Biomimetic 3D Models for Investigating the Role of Monocytes and Macrophages in Atherosclerosis

 ,
,  , ,
, , 
Abstract
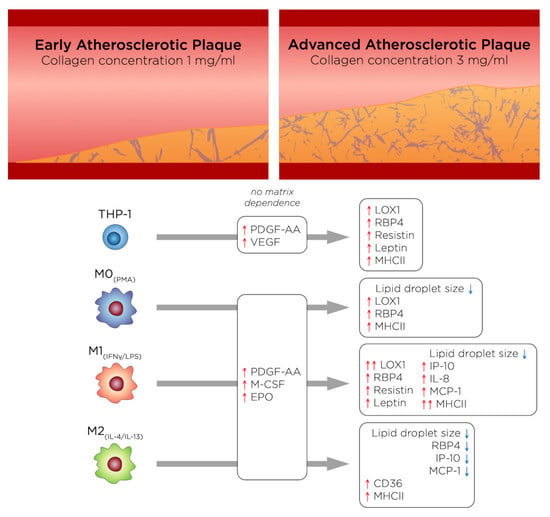
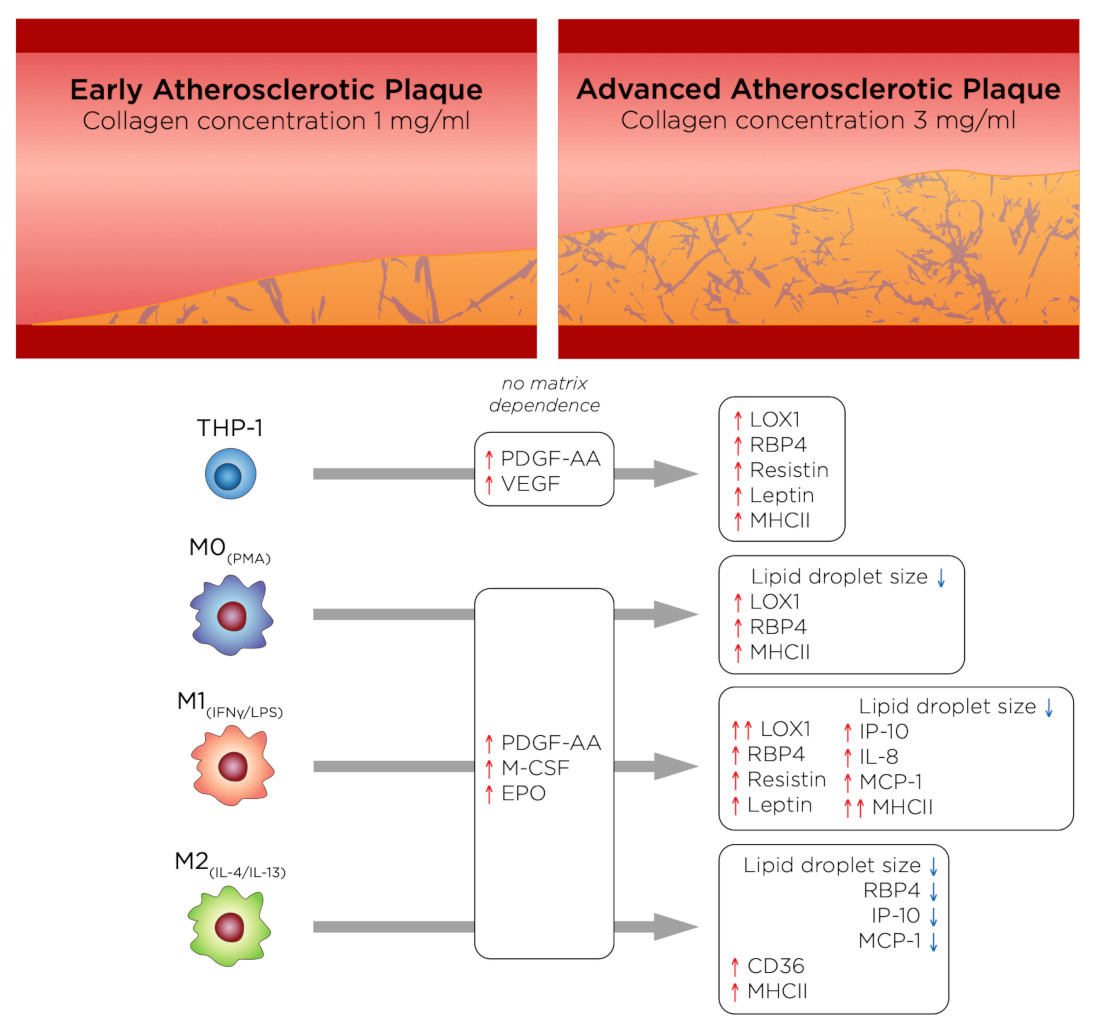{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Embedding and Differentiating THP-1 towards Macrophages in 3D Collagen Matrices
2.3. Topological and Mechanical Characterization of 3D Collagen Matrices
2.4. Treatment with oxLDL
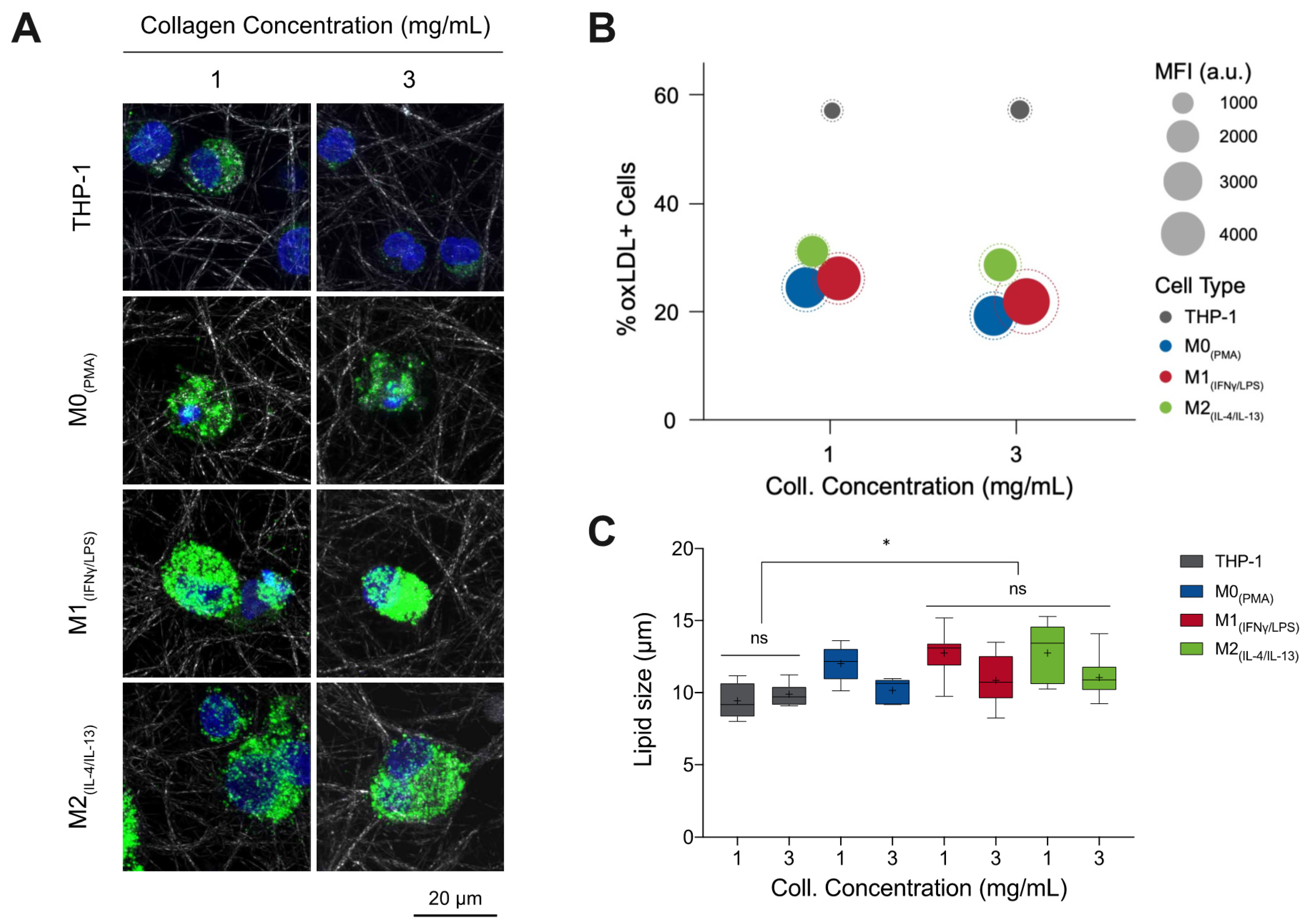
2.5. Quantitative Analysis of oxLDL Uptake
2.6. Quantitative Analysis of Lipid Droplet Size
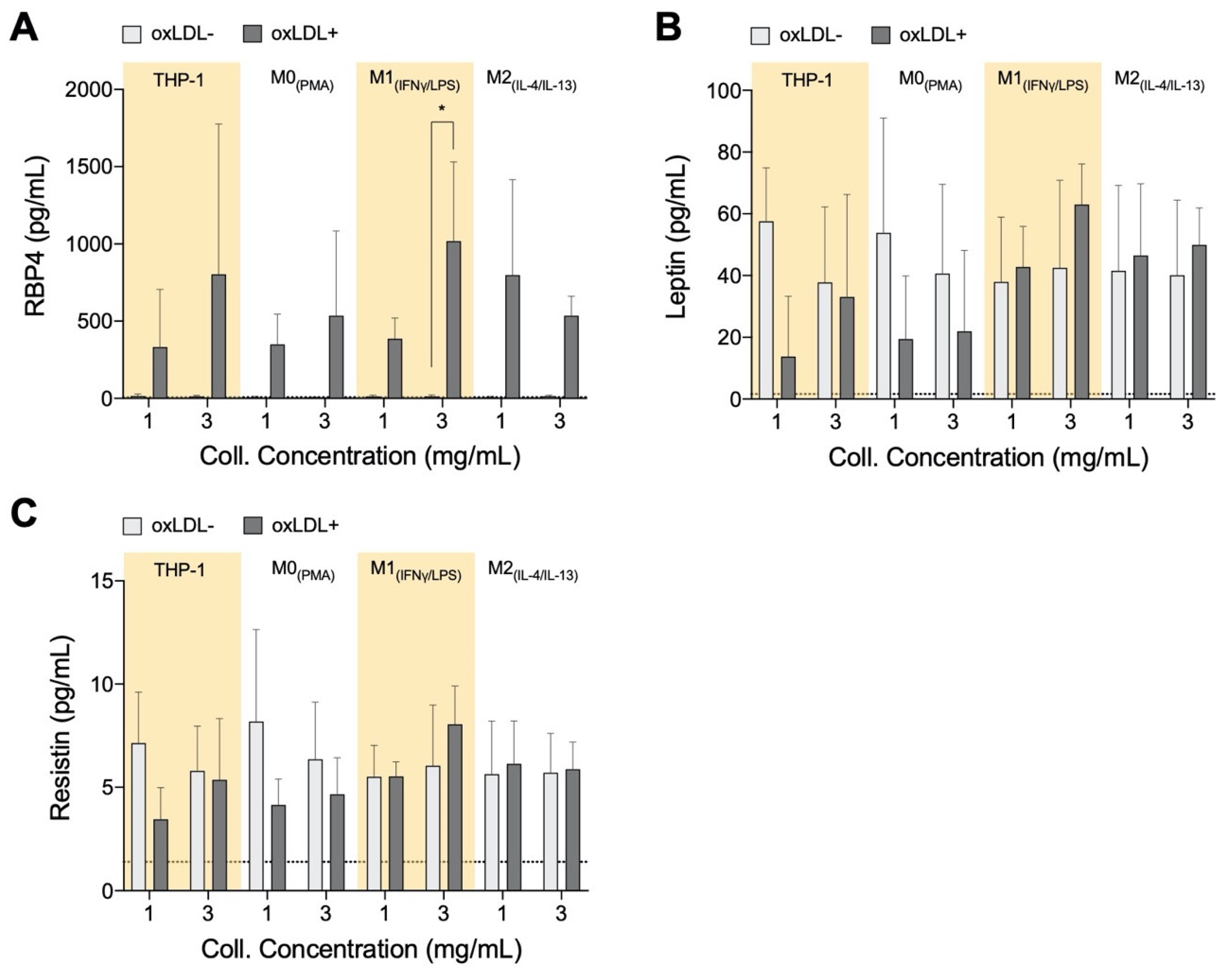
2.7. Quantitative Analysis of Adipokines, Chemoattractants, and Growth Factors
2.8. Gene Expression Analysis
2.9. Data and Statistical Analysis
3. Results
3.1. %oxLDL + Cells Are Greatest in Monocytes and M1 Accumulates the Most oxLDL
3.2. oxLDL Induced LOX1 Expression in Monocytes and Macrophages, while It Enhanced CD36 Expression in M2 Phenotypes
3.3. RBP4 Is Up-Regulated by All Cell Types with the Addition of oxLDL and Enhanced by High-Density Matrices for THP-1+LDL and M1+LDL
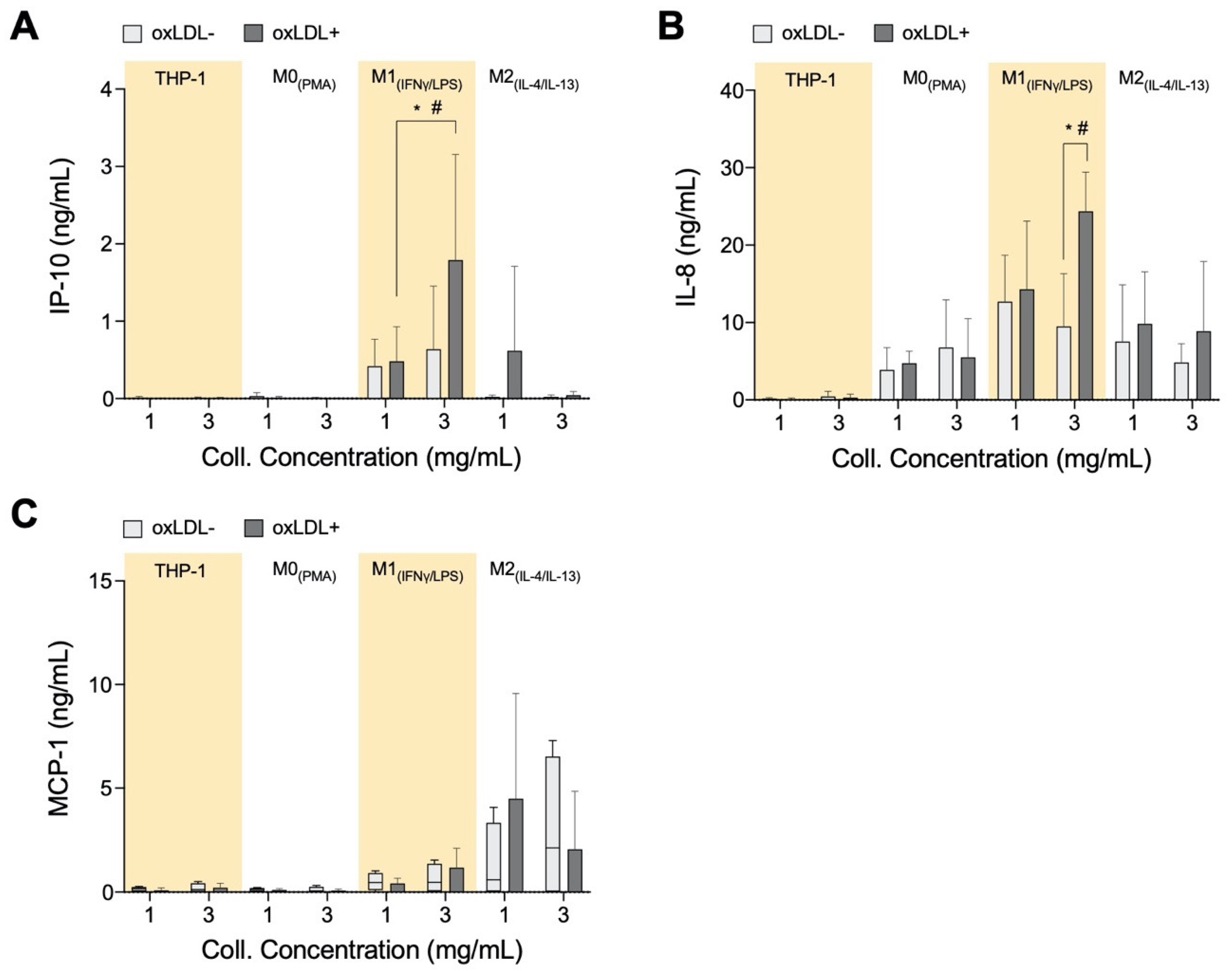
3.4. In High-Density Matrices THP-1+LDL and M1+LDL Secretion of Immune Cell Chemoattractants Are Elevated and Suppressed in M2+LDL
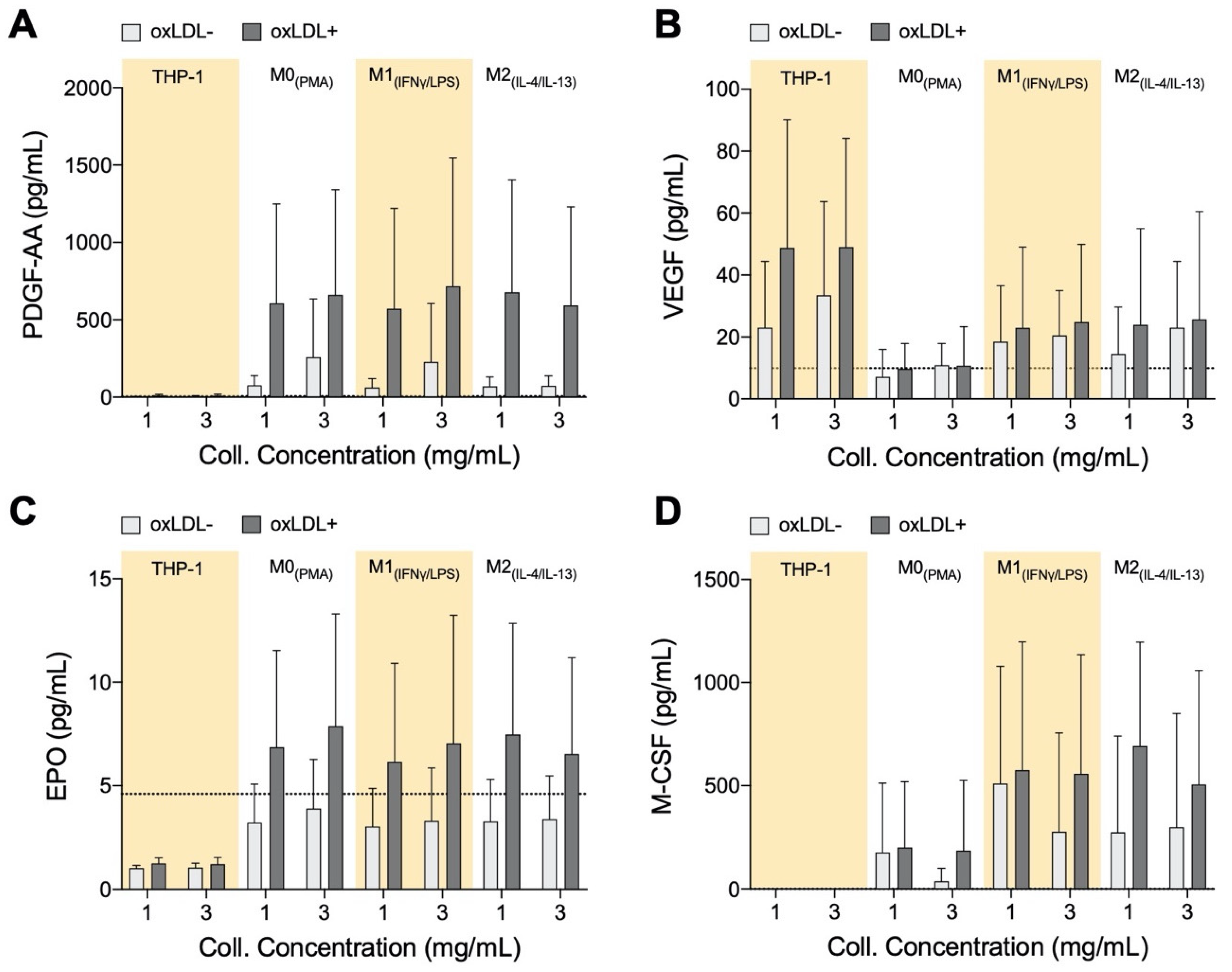
3.5. PDGF-AA, EPO, and M-CSF Are Up-Regulated by Macrophages and VEGF Is Up-Regulated by Monocytes
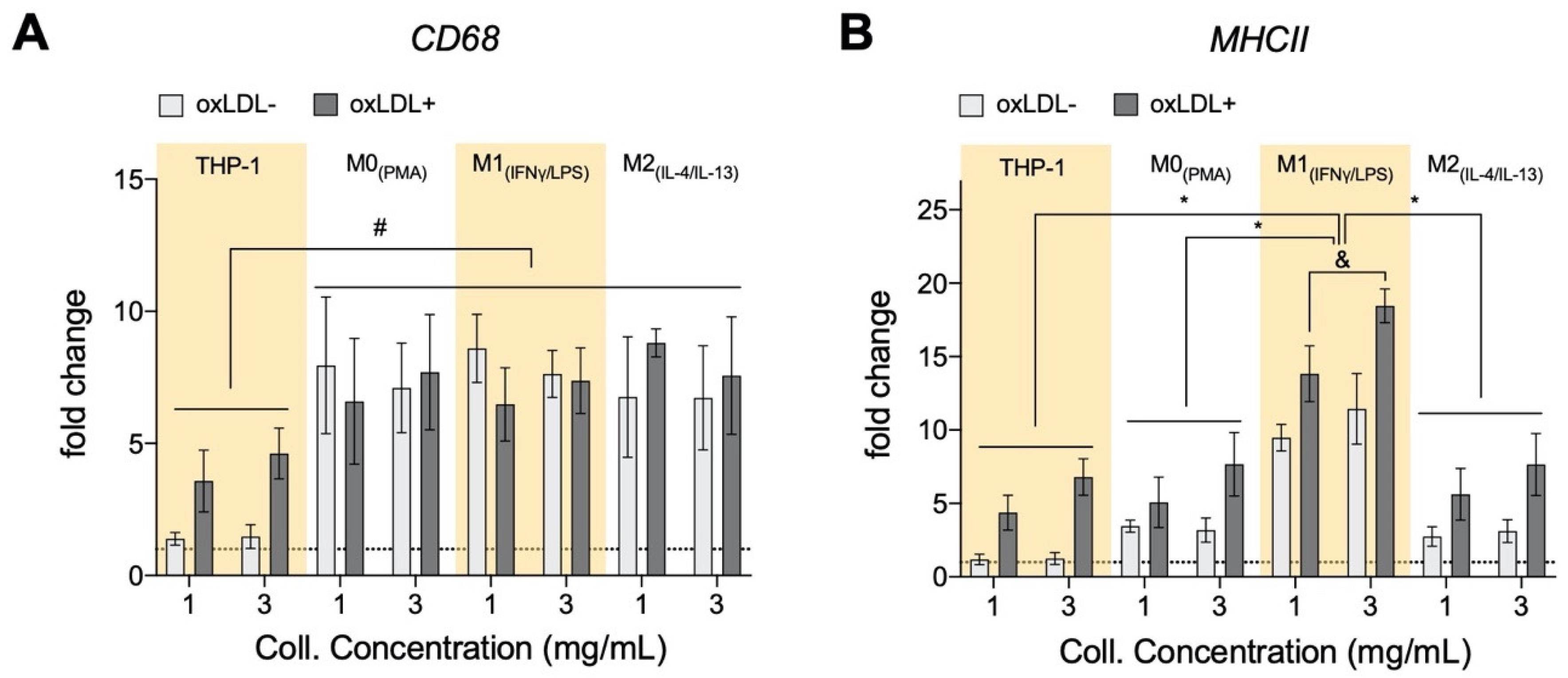
3.6. oxLDL Enhances Expression of CD68 in Monocytes and MHCII Is Enhanced in Dense Matrices for All Cell Types
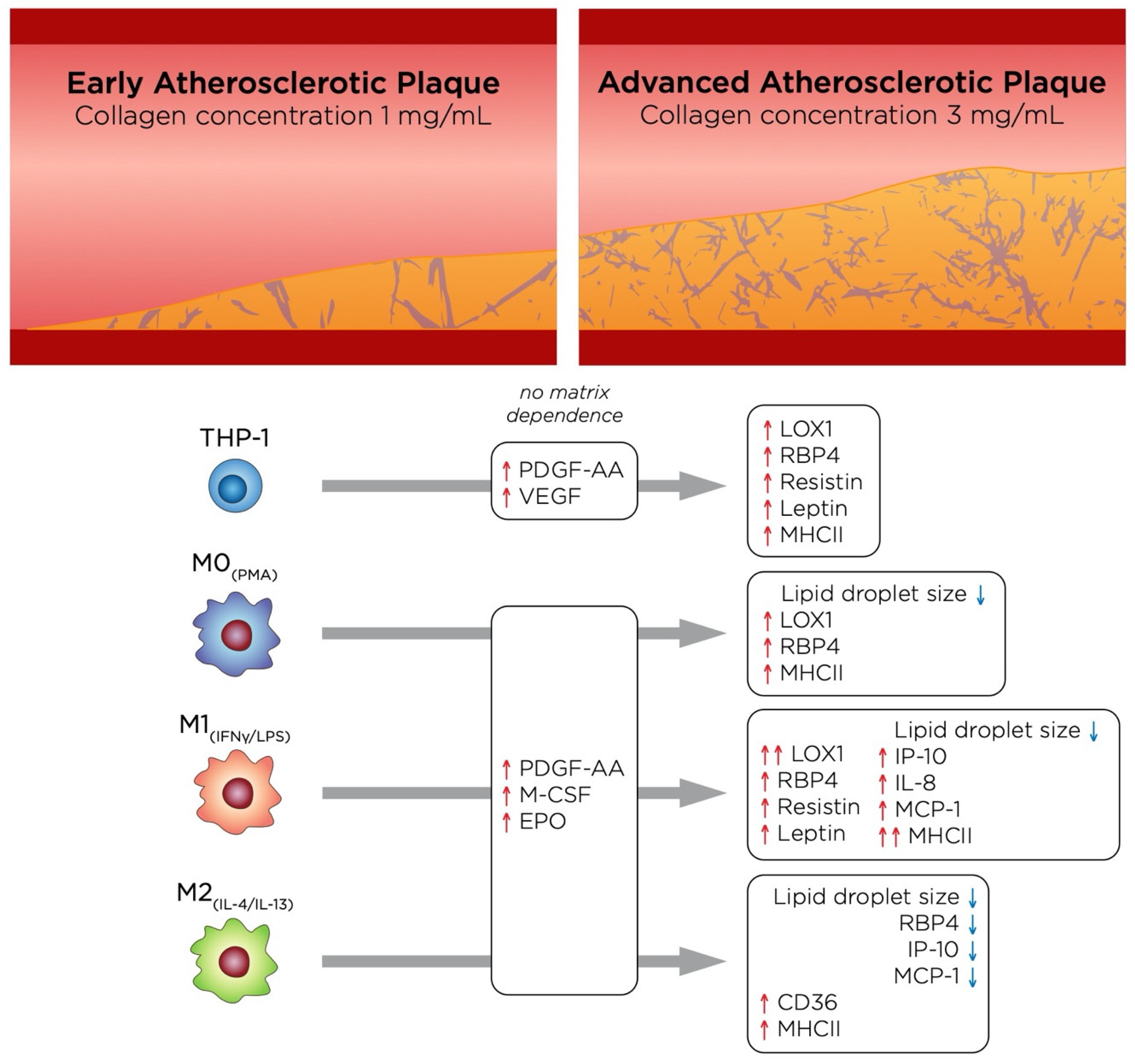
4. General Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Phipps, R.P. Atherosclerosis: The emerging role of inflammation and the CD40-CD40 ligand system. Proc. Natl. Acad. Sci. USA 2000, 97, 6930–6932. [Google Scholar] [CrossRef]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Puré, E. Lipoproteins, macrophage function, and atherosclerosis: Beyond the foam cell? Cell Metab. 2005, 1, 223–230. [Google Scholar] [CrossRef]
- Roma-Lavisse, C.; Tagzirt, M.; Zawadzki, C.; Lorenzi, R.; Vincentelli, A.; Haulon, S.; Juthier, F.; Rauch, A.; Corseaux, D.; Staels, B.; et al. M1 and M2 macrophage proteolytic and angiogenic profile analysis in atherosclerotic patients reveals a distinctive profile in type 2 diabetes. Diabetes Vasc. Dis. Res. 2015, 12, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Kashirskikh, D.A.; Khotina, V.A.; Grechko, A.V.; Orekhov, A.N. Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells. J. Clin. Med. 2019, 8, 1798. [Google Scholar] [CrossRef]
- De Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front. Immunol. 2016, 7, 275. [Google Scholar] [CrossRef] [PubMed]
- Pourcet, B.; Staels, B. Alternative macrophages in atherosclerosis: Not always protective! J. Clin. Investig. 2018, 128, 910–912. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, J.; Hu, F.; Liu, J.; Li, M.; Zhao, L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019, 2019, 6724903. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, L.; de Winther, M.P.J. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol. 2020, 250, 705–714. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Sharma, G.; Fisher, E.A. Atherosclerosis: Making a U turn. Annu. Rev. Med. 2020, 71, 191–201. [Google Scholar] [CrossRef]
- Gater, D.L.; Widatalla, N.; Islam, K.; Alraeesi, M.; Teo, J.C.M.; Pearson, Y.E. Quantification of sterol-specific response in human macrophages using automated imaged-based analysis. Lipids Health Dis. 2017, 16, 242. [Google Scholar] [CrossRef]
- Gualtero, D.F.; Lafaurie, G.I.; Fontanilla, M.R. Two-dimensional and three-dimensional models for studying atherosclerosis pathogenesis induced by periodontopathogenic microorganisms. Mol. Oral Microbiol. 2018, 33, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Dorweiler, B.; Torzewski, M.; Dham, M.; Ochsenhirt, V.; Lehr, H.-A.; Lackner, K.J.; Vahl, C.-F. A novel in vitro model for the study of plaque development in atherosclerosis. Thromb. Haemost. 2006, 95, 182–189. [Google Scholar] [CrossRef]
- Robert, J.; Weber, B.; Frese, L.; Emmert, M.Y.; Schmidt, D.; Von Eckardstein, A.; Rohrer, L.; Hoerstrup, S.P. A three-dimensional engineered artery model for in vitro atherosclerosis research. PLoS ONE 2013, 8, e79821. [Google Scholar] [CrossRef] [PubMed]
- Venugopal Menon, N.; Tay, H.M.; Pang, K.T.; Dalan, R.; Wong, S.C.; Wang, X.; Li, K.H.H.; Hou, H.W. A tunable microfluidic 3D stenosis model to study leukocyte-endothelial interactions in atherosclerosis. APL Bioeng. 2018, 2, 016103. [Google Scholar] [CrossRef]
- Mallone, A.; Stenger, C.; Von Eckardstein, A.; Hoerstrup, S.P.; Weber, B. Biofabricating atherosclerotic plaques: In vitro engineering of a three-dimensional human fibroatheroma model. Biomaterials 2018, 150, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Rekhter, M.D. Collagen synthesis in atherosclerosis: Too much and not enough. Cardiovasc. Res. 1999, 41, 376–384. [Google Scholar] [CrossRef]
- Nadkarni, S.K.; Bouma, B.E.; De Boer, J.; Tearney, G.J. Evaluation of collagen in atherosclerotic plaques: The use of two coherent laser-based imaging methods. Lasers Med. Sci. 2009, 24, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Shirwany, N.A.; Zou, M.H. Arterial stiffness: A brief review. Acta Pharmacol. Sin. 2010, 31, 1267–1276. [Google Scholar] [CrossRef]
- Sapudom, J.; Mohamed, W.K.E.; Garcia-Sabaté, A.; Alatoom, A.; Karaman, S.; Mahtani, N.; Teo, J.C. Collagen Fibril Density Modulates Macrophage Activation and Cellular Functions during Tissue Repair. Bioengineering 2020, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; Van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors-A review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Montagnani, M.; Chandrasekran, S.; Quon, M.J. Role of Lipotoxicity in Endothelial Dysfunction. Heart Fail. Clin. 2012, 8, 589–607. [Google Scholar] [CrossRef]
- Wolf, K.; Alexander, S.; Schacht, V.; Coussens, L.M.; von Andrian, U.H.; van Rheenen, J.; Deryugina, E.; Friedl, P. Collagen-based cell migration models in vitro and in vivo. Semin. Cell Dev. Biol. 2009, 20, 931–941. [Google Scholar] [CrossRef]
- Sapudom, J.; Pompe, T. Biomimetic tumor microenvironments based on collagen matrices. Biomater. Sci. 2018, 6, 2009–2024. [Google Scholar] [CrossRef]
- Sapudom, J.; Rubner, S.; Martin, S.; Kurth, T.; Riedel, S.; Mierke, C.T.; Pompe, T. The phenotype of cancer cell invasion controlled by fibril diameter and pore size of 3D collagen networks. Biomaterials 2015, 52, 367–375. [Google Scholar] [CrossRef]
- Franke, K.; Sapudom, J.; Kalbitzer, L.; Anderegg, U.; Pompe, T. Topologically defined composites of collagen types I and V as in vitro cell culture scaffolds. Acta Biomater. 2014, 10, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Sapudom, J.; Alatoom, A.; Mohamed, W.K.E.; Garcia-Sabaté, A.; McBain, I.; Nasser, R.A.; Teo, J.C.M. Dendritic cell immune potency on 2D and in 3D collagen matrices. Biomater. Sci. 2020, 8, 5106–5120. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Parathath, S.; Rong, J.X.; Mick, S.L.; Vengrenyuk, Y.; Grauer, L.; Young, S.G.; Fisher, E.A. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 2011, 123, 989–998. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, F.Y.; Davis, C.T.; Liu, W.F. Physical and mechanical regulation of macrophage phenotype and function. Cell. Mol. Life Sci. 2015, 72, 1303–1316. [Google Scholar] [CrossRef]
- Van Tits, L.J.H.; Stienstra, R.; van Lent, P.L.; Netea, M.G.; Joosten, L.A.B.; Stalenhoef, A.F.H. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: A crucial role for Krüppel-like factor 2. Atherosclerosis 2011, 214, 345–349. [Google Scholar] [CrossRef]
- De La Paz Sánchez-Martínez, M.; Blanco-Favela, F.; Mora-Ruiz, M.D.; Chávez-Rueda, A.K.; Bernabe-García, M.; Chávez-Sánchez, L. IL-17-differentiated macrophages secrete pro-inflammatory cytokines in response to oxidized low-density lipoprotein. Lipids Health Dis. 2017, 16, 196. [Google Scholar] [CrossRef]
- Adamson, S.; Leitinger, N. Phenotypic modulation of macrophages in response to plaque lipids. Curr. Opin. Lipidol. 2011, 22, 335–342. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Nikiforov, N.G.; Elizova, N.V.; Sobenin, I.A.; Orekhov, A.N. Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int. J. Cardiol. 2015, 184, 436–445. [Google Scholar] [CrossRef]
- De Paoli, F.; Staels, B.; Chinetti-Gbaguidi, G. Macrophage phenotypes and their modulation in atherosclerosis. Circ. J. 2014, 78, 1775–1781. [Google Scholar] [CrossRef]
- Peled, M.; Nishi, H.; Weinstock, A.; Barrett, T.J.; Zhou, F.; Quezada, A.; Fisher, E.A. A wild-type mouse-based model for the regression of inflammation in atherosclerosis. PLoS ONE 2017, 12, e173975. [Google Scholar] [CrossRef]
- Palanisamy, G.S.; Kirk, N.M.; Ackart, D.F.; Obregón-Henao, A.; Shanley, C.A.; Orme, I.M.; Basaraba, R.J. Uptake and accumulation of oxidized low-density lipoprotein during mycobacterium tuberculosis infection in guinea pigs. PLoS ONE 2012, 7, e34148. [Google Scholar] [CrossRef] [PubMed]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger Receptors Class A-I/II and CD36 Are the Principal Receptors Responsible for the Uptake of Modified Low Density Lipoprotein Leading to Lipid Loading in Macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and Its Role in Atherosclerosis. Antioxidants 2019, 8, 218. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Sawamura, T.; Furutani, Y.; Matsuoka, R.; Yoshida, M.C.; Fujiwara, H.; Masaki, T. Structure and chromosomal assignment of the human lectin-like oxidized low-density-lipoprotein receptor-1 (LOX-1) gene. Biochem. J. 1999, 339, 177–184. [Google Scholar] [CrossRef] [PubMed]
- N’Guessan, P.D.; Riediger, F.; Vardarova, K.; Scharf, S.; Eitel, J.; Opitz, B.; Slevogt, H.; Weichert, W.; Hocke, A.C.; Schmeck, B.; et al. Statins control oxidized ldl-mediated histone modifications and gene expression in cultured human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 380–386. [Google Scholar] [CrossRef]
- Xu, S.; Ogura, S.; Chen, J.; Little, P.J.; Moss, J.; Liu, P. LOX-1 in atherosclerosis: Biological functions and pharmacological modifiers. Cell. Mol. Life Sci. 2013, 70, 2859–2872. [Google Scholar] [CrossRef]
- Peled, M.; Fisher, E.A. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front. Immunol. 2014, 5, 579. [Google Scholar] [CrossRef]
- Shioi, A.; Ikari, Y. Plaque calcification during atherosclerosis progression and regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef]
- Di Gregoli, K.; Somerville, M.; Bianco, R.; Thomas, A.C.; Frankow, A.; Newby, A.C.; George, S.J.; Jackson, C.L.; Johnson, J.L. Galectin-3 Identifies a Subset of Macrophages with a Potential Beneficial Role in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1491–1509. [Google Scholar] [CrossRef]
- Moore, K.J.; Freeman, M.W. Scavenger receptors in atherosclerosis: Beyond lipid uptake. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1702–1711. [Google Scholar] [CrossRef]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [PubMed]
- Tang, H.; Husch, J.F.A.; Zhang, Y.; Jansen, J.A.; Yang, F.; van den Beucken, J.J.J.P. Coculture with monocytes/macrophages modulates osteogenic differentiation of adipose-derived mesenchymal stromal cells on poly(lactic-co-glycolic) acid/polycaprolactone scaffolds. J. Tissue Eng. Regen. Med. 2019, 13, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Le Master, E.; Fancher, I.S.; Lee, J.; Levitan, I. Comparative analysis of endothelial cell and sub-endothelial cell elastic moduli in young and aged mice: Role of CD36. J. Biomech. 2018, 76, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Xu, X.; Grijalva, A.; Skowronski, A.; Van Eijk, M.; Serlie, M.J.; Ferrante, A.W. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013, 18, 816–830. [Google Scholar] [CrossRef]
- Boden, G.; Chen, X.; Ruiz, J.; White, J.V.; Rossetti, L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J. Clin. Investig. 1994, 93, 2438–2446. [Google Scholar] [CrossRef]
- Boden, G. Obesity and Free Fatty Acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef]
- Moraes-Vieira, P.M.; Yore, M.M.; Dwyer, P.M.; Syed, I.; Aryal, P.; Kahn, B.B. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab. 2014, 19, 512–526. [Google Scholar] [CrossRef]
- Silverstein, R.L. Linking Metabolic Dysfunction to Atherosclerosis Via Activation of Macrophage CD36 Gene Transcription by Retinol Binding Protein-4. Circulation 2017, 135, 1355–1357. [Google Scholar] [CrossRef][Green Version]
- Liu, Y.; Zhong, Y.; Chen, H.; Wang, D.; Wang, M.; Ou, J.S.; Xia, M. Retinol-binding protein-dependent cholesterol uptake regulates macrophage foam cell formation and promotes atherosclerosis. Circulation 2017, 135, 1339–1354. [Google Scholar] [CrossRef]
- Monteiro, L.; Pereira, J.A.d.S.; Palhinha, L.; Moraes-Vieira, P.M.M. Leptin in the regulation of the immunometabolism of adipose tissue-macrophages. J. Leukoc. Biol. 2019, 106, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Park, K.H.; Cho, Y.M.; Chung, S.S.; Cho, H.J.; Cho, S.Y.; Kim, S.J.; Kim, S.Y.; Lee, H.K.; Park, K.S. Resistin is secreted from macrophages in atheromas and promotes atherosclerosis. Cardiovasc. Res. 2006, 69, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Kwak, M.K.; Kim, H.J.; Ahima, R.S. Linking resistin, inflammation, and cardiometabolic diseases. Korean J. Intern. Med. 2017, 32, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Curat, C.A.; Miranville, A.; Sengene, C.; Diehl, M.; Tonus, C.; Busse, R.; Bouloumie, A. From Blood Monocytes to Adipose Tissue-Resident Macrophages. Diabetes 2004, 53, 1285–1292. [Google Scholar] [CrossRef]
- Cho, Y.; Lee, S.E.; Lee, H.C.; Hur, J.; Lee, S.; Youn, S.W.; Lee, J.; Lee, H.J.; Lee, T.K.; Park, J.; et al. Adipokine resistin is a key player to modulate monocytes, endothelial cells, and smooth muscle cells, leading to progression of atherosclerosis in rabbit carotid artery. J. Am. Coll. Cardiol. 2011, 57, 99–109. [Google Scholar] [CrossRef]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Lin, J.; Kakkar, V.; Lu, X. Impact of MCP -1 in Atherosclerosis. Curr. Pharm. Des. 2014, 20, 4580–4588. [Google Scholar] [CrossRef]
- Van Den Borne, P.; Quax, P.H.A.; Hoefer, I.E.; Pasterkamp, G. The multifaceted functions of CXCL10 in cardiovascular disease. Biomed. Res. Int. 2014, 2014, 893106. [Google Scholar] [CrossRef]
- Apostolopoulos, J.; Davenport, P.; Tipping, P.G. Interleukin-8 Production by Macrophages from Atheromatous Plaques. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1007–1012. [Google Scholar] [CrossRef]
- Apostolakis, S.; Vogiatzi, K.; Amanatidou, V.; Spandidos, D.A. Interleukin 8 and cardiovascular disease. Cardiovasc. Res. 2009, 84, 353–360. [Google Scholar] [CrossRef]
- Zernecke, A.; Shagdarsuren, E.; Weber, C. Chemokines in atherosclerosis an update. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1897–1908. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, A.; Vorobyeva, D.; Vagida, M.; Ivanova, O.; Felker, E.; Fitzgerald, W.; Danilova, N.; Gontarenko, V.; Shpektor, A.; Vasilieva, E.; et al. Ex vivo culture of human atherosclerotic plaques: A model to study immune cells in atherogenesis. Atherosclerosis 2017, 267, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Burdick, M.D.; Gomperts, B.N.; Belperio, J.A.; Keane, M.P. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 593–609. [Google Scholar] [CrossRef]
- Du, F.; Zhou, J.; Gong, R.; Huang, X.; Pansuria, M.; Virtue, A.; Li, X.; Wang, H.; Yang, X.-F. Endothelial progenitor cells in atherosclerosis. Front. Biosci. 2012, 17, 2327–2349. [Google Scholar] [CrossRef]
- Yang, J.X.; Pan, Y.Y.; Wang, X.X.; Qiu, Y.G.; Mao, W. Endothelial progenitor cells in age-related vascular remodeling. Cell Transplant. 2018, 27, 786–795. [Google Scholar] [CrossRef]
- Fadini, G.P.; Sartore, S.; Albiero, M.; Baesso, I.; Murphy, E.; Menegolo, M.; Grego, F.; De Kreutzenberg, S.V.; Tiengo, A.; Agostini, C.; et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2140–2146. [Google Scholar] [CrossRef]
- Dimmeler, S.; Zeiher, A.M. Vascular repair by circulating endothelial progenitor cells: The missing link in atherosclerosis? J. Mol. Med. 2004, 82, 671–677. [Google Scholar] [CrossRef]
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.W.; Chen, W.L.; Huang, S.M.; Chan, J.Y.H. Platelet-derived growth factor-AA is a substantial factor in the ability of adipose-derived stem cells and endothelial progenitor cells to enhance wound healing. FASEB J. 2019, 33, 2388–2395. [Google Scholar] [CrossRef] [PubMed]
- Van Der Veken, B.; De Meyer, G.; Martinet, W. Inhibition of VEGF receptor signaling attenuates intraplaque angiogenesis and plaque destabilization in a mouse model of advanced atherosclerosis. Atherosclerosis 2017, 263, e33–e34. [Google Scholar] [CrossRef]
- Korn, C.; Augustin, H.G. Mechanisms of Vessel Pruning and Regression. Dev. Cell 2015, 34, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.K.; Tsai, T.H.; Lin, H.S.; Chen, S.F.; Sun, C.K.; Leu, S.; Yuen, C.M.; Tan, T.Y.; Lan, M.Y.; Liou, C.W.; et al. Effect of erythropoietin on level of circulating endothelial progenitor cells and outcome in patients after acute ischemic stroke. Crit. Care 2011, 15, R40. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tian, Y.; Wei, H.J.; Chen, J.; Dong, J.F.; Zacharek, A.; Zhang, J.N. Erythropoietin increases circulating endothelial progenitor cells and reduces the formation and progression of cerebral aneurysm in rats. Neuroscience 2011, 181, 292–299. [Google Scholar] [CrossRef]
- Lu, K.Y.; Ching, L.C.; Su, K.H.; Yu, Y.B.; Kou, Y.R.; Hsiao, S.H.; Huang, Y.C.; Chen, C.Y.; Cheng, L.C.; Pan, C.C.; et al. Erythropoietin suppresses the formation of macrophage foam cells: Role of liver X receptor α. Circulation 2010, 121, 1828–1837. [Google Scholar] [CrossRef]
- Viktorinova, A. Potential Clinical Utility of Macrophage Colony-stimulating Factor, Monocyte Chemotactic Protein-1, and Myeloperoxidase in Predicting Atherosclerotic Plaque Instability. Discov. Med. 2019, 28, 237–245. [Google Scholar]
- Zhang, Y.H.; He, M.; Wang, Y.; Liao, A.H. Modulators of the balance between M1 and M2 macrophages during pregnancy. Front. Immunol. 2017, 8, 120. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Gordon, S.; Mantovani, A. Diversity and plasticity of mononuclear phagocytes. Eur. J. Immunol. 2011, 41, 2470–2472. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.C.; Kim, W.-K. Editorial: Identification of in vivo markers for human polarized macrophages: A need that’s finally met. J. Leukoc. Biol. 2015, 98, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, L.S.; Mogensen, C.K.; Rosendahl, A.; Cucak, H.; Nielsen, L.B.; Rasmussen, S.E.; Pedersen, T.X. Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression—Implications for atherosclerosis research. Sci. Rep. 2016, 6, 35234. [Google Scholar] [CrossRef] [PubMed]
- Ramprasad, M.P.; Terpstra, V.; Kondratenko, N.; Quehenberger, O.; Steinberg, D. Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1996, 93, 14833–14838. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Gonen, A.; Diehl, C.J.; Kim, J.; Almazan, F.; Witztum, J.L.; Miller, Y.I. SYK regulates macrophage MHC-II expression via activation of autophagy in response to oxidized LDL. Autophagy 2015, 11, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Witzel, I.-I.; Nasser, R.; Garcia-Sabaté, A.; Sapudom, J.; Ma, C.; Chen, W.; Teo, J.C.M. Deconstructing Immune Microenvironments of Lymphoid Tissues for Reverse Engineering. Adv. Healthc. Mater. 2018, 8, 1801126. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Vinchi, F.; Muckenthaler, M.U.; Da Silva, M.C.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis and iron: From epidemiology to cellular level. Front. Pharmacol. 2014, 5, 94. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Sabaté, A.; Mohamed, W.K.E.; Sapudom, J.; Alatoom, A.; Al Safadi, L.; Teo, J.C.M. Biomimetic 3D Models for Investigating the Role of Monocytes and Macrophages in Atherosclerosis. Bioengineering 2020, 7, 113. https://doi.org/10.3390/bioengineering7030113
Garcia-Sabaté A, Mohamed WKE, Sapudom J, Alatoom A, Al Safadi L, Teo JCM. Biomimetic 3D Models for Investigating the Role of Monocytes and Macrophages in Atherosclerosis. Bioengineering. 2020; 7(3):113. https://doi.org/10.3390/bioengineering7030113
Chicago/Turabian StyleGarcia-Sabaté, Anna, Walaa Kamal E. Mohamed, Jiranuwat Sapudom, Aseel Alatoom, Layla Al Safadi, and Jeremy C. M. Teo. 2020. "Biomimetic 3D Models for Investigating the Role of Monocytes and Macrophages in Atherosclerosis" Bioengineering 7, no. 3: 113. https://doi.org/10.3390/bioengineering7030113
APA StyleGarcia-Sabaté, A., Mohamed, W. K. E., Sapudom, J., Alatoom, A., Al Safadi, L., & Teo, J. C. M. (2020). Biomimetic 3D Models for Investigating the Role of Monocytes and Macrophages in Atherosclerosis. Bioengineering, 7(3), 113. https://doi.org/10.3390/bioengineering7030113