1. Introduction
Standard cell culture methodology involves growing cells in a flask with a flat surface and letting them attach and proliferate on this area. While simple and reproducible, this technique does not represent the actual physiological condition of the cells in the human body. In recent years, 3D cell culture has been able to replicate many aspects of in vivo cell biology [
1,
2,
3]. There are a range of approaches to 3D cell culture, each with their own specific properties. Cells can be maintained in suspension and driven to form a spheroid, or using tissue samples and stem cell biology they can be differentiated to form organoids that replicate the cell diversity found in organs like the kidney. Both of these systems have been shown to be highly effective for testing both basic biological function and drug interactions. For example, when drugs are tested in 3D, it has been shown that the expression levels of drug-metabolising enzymes resemble that of native tissue [
2,
3]. A third approach involves the use of a scaffold or matrix to support cell attachment and engineer the tissue architecture. The choice of matrix in which the cells grow is critical in determining cell morphology and function [
2,
4]. These systems can be designed to replicate the spatial organisation, cell population, cell diversity and function of human tissues in a way that cannot be readily achieved in 2D monolayer cultures and create larger tissue samples than can be achieved readily with organoids and spheroid cultures.
However, as 3D cell culture makes its way into laboratories, the majority of monitoring tools are still designed for 2D culture systems. Bright-field microscopy, a workhorse for cell monitoring in 2D, is not suitable for 3D cultures where the cells are usually embedded in a less-transparent extracellular matrix such as collagen and hydrogel matrices. End-point histology and fluorescence microscopy are currently the most commonly used tools in 3D cell culture [
5]. Even though these techniques are gold standards, they are time-consuming and, in most cases, require the destruction of the sample for sectioning and staining. While live cell approaches abound, reporter constructs and stains have a limited lifespan and the architecture and scattering associated with 3D cultures makes the adaptation of traditional detection techniques challenging [
6].
An alternative in situ approach for monitoring cell cultures involves the characterisation of electrical properties of cells in culture. Electrical impedance spectroscopy (EIS) is a well-established technique that is widely used to monitor cell proliferation, migration, and viability in 2D cultures [
7,
8,
9,
10]. This is a low-cost, label-free and fast-readout technique where electrodes are usually positioned beneath the culture and changes in current/voltage through the cells are measured as a frequency sweep is undertaken. This process allows the electrical resistance and capacitance of the cells to be monitored across the frequency spectrum [
11,
12]. Moreover, the whole spectrum can be studied and correlated with different parts of the cell culture system. Researchers have started applying this technique with 3D cultures [
12,
13,
14,
15,
16,
17,
18,
19]. Lei et al. and Pan et al. successfully detected cell proliferation and death of cells within 3D spheroids [
15,
16,
17,
18]. Further, Lee et al. were able to monitor single cells embedded in a hydrogel using vertical parallel electrodes positioned on either side of the culture [
14]. Nonetheless, the application of EIS in 3D systems is complex. These models have several components that are continuously changing over the culture time (cells, matrix and medium), and one should be careful when interpreting EIS data as there is not yet a clear understanding of the effects of these changes on the impedance. Detailed reviews of the challenges involved in implementing EIS in 3D cultures have been recently written by our group and Gerasimenko et al. [
20,
21].
The cell index (CI) has been introduced by scientists to normalise the impedance values for data analysis. This allows for better analysis of the cell data as the background from control measurements is subtracted [
18,
20]. Further, it gives biologists an easier readout from their 2D systems that they can then correlate with cell proliferation and death. Even though application of the CI seems to be straight forward in 2D cultures, there are still some drawbacks in the use of the CI to analyse impedance based sensing. When CI is used to normalised the results the most sensitive frequency is singled out and the whole spectrum is discriminated as a result, this can lead to biases when choosing the most sensitive frequency. Further, in 3D cultures the lack of understanding of the individual components in the impedance spectrum means use of the CI to analyse 3D measurements is not straight forward and could lead to over-interpretation of results. The CI relies on the use of reproducible control samples and in 3D this become more challenging due to the nature of the matrix, cell distribution and the growth behaviour of the specific cell type. If the matrix in the absence of cells is used as the control one risks misinterpreting results if the matrix is not stable over-time or if its fundamental properties change in the presence of cells (e.g., contraction, deposition or turnover of extracellular matrix and/or matrix degradation).
The goal of this study was to understand how the distribution and architecture of cells in 2D and 3D affects impedance measurements gathered with an in-plane electrode. The 3D culture used in this study was a dermis model with fibroblast cells embedded in a collagen matrix [
22]. We analysed and compared the changes observed in both 2D and 3D cultures and studied their relationship to the cells and components present in the system. X-ray photoelectron spectroscopy (XPS) was used to examine the surfaces of the electrodes and bright field or fluorescence microscopy to examine the cells’ location during culture, allowing these parameters to be correlated with changes in the impedance spectrum and the associated CI.
2. Materials and Methods
2.1. Cell Culture
Human telomerase reverse transcriptase (hTERT) immortalised foreskin human fibroblast cells (BJ-5Ta, CRL-4001) were purchased from ATCC (American Type Culture Collection) (Manassas, VA, USA). The cells were cultivated in 4:1 mixture of Gibco Dulbecco’s modified Eagle’s medium (DMEM) and Gibco Medium 199 supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, Darmstadt, Germany) and 0.01mg/mL hygromycin B under standard conditions (37 C, 5% CO). Sub-cultivation was done in a 1:10 ratio. Cells were detached from culture flasks by treatment with trypsin-EDTA (0.25%, Life Technologies, Darmstadt, Germany) for 5 min. After detachment, cells were re-suspended in their cultivation medium to stop any remaining trypsin activity. After centrifugation at 250× g for 5 min, the supernatant was removed and the cells were re-suspended in the cultivation medium. Cells were used in passage 5 to 20.
2.2. Impedance Measurement System
A commercially available electric cell-substrate impedance sensing (ECIS) electrode array with integrated wells was purchased (ECIS-8W1E PET, Applied BioPhysics, NY, USA). The system consists of an array of gold electrodes deposited on a polyethylene terephthalate (PET) sheet, which is glued to 8 wells with a surface area of 0.8 cm. The working electrode has a diameter of 250 m. The wells were supplied sterile by the manufacturer. Before every experiment, each electrode was treated by adding 200 L of 10 mM L-cysteine (Sigma-Aldrich) in reverse osmosis (RO) water for 10 min at room temperature, then rinsed twice with RO water as recommended by the manufacturer.
The impedance spectrum was recorded every 24 h. To stabilise the temperature of the medium and cells, before every measurement the well was left at room temperature for 30 min. The impedance spectrum was collected from 10 Hz to 5 MHz. To obtain the impedance of the system, a small sinusoidal voltage (10 mV) was applied for a few milliseconds. Electrical impedance spectroscopy measurements were performed using a MFIA Impedance Analyser (Zurich Instruments, Zurich, Switzerland). The instrument was controlled using custom software initially developed by Dr. Steve Beguin and later adapted to this work in MATLAB R2016b (MathWorks, USA). The data was analysed using Python 3 in a Jupyter notebook. Here, the cell index (CI) is defined as
where
is the impedance magnitude of the samples containing cells at time
t and
is the impedance magnitude of the samples used as a control at time
t. The controls were DMEM + FBS for the measurements in 2D and collagen in DMEM + FBS for the measurements in 3D.
2.3. Cell Culture on Electrodes
For the 2D measurements, fibroblast cells were suspended in 500 L of their culture medium and seeded on top of the electrodes. Cells were allowed to attach to the electrode surface and then measurements were collected every 24 h over a total of 7 to 10 days.
The collagen matrix was prepared so that the final concentration of collagen was 3 mg/mL. The application note for KerCT Immortalized Keratinocyte Cells (CRL–4048
TM, ATCC, Manassas, VA, USA) was used to prepare the collagen matrix. Stock reagents and final concentrations are shown in
Table 1.
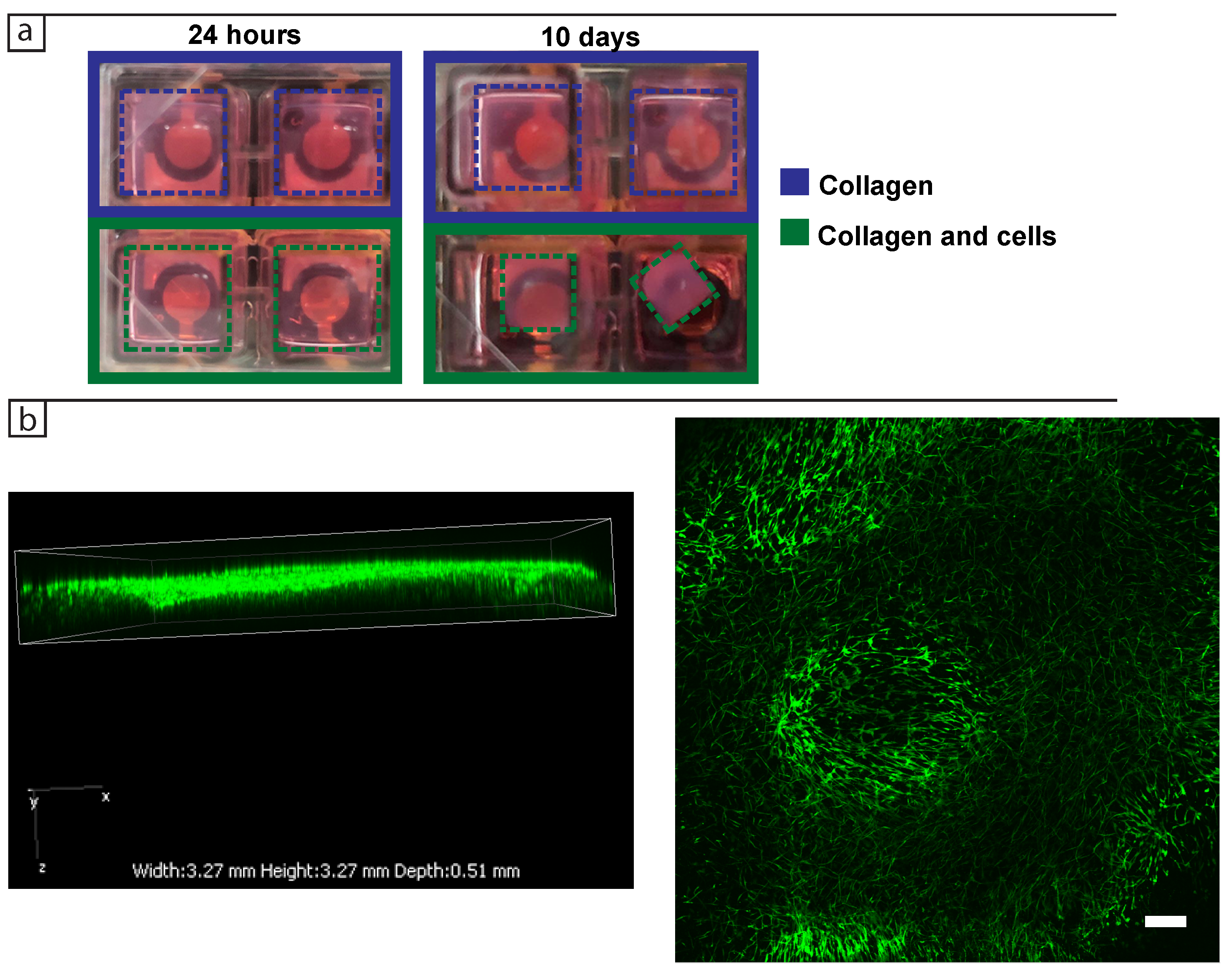
For the 3D dermis model, 200 L of the collagen matrix solution was prepared as described above with fibroblast cells mixed into the collagen prior to gelling for 30 min in standard cell culture conditions (37 C, 5% CO) and then topped up with cell culture medium. For each experiment two or three wells were seeded with cells in collagen and topped up with DMEM, while two or three wells were seeded with cell-free collagen and topped up with DMEM to serve as controls. The impedance spectrum was then collected every 24 h for 10 days in a frequency range from 50 Hz to 5 MHz for the three biological replicates.
2.4. Cell Imaging
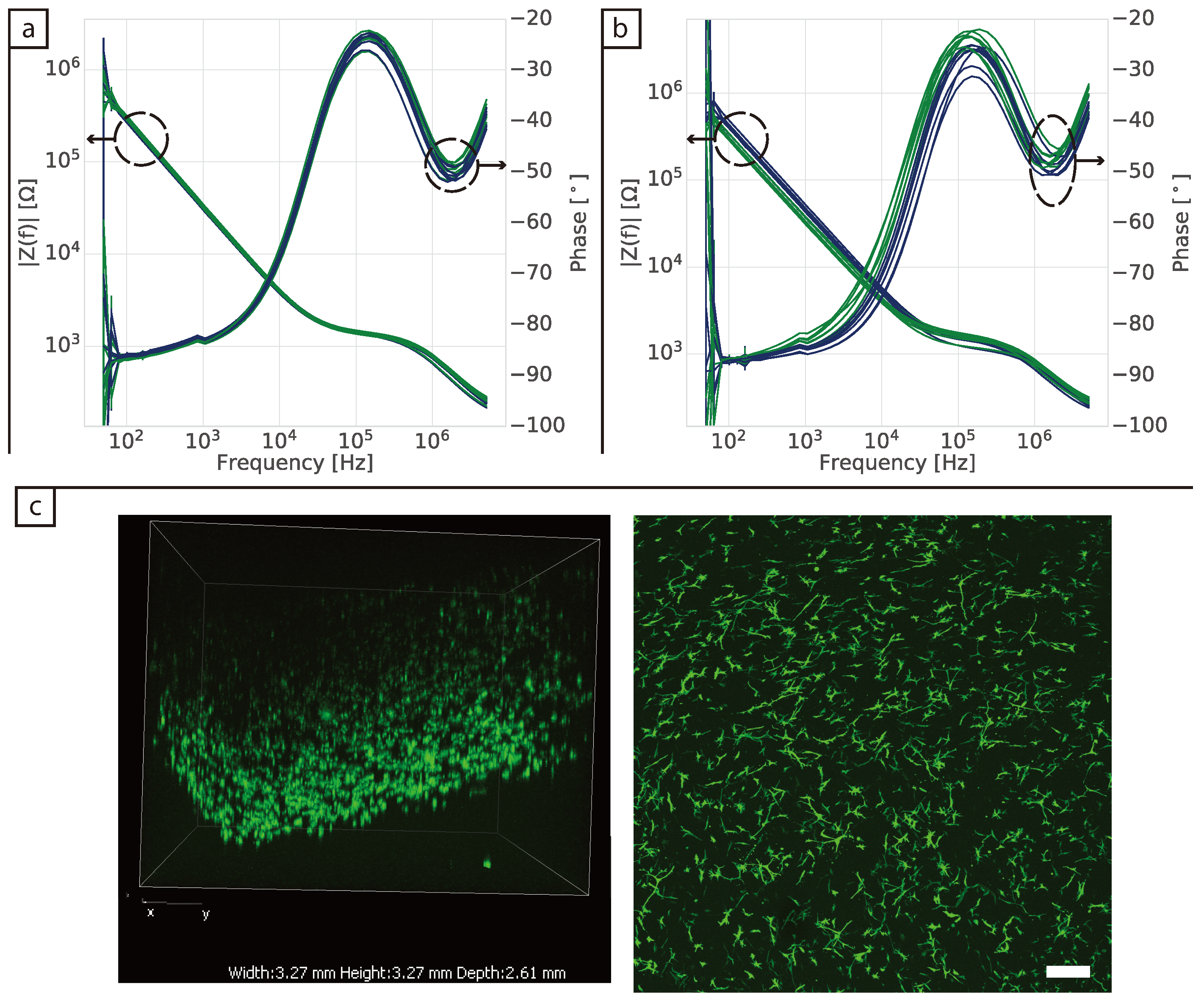
Bright field images were taken using either ZEISS Axio Vert.A1 and ZEN 2010 (Carl Zeiss Microscopy GmbH, 2011) or Nikon Eclipse Ti with NIS-Elements Advanced solutions (Nikon). For the phalloidin staining the 3D samples were first fixed in formaldehyde for 30 min on a shaker, followed by washing three times in 1× PBS, each time for 5 min on a shaker. They were then permeabilised using Triton X-100 for 30 min at room temperature on a shaker. The tissues were washed three times for 5 min with PBS on a shaker and incubated with Alexa FluorTM 488 Phalloidin (Invitrogen, Carlsbad, CA, USA) for 30 min for F-actin staining. Confocal images of 3D tissues were taken using Nikon A1 motorised inverted confocal microscope and NIS-Elements Advanced Research (Nikon, Tokio, Japan) with an emission wavelength of 525 nm and excitation of 488 nm.
2.5. X-ray Photoelectron Spectroscopy (XPS) Measurements
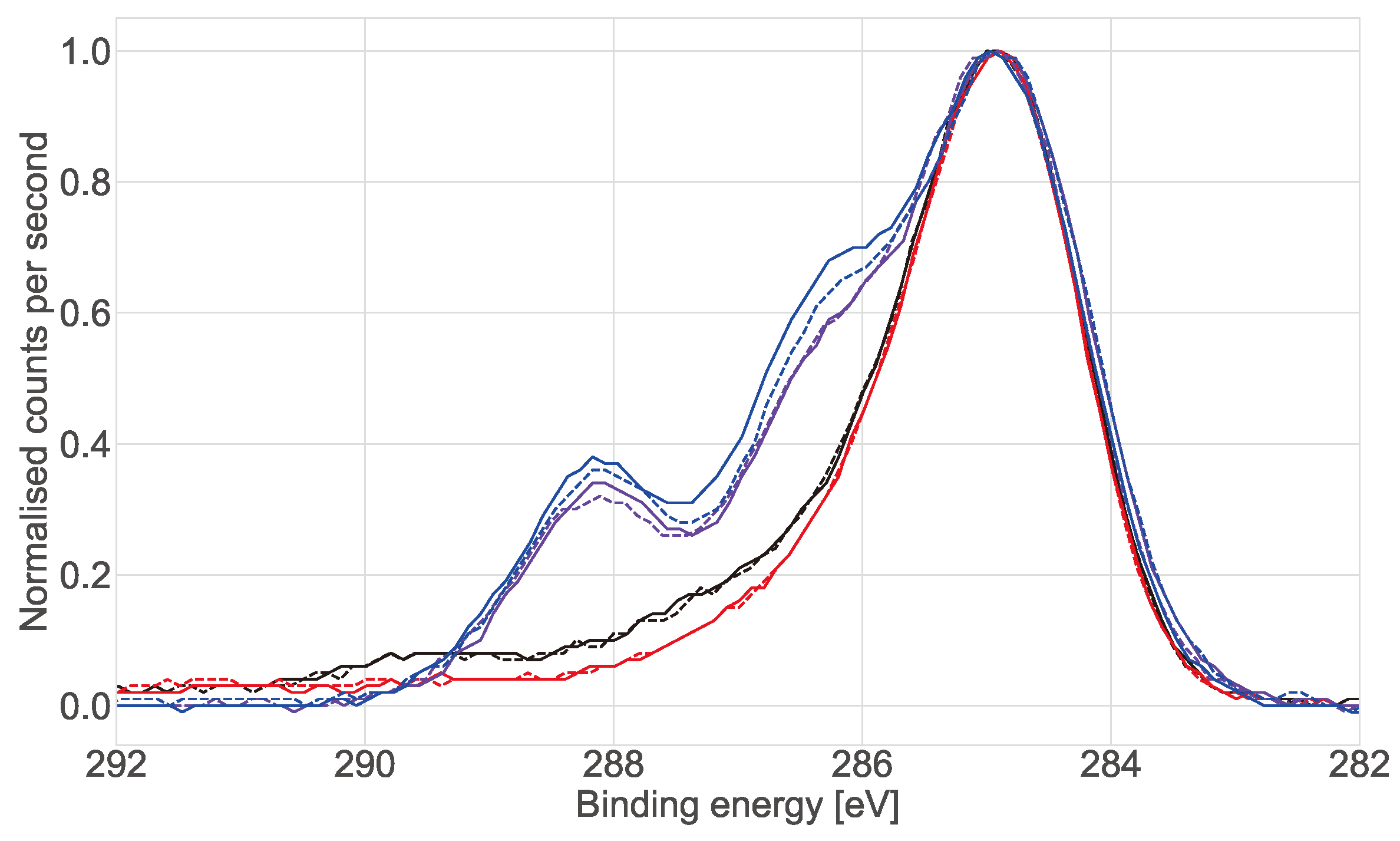
X-ray photoelectron spectroscopy (XPS) analysis was carried out on the commercial (ECIS) wells. Two working electrodes were left intact supplied by the manufacturer. Six wells were treated with L-Cysteine with two then incubated in Dubelcco’s phosphate-buffered saline (DPBS), two with cell medium and two with a collagen gel topped up with cell medium. All the electrodes were incubated for 7 days under standard cell culture conditions (37 C, 5% CO). After 7 days the collagen and medium were removed and the wells were washed with reverse osmosis (RO) water three times, the wells were then dried under a stream of nitrogen before XPS measurements were taken.
X-ray photoelectron spectroscopy (XPS) analysis was performed using an AXIS Nova spectrometer (Kratos Analytical Inc., Manchester, UK) with a monochromated Al K source at a power of 180 W (15 kV × 12 mA) and a hemispherical analyser operating in the fixed analyser transmission mode. The total pressure in the main vacuum chamber during analysis was typically between 10 and 10 mbar. Survey spectra were acquired at a pass energy of 160 eV and step size 0.5 eV. To obtain more detailed information about chemical structure, oxidation states etc., high resolution spectra were recorded from individual peaks at 40 eV pass energy and a step size of 0.1 eV, typically yielding a FWHM of <0.8 eV for the Ag 3d peak and <0.85 eV for the carbon 1s ester peak of polyethylene terephthalate (PET) during performance tests.
The samples were analysed at a nominal photoelectron emission angle of 0 w.r.t. the surface normal. Since the actual emission angle is ill-defined in the case of rough surfaces and powders as in the present case (ranging from 0 to 90) the sampling depth may range from 0 nm to approx. 10 nm. In a first step the standard aperture slot was used for analysis in which case the area analysed on the sample is elliptical in shape with approximate dimensions of 0.3 mm × 0.7 mm. In a second step (repeat analysis) the sampling area was reduced to a 220 m spot using an aperture.
Data processing was performed using CasaXPS processing software version 2.3.21 (Casa Software Ltd., Teignmouth, UK). All elements present were identified from survey spectra. The atomic concentrations of the detected elements were calculated using integral peak intensities and the sensitivity factors supplied by the manufacturer. Binding energies were referenced to the C 1s peak at 285.0 eV (“neutral” hydrocarbon). The accuracy associated with quantitative XPS is ca. 10–15%. Precision (ie. reproducibility) depends on the signal/noise ratio but is usually much better than 5%. The latter is relevant when comparing similar samples.
4. Discussion
Impedance spectroscopy has been widely characterised and studied for its use in cell culture since the 90s [
7,
8]. Research using small working electrodes and interdigitated electrodes has shown that it is possible to monitor cells in two dimensional (2D) cultures using this technique. While small working electrodes have been used to monitor confluent cell cultures challenged with drugs, interdigitated working and reference electrodes have been shown to be more effective at monitoring cell proliferation across the well surface over time [
7,
10,
25,
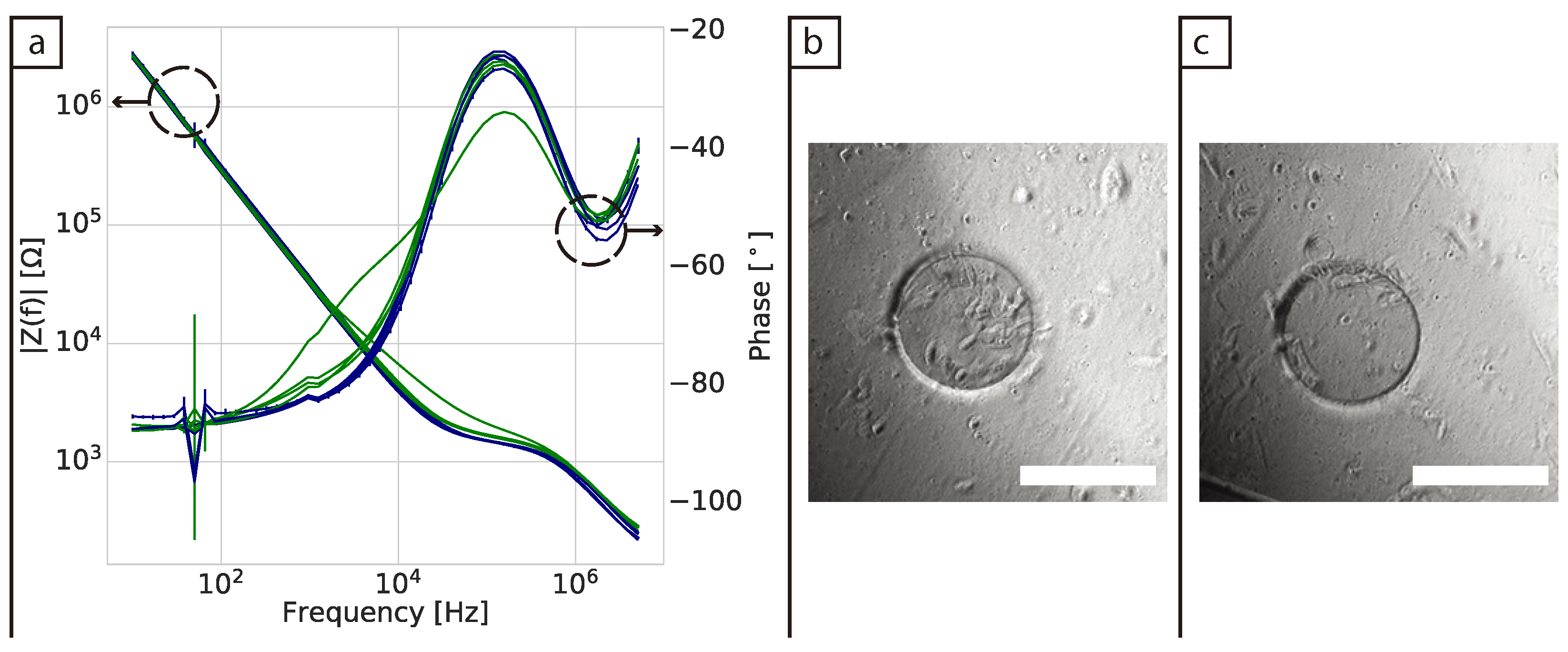
30]. This highlights the importance of electrode configuration for monitoring cell cultures and the differences in their potential applications. We have studied the use of a commercially available well with a small working electrode to monitor fibroblast cells in 2D and 3D. We have also measured the impedance across a broad frequency range (10 Hz to 5 MHz), rather than just the typically sensitive frequency of ≈10 kHz used in 2D cultures.
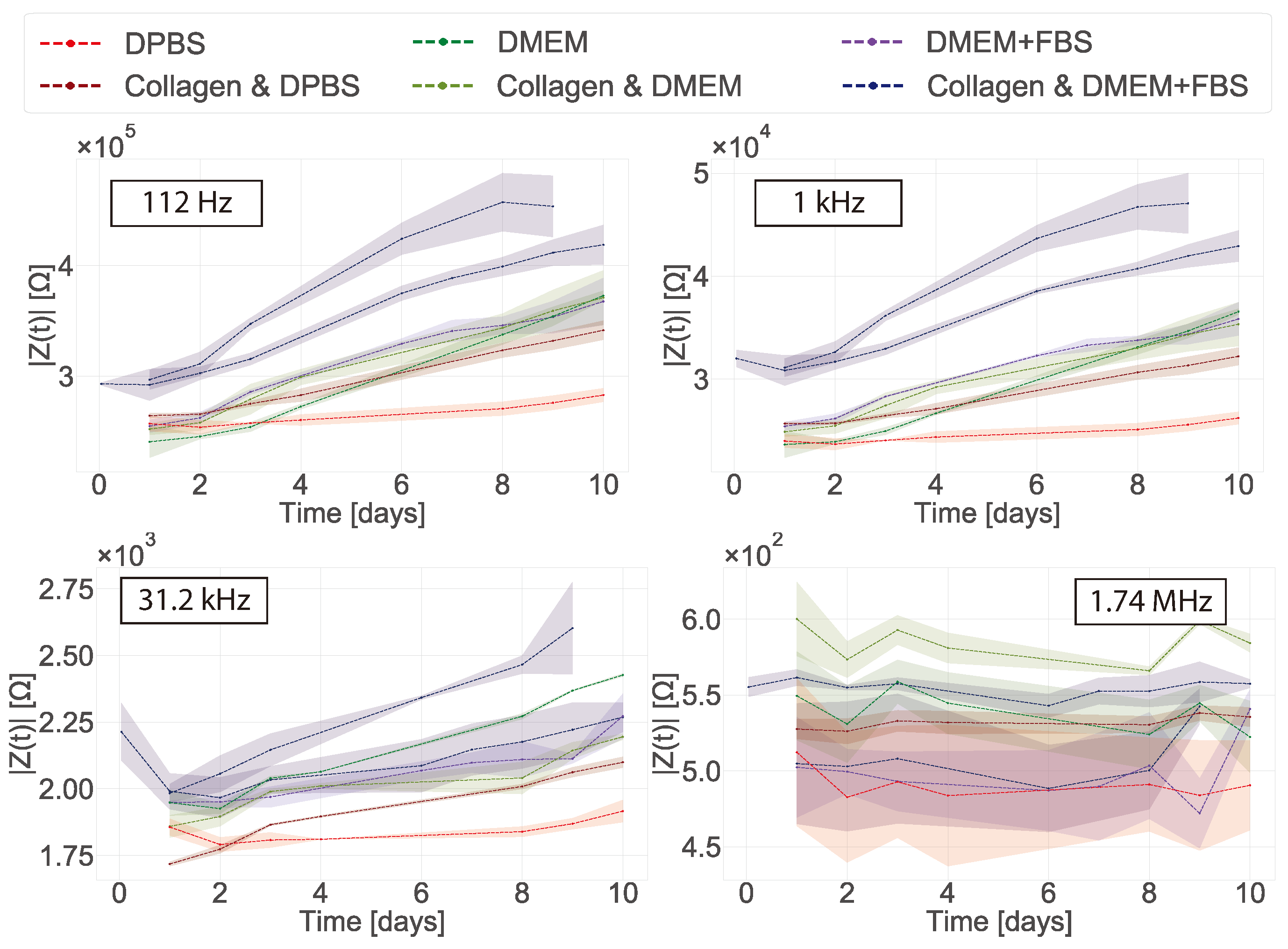
In 2D cultures the impedance magnitude of the control samples (DMEM + FBS only) increased over time in the low frequency range (10 Hz to 1 kHz), reflecting an increase in protein adsorption at the surface of the electrodes. In contrast, a stable signal was observed for the samples with cells. This decrease of impedance magnitude of cell samples relative to cell-free controls at low frequencies has been observed in 2D before [
31]. However, no clear explanation was given to this phenomenon. Here we proposed that protein adsorption from the medium was the cause and confirmed it with XPS measurements. This effect is most visible in the lower frequency range (<10 kHz). We hypothesise that the presence of the cells in the culture serves to stabilise the surface of the electrode, hence controlling protein adsorption (though not eliminating it). At mid-frequencies (1 kHz to 100 kHz) an increase related to the presence and proliferation of the cells was observed. However, there was variation between replicates and the increases reflected the number of cells on the surface of the individual working electrode. This highlights why confluent layers are more commonly used as controls when using impedance in 2D as they ensure coverage of the whole electrode and well area [
10,
30]. Furthermore, validation of impedance measurements in 2D is relatively straightforward, as the surface of the electrodes can usually be viewed directly with a brightfield microscope. This adds to the challenges of translating the technique to 3D cultures, where validation is done mainly via the analysis of replicates, rather than measuring the same cultures post impedance measurement due to the need to fix and/or stain samples prior to imaging.
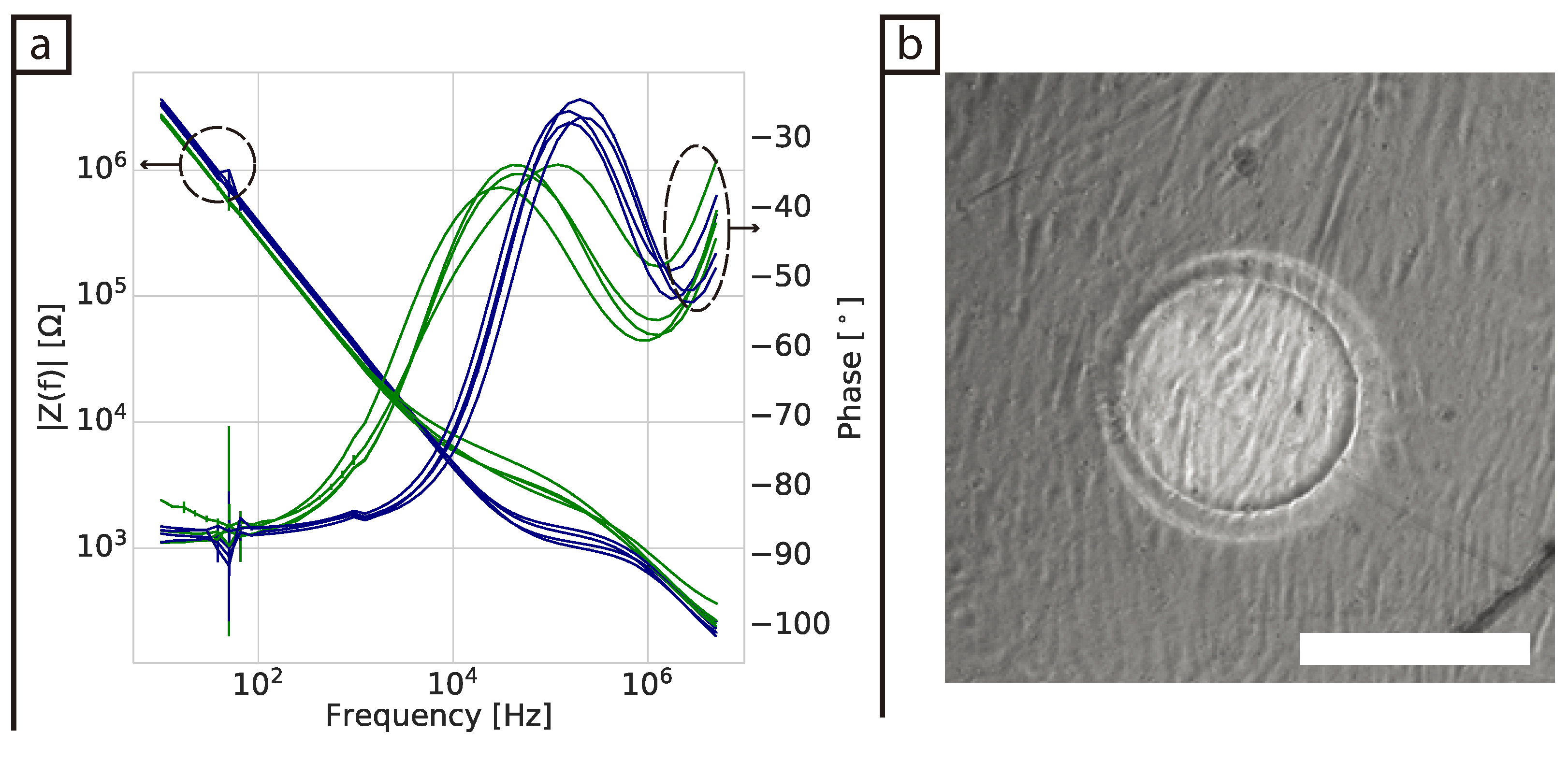
Some challenges were encountered when the in-plane ECIS device was used to monitor fibroblast cells in a collagen gel (3D model). First, the collagen contracts as the cells proliferate in the gel and this appears to generate additional variability in the measurements. However, at low frequencies protein adsorption was still observed for the control samples (DMEM and collagen gel), but not for the samples containing cells. It is not clear why no protein adsorption is observed in the samples containing cells as no cells were present at the surface of the electrodes. To the best of our knowledge there have been no impedance studies of fibroblast cells immerse in a collagen gel before. Pan et al. [
17,
18] and Lei et al. [
16] were able to monitor cell proliferation of 3D spheroids using parallel and interdigitated electrodes, respectively. However, their 3D cell culture model was fundamentally different as it consisted of multiple spheroids with each spheroid consisting of a cluster of cells connected via tight junctions. Thus, one could expect this system to have a higher resistance compared to our system where we are attempting to monitor dispersed single cells spread throughout a gel.
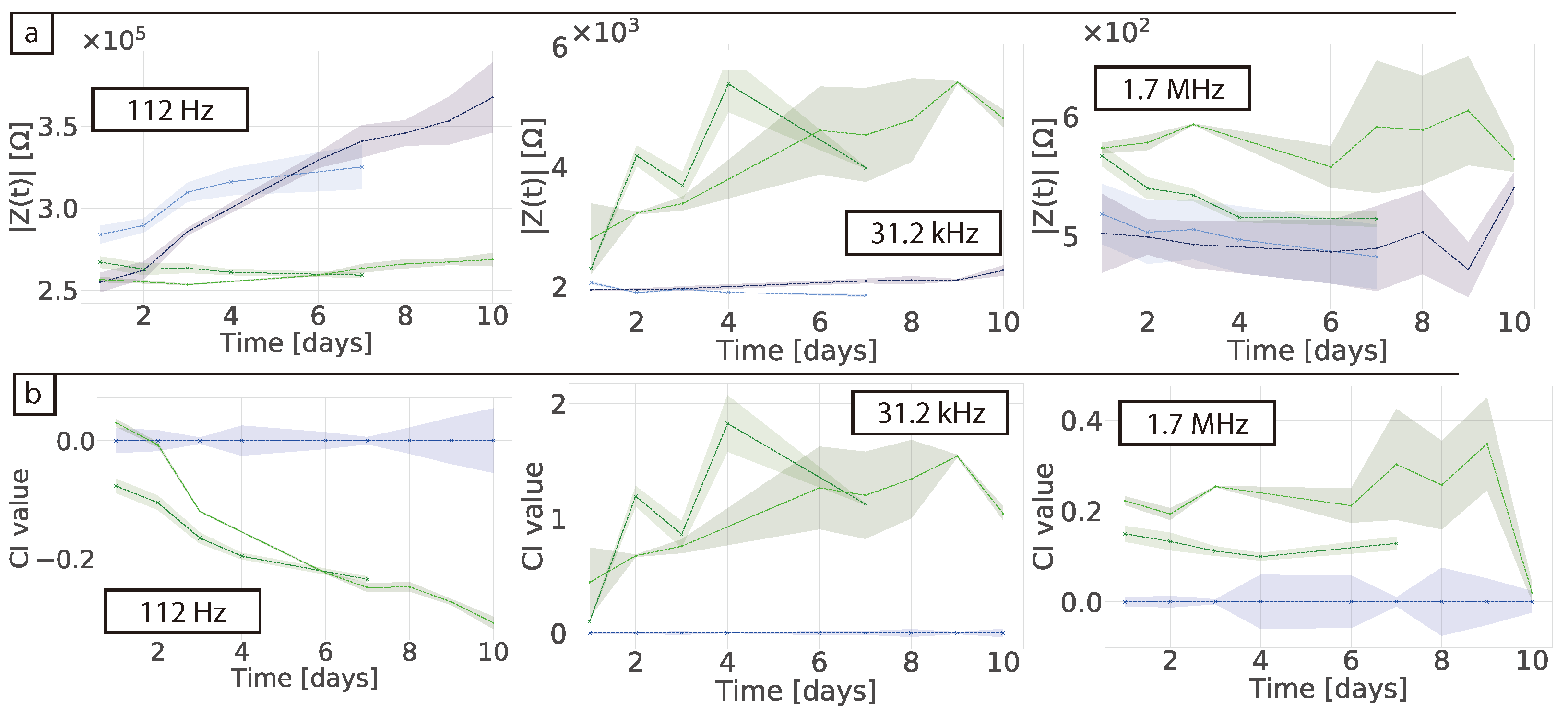
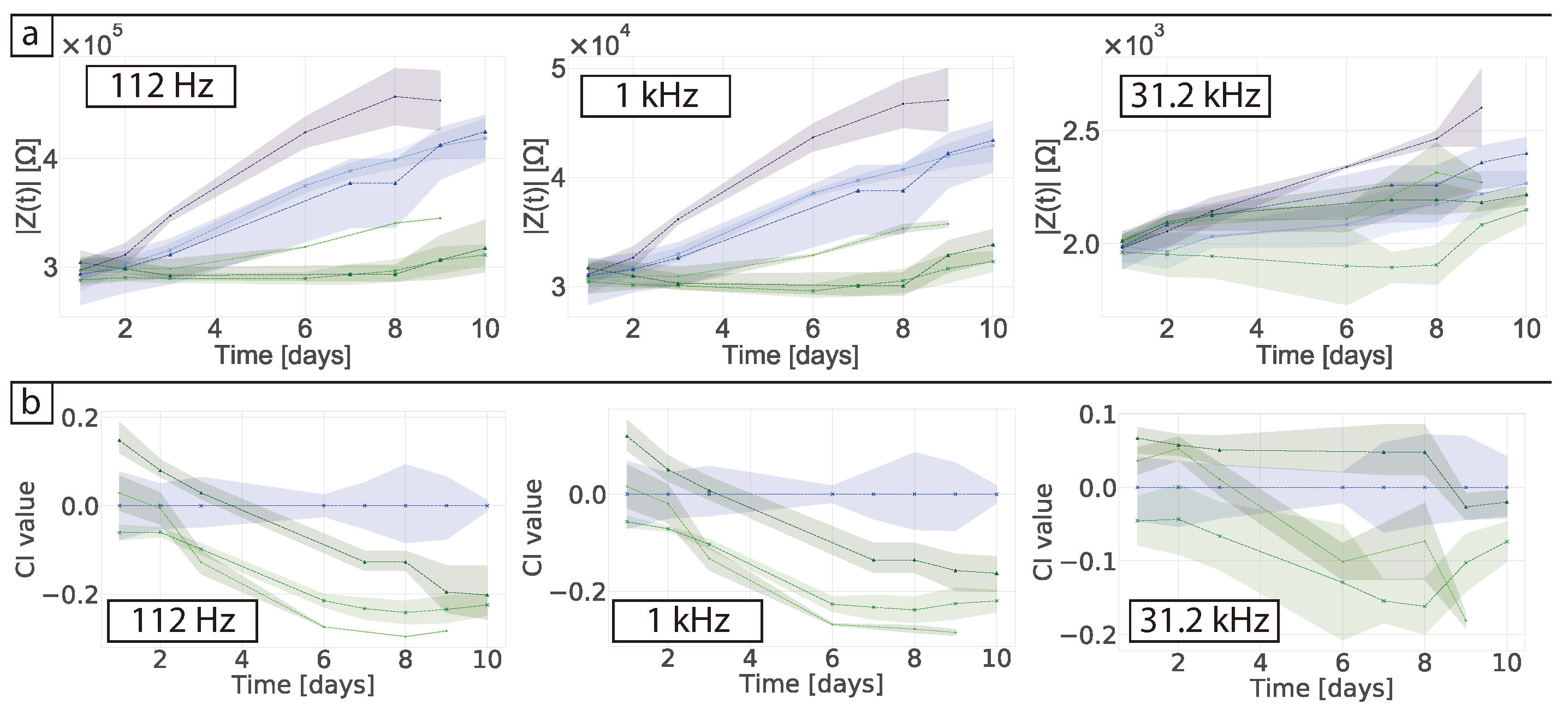
In their study Pan et al. used an absolute value of the CI to determine the difference between their controls and the samples containing cells, ascribing growth with CI value differences no larger than 0.6 [
17,
18]. Here we obtained a CI of 0.3 that can be attributed to protein adsorption. When the cells were measured in 2D a clear CI of ≈2 was obtained in our studies and ≈1.5 in Pan et al. This shows that overall the sensing system is not as sensitive in 3D as they are in 2D. Furthermore, the authors found that the proliferation of the cells decreased the impedance magnitude, which is the same effect we saw in our normalised data and showed the effect of protein adsorption. Nonetheless, the authors measured the stability of their matrigel alone for a period of 3 days and the CI variability was of around 0.02. The authors also found a sensitive frequency of 25 kHz which is not related to protein adsorption. This suggests that the small differences they observed were indeed from the cells. When our data was normalised to obtain the CI, the effects of protein adsorption in the cell-free controls were lost and results could be misinterpreted as measurements of the cells themselves. One should be careful when normalising data so no valuable information from the system is lost and data is not over-interpreted. In this regard, choosing and monitoring the temporal changes of the right controls is crucial when performing impedance measurements. Tonello et al. used a different equation to calculate the CI, namely
[
32]. In this case they were looking at time point variations by subtracting the impedance of the cells at the different time points and comparing them to the gel in the absence of cells. They found their CI increased over-time with as the cells proliferated. When our data was normalised using this equation no clear trend was observed and a negative CI value was obtained (see
Figure S4).
Lee et al. have used parallel electrodes to monitor single human breast cancer cells (GFP-MCF-7) encapsulated in alginate hydrogel [
14]. However, they did not use the full impedance components of the spectrum but only the capacitance, and monitored the cells over-time using 1 kHz as their sensitive frequency. This is interesting because we were still observing the effects of protein adsorption in our system at 1 kHz, which makes it unclear if the changes observed in the earlier study were due to cell proliferation itself or protein adsorption. Note that it is also not clear whether Lee et al. immersed the cell-free hydrogels in cell medium or protein-free PBS, in which case the changes could be more easily correlated to cell proliferation. More recently, Tonello et al. measured the impedance of human mesenchymal stromal cells in gelatin-chitosan hybrid hydrogel scaffolds [
32]. These cells did not form spheroids but rather individually proliferated within the hydrogel. Similar to our results, the authors found that the impedance of the cell-free hydrogels was changing over time and this phenomenon was different when the cells were present in the system. They attributed this behaviour to the medium hydrating the hydrogel scaffold and increasing the conductivity, hence decreasing the impedance of the system over time. This effect is opposite to the results observed in the present study, where we saw the impedance of the gel increasing over time, whereas in our case this effect was not related to the gel itself but to the medium. Moreover, Tonello et al. used 4 kHz and were able to monitor the cells at this frequency, whereas that was not the case in our study. The key difference between their setup and the one in this study is that they used parallel electrodes placed on each side of the gel, which may improve the sensitivity.
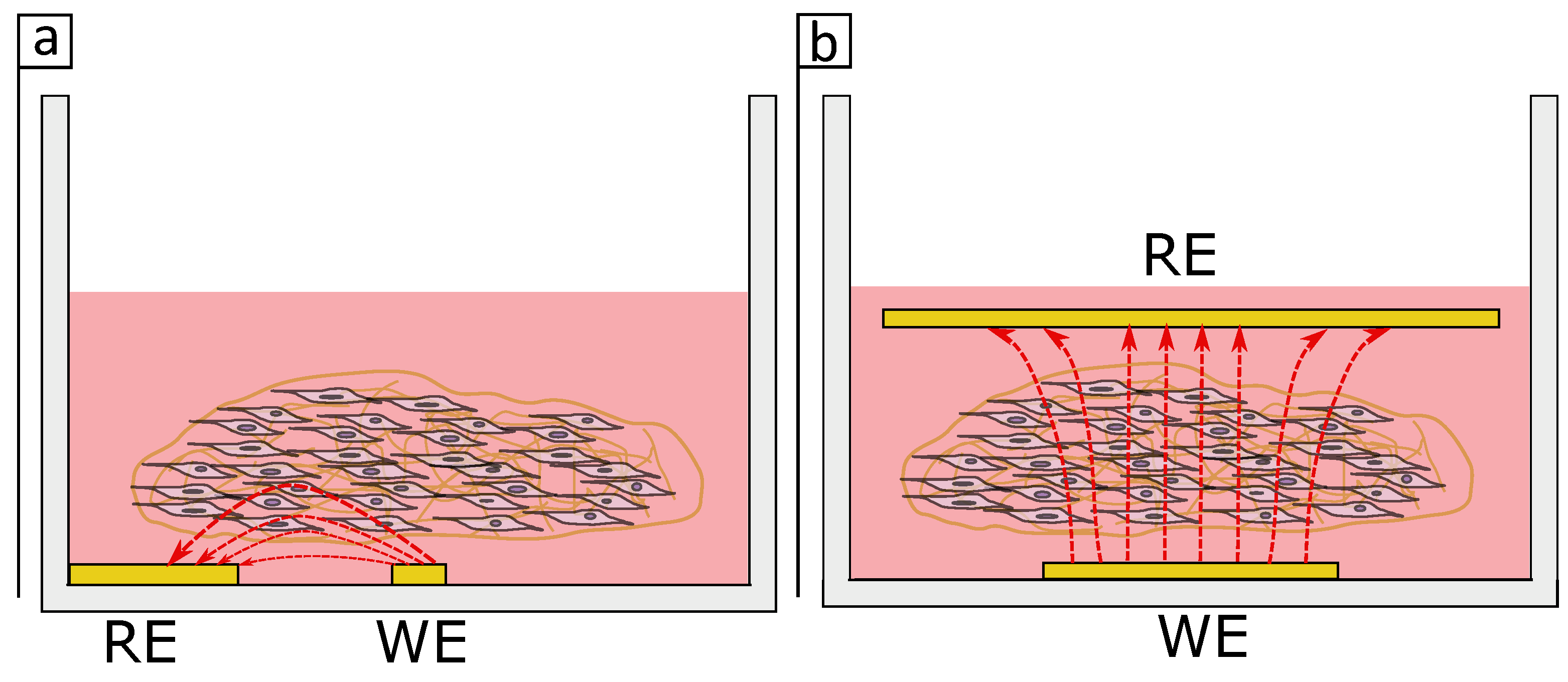
Figure 9 shows a schematic drawing of the current flow in an in-plane and parallel electrodes setup. While for the in-plane setup (
Figure 9a) the current may penetrate only a small volume of the tissue when the electrodes are placed in parallel (
Figure 9b) a higher volume of the tissue is penetrated by the current which in hand may have a bigger impact in the impedance measurements. It appears the electrodes dimensions and design are critical for monitoring distributed cells in a 3D tissue.
Overall, this study shows that commercially available in-plane electrodes are not suitable for monitoring 3D cultures. The complexity of the cell distribution in addition to the penetration of the electrical signals into the 3D culture represents a challenge for the direct translation of conventional impedance systems to work with 3D cultures. While researchers have been able to monitor 3D spheroids, the CI differences are still small and could be misleading if normalisation is done without caution. However, in our system it was not possible to monitor single fibroblast cells embedded in a collagen gel using electrodes on the surface of the culture well. There are too many components changing in this 3D systems (matrix, cells and medium) which makes it even more difficult to understand what is happening in the system. Different electrodes architectures have been shown to be significant in the monitoring of cells in 3D cultures, and may aid with the monitoring of the 3D model presented in this work. Furthermore, the contraction of the tissue represents a challenge for the reproducibility of both the 3D model itself and its consequent impedance measurement. Future work on modelling different electrode configurations should provide insight into how changes in electrode design will influence the measurement of 3D cultures and determine how electrodes can be designed to ensure current penetrates through the tissue and in turn improve the sensitivity.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}