.; Ruggiero, M.; Varesio, L.; Wang, L.M.; Choudhury, G.C.; Sakaguchi, A.Y.; Di Fiore, P.P. Macrophage-colony-stimulating factor (CSF-1) induces proliferation, chemotaxis, and reversible monocytic differentiation in myeloid progenitor cells transfected with human c-fms/CSF-1 receptor cDNA. Proc. Natl. Acad. Sci. USA 1990, 87, 5613–5617. [Google Scholar] [CrossRef]
- Gomez-Cambronero, J.; Horn, J.; Paul, C.C.; Baumann, M.A. Granulocyte-macrophage colony-stimulating factor is a chemoattractant cytokine for human neutrophils: Involvement of the ribosomal p70 S6 kinase signalling pathway. J. Immunol. 2003, 171, 6846–6855. [Google Scholar] [CrossRef] [PubMed]
- Bussolino, F.; Wang, J.M.; Defilippi, P.; Turrini, F.; Sanavio, F.; Edgell, J.; Aglietta, M.; Arese, P.; Mantovani, A. Granulocyte- and granulocyte-macrophage-colony stimulating factors induce human endothelial cells to migrate and proliferate. Nature 1989, 337, 471–473. [Google Scholar] [CrossRef]
- Vaillant, P.; Muller, V.; Martinet, Y.; Martinet, N. Human granulocyte and granulocyte-macrophage-colony stimulating factors are chemotactic and “competence” growth factors for human mesenchymal cells. Biochem. Biophys. Res. Commun. 1993, 192, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Ceredig, R. Modelling the hematopoietic landscape. Front. Cell. Dev. Biol. 2019, 7, 104. [Google Scholar] [CrossRef]
- Sugima, R.; Jha, D.K.; Han, A.; Soria-Vallas, C.; da Rocha, E.L.; Lu, Y.-F.; Goettel, J.A.; Serro, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoetic stem and progenitor cells from human pluripotent cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Torisawa, Y.-S.; Spina, C.S.; Mammoto, T.; Mammoto, A.; Weaver, J.C.; Tat, T.; Collins, J.J.; Ingbar, D.E. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat. Methods 2014, 11, 663–669. [Google Scholar] [CrossRef]
- Bourgine, P.E.; Klein, T.; Paczulla, A.M.; Shimizu, T.; Kunz, L.; Kokkaliaris, K.D.; Coutu, D.L.; Lengerke, C.; Skoda, R.; Schroeder, T.; et al. In vitro biomimetic engineering of a human hematopoietic niche with functional properties. Proc. Natl. Acad. Sci. USA 2018, 115, E5688–E5695. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukaemia is organised as a hierarchy that originates from a primitive haematopoietic cell. Nat. Med. 1997, 197, 461–463. [Google Scholar]
- Vicente-Duenas, C.; Perez-Caro, M.; Abollo-Jimenez, F.; Cobaleda, C.; Sanchez-Garcia, I. Stem-cell driven cancer: “hands-off” regulation of cancer development. Cell Cycle 2009, 8, 1314–1318. [Google Scholar] [CrossRef]
- Agiaz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schnieder, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unravelling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic luekemia using genome-wide analysis. Haematologica 2019, 104, 1176–1188. [Google Scholar] [CrossRef]
- Cox, C.V.; Blair, A. A primitive cell origin for B-cell precursor ALL? Stem Cell Rev. 2005, 1, 189–196. [Google Scholar] [CrossRef]
- Edwards, R.H.; Wasik, M.A.; Finan, J.; Rodriguez, R.; Moore, J.; Kamoun, M.; Rennert, H.; Bird, J.; Nowell, P.C.; Salhang, K.E. Evidence for early hematopoietic progenitor cell involvement in acute promyelocytic leukemia. Am. J. Clin. Pathol. 1999, 112, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ramirez, I.; Bhatia, S.; Rodriguez-Hernandez, G.; Gonzalez-Herrero, I.; Walter, C.; Gonzalez de Tena-Davila, S.; Parvin, S.; Haas, O.; Woessmann, W.; Stanulla, M.; et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 2018, 37, e98783. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lorenzo, A.; Auer, F.; Chan, L.N.; Garcia-Ramirez, I.; Gonzalez-Herrero, I.; Rodriguez-Hernandez, G.; Bartenhagen, C.; Dugas, M.; Gombert, M.; Ginzel, S.; et al. Loss of Pax5 Exploits Sca1-BCR-ABL(p190) susceptibility to confer the metabolic shift essential for pB-ALL. Cancer Res. 2018, 78, 2669–2679. [Google Scholar] [CrossRef]
- Perez-Caro, M.; Cobaleda, C.; Gonzalez-Herrero, I.; Vicente-Duenas, C.; Bermejo-Rodriguez, C.; Sanchez-Beato, M.; Orfao, A.; Pintado, B.; Flores, T.; Sanchez-Martin, M.; et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009, 28, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Sanchez-Garcia, I. Is lineage decision-making restricted during tumoral reprograming of haematopoietic stem cells? Oncotarget 2015, 6, 43326–43341. [Google Scholar] [CrossRef]
- Nishi, K.; Katayama, N.; Miwa, H.; Shikami, M.; Masuya, M.; Shiki, H.; Kila, K. The survival of human leukemic B-cell precursors is supported by stromal cells and cytokines associated with the expression of bcl-2 protein. Br. J. Haematol. 1995, 105, 701–710. [Google Scholar] [CrossRef]
- Hiwase, D.K.; White, D.L.; Powell, J.A.; Saunders, V.A.; Zrim, S.A.; Frede, A.K.; Guthridge, M.A.; Lopez, A.F.; D’Andrea, R.J.; To, L.B.; et al. Blocking cytokine signalling along with intense bcr-Abl kinase inhibition induces apopotosis in primary CML progenitors. Leukemia 2010, 24, 771–778. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Guttorp, P.; Abkowitz, J.L. Visualizing hematopoiesis as a stochastic process. Blood Adv. 2018, 2, 2637–2645. [Google Scholar] [CrossRef]
- Chasis, J.A.; Mohandas, N. Erythroblastic islands: Niches for erythropoiesis. Blood 2008, 112, 470–478. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, G. Hematopoietic Stem Cells: Nature and Niche Nurture. Bioengineering 2021, 8, 67. https://doi.org/10.3390/bioengineering8050067
Brown G. Hematopoietic Stem Cells: Nature and Niche Nurture. Bioengineering. 2021; 8(5):67. https://doi.org/10.3390/bioengineering8050067
Chicago/Turabian StyleBrown, Geoffrey. 2021. "Hematopoietic Stem Cells: Nature and Niche Nurture" Bioengineering 8, no. 5: 67. https://doi.org/10.3390/bioengineering8050067
APA StyleBrown, G. (2021). Hematopoietic Stem Cells: Nature and Niche Nurture. Bioengineering, 8(5), 67. https://doi.org/10.3390/bioengineering8050067