Preclinical Models of Pediatric Brain Tumors—Forging Ahead

Abstract
1. Introduction
2. History of Preclinical Animal Models of Pediatric Brain Cancer
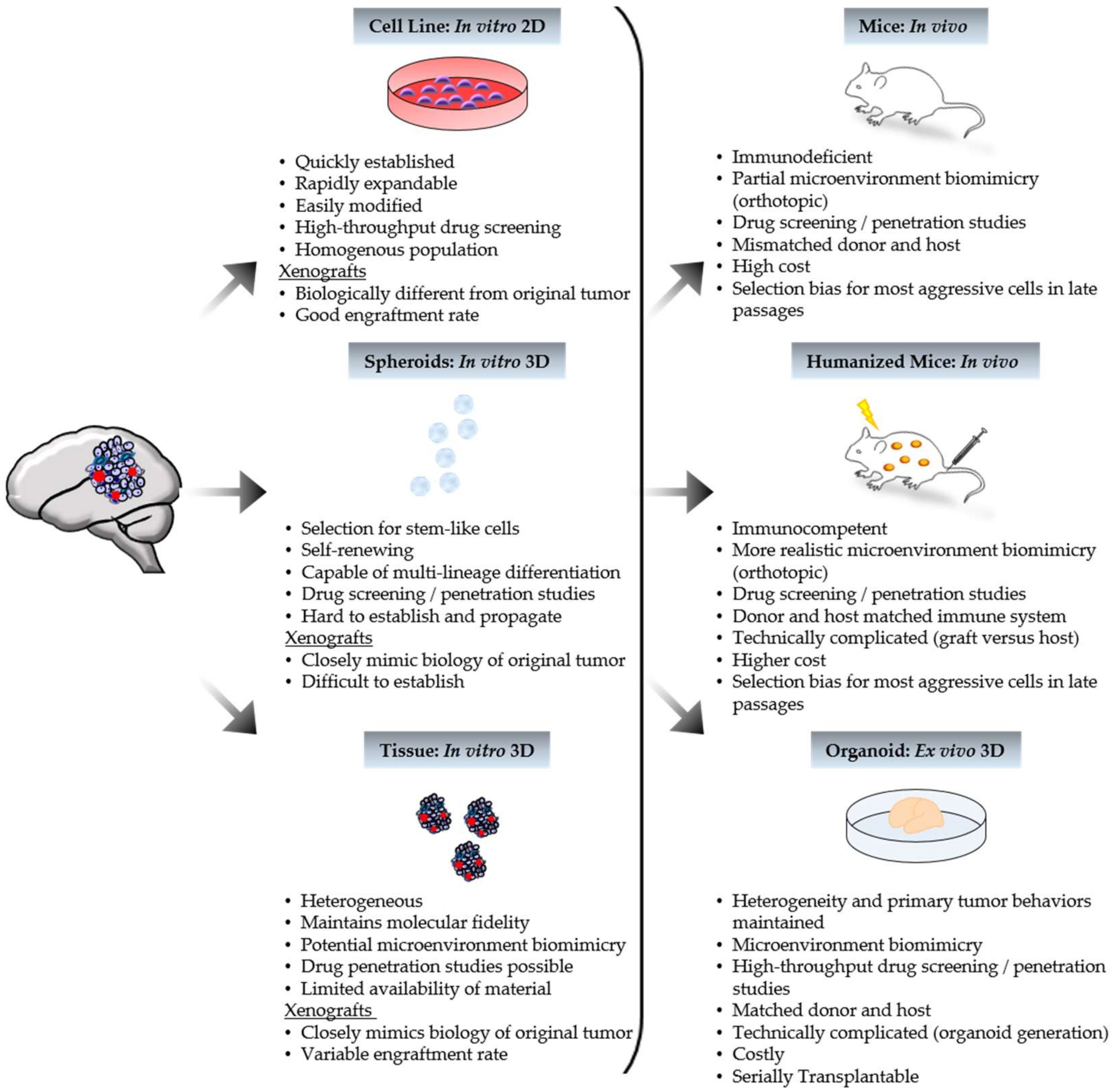
3. Applications to the Clinic: The Good, the Bad, and the Ugly
4. Promising Progress
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2018, 00, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Strand, A.D.; Girard, E.; Olson, J.M. Patient Derived Tumor Xenograft Models: Promise, Potential and Practice; Uthamanthil, R., Tinkey, P., Eds.; Elsvier Inc.: London, UK, 2017. [Google Scholar]
- Davis, C.; Naci, H.; Gurpinar, E.; Poplavska, E.; Pinto, A.; Aggarwal, A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: Retrospective cohort study of drug approvals 2009–13. BMJ 2017, 359, 0959–8138. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.L.; Sheridan, P.J.; Brown, W.E., Jr. Animal models for brain tumors: Historical perspectives and future directions. J. Neurosurg. 1994, 80, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Seligman, A.M.; Shear, M.J.; Alexander, L. Studies in carcinogenesis: VIII. Experimental production of brain tumors in mice with methylcholanthrene. Am. J. Cancer 1939, 37, 364–395. [Google Scholar]
- Druckrey, H.; Ivankovic, S.; Preussmann, R. Teratogenic and carcinogenic effects in the offspring after single injection of ethylnitrosourea to pregnant rats. Nature 1966, 210, 1378–1379. [Google Scholar] [CrossRef] [PubMed]
- Koestner, A.; Swenberg, J.A.; Wechsler, W. Transplacental production with ethylnitrosourea of neoplasms of the nervous system in Sprague-Dawley rats. Am. J. Pathol. 1971, 63, 37–56. [Google Scholar] [PubMed]
- Vazquez-Lopez, E. On the growth of Rous sarcoma inoculated into the brain. Am. J. Cancer 1939, 29, 29–55. [Google Scholar] [CrossRef]
- Rabotti, G.F.; Raine, W.A. Brain tumours induced in hamsters inoculated intracerebrally at birth with rous sarcoma virus. Nature 1964, 204, 898–899. [Google Scholar] [CrossRef] [PubMed]
- Cuatico, W.; Cho, J.R.; Spiegelman, S. Molecular evidence for a viral etiology of human CNS tumors. Acta Neurochir. 1976, 35, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, J.; Cravioto, H. Nitrosourea-induced brain tumors: An in vivo and in vitro tumor model system. J. Natl. Cancer Inst. 1978, 61, 365–374. [Google Scholar] [PubMed]
- Huszthy, P.C.; Daphu, I.; Niclou, S.P.; Stieber, D.; Nigro, J.M.; Sakariassen, P.O.; Miletic, H.; Thorsen, F.; Bjerkvig, R. In vivo models of primary brain tumors: Pitfalls and perspectives. Neuro. Oncol. 2012, 14, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, I.; Huillard, E. In vivo models of brain tumors: Roles of genetically engineered mouse models in understanding tumor biology and use in preclinical studies. Cell. Mol. Life Sci. 2014, 71, 4007–4026. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.E.; Ballard, E.T.; Britt, A.L.; Workman, L. Characteristics of 85 pediatric tumors heterotransplanted into nude mice. Exp. Cell. Biol. 1983, 51, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Burger, P.C.; Bigner, S.H.; Trojanowski, J.Q.; Wikstrand, C.J.; Halperin, E.C.; Bigner, D.D. Establishment and characterization of the human medulloblastoma cell line and transplantable xenograft D283 Med. J. Neuropathol. Exp. Neurol. 1985, 44, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, P.F.; Jenkyn, D.J.; Papadimitriou, J.M. Establishment of a human medulloblastoma cell line and its heterotransplantation into nude mice. J. Neuropathol. Exp. Neurol. 1985, 44, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Keles, G.E.; Berger, M.S.; Srinivasan, J.; Kolstoe, D.D.; Bobola, M.S.; Silber, J.R. Establishment and characterization of four human medulloblastoma-derived cell lines. Oncol. Res. 1995, 7, 493–503. [Google Scholar] [PubMed]
- Yachnis, A.T.; Neubauer, D.; Muir, D. Characterization of a primary central nervous system atypical teratoid/rhabdoid tumor and derivative cell line: Immunophenotype and neoplastic properties. J. Neuropathol. Exp. Neurol. 1998, 57, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Burger, P.C.; Bigner, S.H.; Trojanowski, J.Q.; Brodeur, G.M.; He, X.M.; Wikstrand, C.J.; Kurtzberg, J.; Berens, M.E.; Halperin, E.C.; et al. Phenotypic and genotypic analysis of a human medulloblastoma cell line and transplantable xenograft (D341 Med) demonstrating amplification of c-myc. Am. J. Pathol. 1988, 130, 472–484. [Google Scholar] [PubMed]
- Wasson, J.C.; Saylors, R.L.; Zeltzer, P.; Friedman, H.S.; Bigner, S.H.; Burger, P.C.; Bigner, D.D.; Look, A.T.; Douglass, E.C.; Brodeur, G.M. Oncogene amplification in pediatric brain tumors. Cancer Res. 1990, 50, 2987–2990. [Google Scholar] [PubMed]
- Pietsch, T.; Scharmann, T.; Fonatsch, C.; Schmidt, D.; Ockler, R.; Freihoff, D.S.; Albrecht, O.D.; Wiestler, P.; Zeltzer, H. Characterization of five new cell lines derived from human primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1994, 54, 3278–3287. [Google Scholar] [PubMed]
- Goodrich, L.V.; Milenkovic, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; McKinnon, P.J. DNA ligase IV suppresses medulloblastoma formation. Cancer Res. 2002, 62, 6395–6399. [Google Scholar] [PubMed]
- Marino, S.; Vooijs, M.H.; Der Gulden, V.; Jonkers, J.; Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Gene. Dev. 2000, 14, 994–1004. [Google Scholar] [PubMed]
- Wetmore, C.; Eberhart, D.E.; Curran, T. The normal patched allele is expressed in medulloblastomas from mice with heterozygous germ-line mutation of patched. Cancer Res. 2000, 60, 2239–2246. [Google Scholar] [PubMed]
- Perrin, S. Preclinical research: Make mouse studies work. Nature 2014, 507, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar] [CrossRef]
- Li, A.P. Preclinical in vitro screening assays for drug-like properties. Drug Discov. Today Technol. 2005, 2, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-derived cell lines as molecular models of cancer pharmacogenomics. Mol. Cancer Res. 2016, 14, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Suslov, O.N.; Kukekov, V.G.; Ignatova, T.N.; Steindler, D.A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. USA 2002, 99, 14506–14511. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Erdreich-Epstein, A.; Gonzalez-Gomez, I.; Melendez, E.Y.; Smbatyan, G.; Moats, R.A.; Rosol, M.; Biegel, J.A.; Reynolds, C.P. Novel cell lines established from pediatric brain tumors. J. Neurooncol. 2012, 107, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Morton, C.L.; Tucker, C.D.; Payne, E.; Favours, C.; Cole, R.; Gorlick, E.A.; Kolb, W.; Zhang, R.; Lock, H.; et al. The pediatric preclinical testing program: Description of models and early testing results. Pediatr. Blood Cancer 2007, 49, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Morfouace, M.; Nimmervoll, B.; Boulos, N.; Patel, Y.T.; Shelat, A.; Freeman, B.B.; Robinson, G.W.; Wright, K.; Gajjar, A.; Stewart, C.F.; et al. Preclinical studies of 5-fluoro-2′-deoxycytidine and tetrahydrouridine in pediatric brain tumors. J. Neurooncol. 2016, 126, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Sewing, A.C.P.T.; Lagerweij, D.G.; van Vuurden, M.H.; Meel, S.J.E.; Veringa, A.M.; Carcaboso, P.J.; Gaillard, W.; Peter Vandertop, P.; Wesseling, D.; Noske, G.J.L.; et al. Preclinical evaluation of convection-enhanced delivery of liposomal doxorubicin to treat pediatric diffuse intrinsic pontine glioma and thalamic high-grade glioma. J. Neuros-Pediatr. 2017, 19, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.M.; Won, J.K.; Park, S.H. Recent advancement of the molecular diagnosis in pediatric brain tumor. J. Korean Neurosurg. Soc. 2018, 61, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Pfister, S.M.; Taylor, M.D.; Gilbertson, R.J. Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin. Cancer Res. 2014, 20, 5630–5640. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Pomeroy, S.L.; Tamayo, P.; Gaasenbeek, M.; Sturla, L.M.; Angelo, M.; McLaughlin, M.E.; Kim, J.Y.; Goumnerova, L.C.; Black, P.M.; Lau, C.; et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 2002, 415, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Bez, A.; Corsini, E.; Curti, D.; Biggiogera, M.; Colombo, A.; Nicosia, R.F.; Pagano, S.F.; Parati, E.A. Neurosphere and neurosphere-forming cells: Morphological and ultrastructural characterization. Brain Res. 2003, 993, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; Park, J.K.; Fine, H.A. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.E.; Swartling, F.J.; Schuller, U. Medulloblastoma: Experimental models and reality. Acta Neuropathol. 2017, 134, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Gendoo, D.M.; Smirnov, P.; Lupien, M.; Haibe-Kains, B. Personalized diagnosis of medulloblastoma subtypes across patients and model systems. Genomics 2015, 106, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.P.; Coyle, B.; Walker, D.A.; Grabowska, A.M. In vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol. 2016, 236, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Dietl, S.; Schwinn, S.; Dietl, S.; Riedel, S.; Deinlein, F.; Rutkowski, S.; von Bueren, A.O.; Krauss, J.; Monoranu, T.; Schweitzer, G.H.; et al. Wolfl, MB3W1 is an orthotopic xenograft model for anaplastic medulloblastoma displaying cancer stem cell- and Group 3-properties. BMC Cancer 2016, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Milde, T.; Lodrini, M.; Savelyeva, L.; Korshunov, A.; Kool, M.; Brueckner, L.M.; Antunes, A.S.; Oehme, I.; Pekrun, A.; Pfister, S.M.; et al. HD-MB03 is a novel Group 3 medulloblastoma model demonstrating sensitivity to histone deacetylase inhibitor treatment. J. Neurooncol. 2012, 110, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Sandén, E. Experimental models of pediatric brain tumors. Establishment, immunophenotyping and clinical implications. Ph.D. Thesis, Lund University, Scania, Sweden, January 2016. [Google Scholar]
- Zhou, Z.; Luther, N.; Singh, R.; Boockvar, J.A.; Souweidane, M.M.; Greenfield, J.P. Glioblastoma spheroids produce infiltrative gliomas in the rat brainstem. Childs Nerv. Syst. 2017, 33, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.; Mitra, S.S.; Freret, M.E.; Raveh, T.B.; Kim, J.; Masek, M.; Attema, J.L.; Li, G.; Haddix, T.; Edwards, M.S.; et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl. Acad Sci. USA 2011, 108, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. 2012, 21, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Margol, A.S.; Shukla, A.; Ren, X.; Finlay, J.L.; Krieger, M.D.; Gilles, F.H.; Couch, F.J.; Aziz, M.; Fung, E.T.; et al. Disseminated Medulloblastoma in a Child with Germline BRCA2 6174delT Mutation and without Fanconi Anemia. Front. Oncol. 2015, 5, 191. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.; Larsson, S.; Danielsson, A.; Elbaek, K.J.; Kettunen, P.; Tisell, M.; Sabel, M.; Lannering, B.; Nordborg, C.; Schepke, E.; et al. Stem cell cultures derived from pediatric brain tumors accurately model the originating tumors. Oncotarget 2017, 8, 18626–18639. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.E. In vitro studies are sometimes better than conventional human pharmacokinetic in vivo studies in assessing bioequivalence of immediate-release solid oral dosage forms. AAPS J. 2008, 10, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Sreedharan, S.; Maturi, N.P.; Xie, Y.; Sundstrom, A.; Jarvius, M.; Libard, S.; Alafuzoff, I.; Weishaupt, H.; Fryknas, M.; Larsson, R.; et al. Uhrbom, Mouse Models of Pediatric Supratentorial High-grade Glioma Reveal How Cell-of-Origin Influences Tumor Development and Phenotype. Cancer Res. 2017, 77, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.A.; Lin, Q.; Mao, H.; Kogiso, M.; Zhao, X.; Liu, Z.; Huang, Y.; Voicu, H.; Gurusiddappa, S.; Su, J.M.; et al. Silencing BMI1 eliminates tumor formation of pediatric glioma CD133+ cells not by affecting known targets but by down-regulating a novel set of core genes. Acta Neuropathol. Com. 2014, 2, 160. [Google Scholar] [CrossRef] [PubMed]
- Girard, E.; Ditzler, S.; Lee, D.; Richards, A.; Yagle, K.; Park, J.; Eslamy, H.; Bobilev, D.; Vrignaud, P.; Olson, J. Efficacy of cabazitaxel in mouse models of pediatric brain tumors. Neuro. Oncol. 2015, 17, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Pre-Clinical Study of Panobinostat in Xenograft and Genetically Engineered Murine Diffuse Intrinsic Pontine Glioma Models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhao, Y.J.; Lin, Q.; Yu, L.; Liu, Z.; Lindsay, H.; Kogiso, M.; Rao, P.; Li, X.N.; Lu, X. Cytogenetic landscape of paired neurospheres and traditional monolayer cultures in pediatric malignant brain tumors. Neuro Oncol. 2015, 17, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Saponara, M.; Frapolli, R.; Tortoreto, M.; Cominetti, D.; Provenzano, S.; Negri, T.; Dagrada, G.P.; Gronchi, A.; Colombo, C.; et al. Patient-derived solitary fibrous tumour xenografts predict high sensitivity to doxorubicin/dacarbazine combination confirmed in the clinic and highlight the potential effectiveness of trabectedin or eribulin against this tumour. Eur. J. Cancer 2017, 76, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Ayrault, O.; Zindy, F.; Rehg, J.; Sherr, C.J.; Roussel, M.F. Two tumor suppressors, p27Kip1 and patched-1, collaborate to prevent medulloblastoma. Mol. Cancer Res. 2009, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Poschl, J.; Stark, S.; Neumann, P.; Grobner, S.; Kawauchi, D.; Jones, D.T.; Northcott, P.A.; Lichter, P.; Pfister, S.M.; Kool, M.; et al. Genomic and transcriptomic analyses match medulloblastoma mouse models to their human counterparts. Acta Neuropathol. 2014, 128, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Zindy, F.; Xie, S.; Lee, Y.; Forget, A.; Magdaleno, S.; Rehg, J.E.; Calabrese, C.; Solecki, D.; Eberhart, C.G.; et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005, 19, 2656–2667. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.M.; Kuijper, S.; Lindsey, J.C.; Petrie, K.; Schwalbe, E.C.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell. 2015, 27, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.G.; Peters, K.B.; Vredenburgh, J.J.; Desjardins, A.; Friedman, H.S.; Reardon, D.A. Everolimus tablets for patients with subependymal giant cell astrocytoma. Expert Opin. Pharmacother. 2011, 12, 2265–2269. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.; Jager, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.I.; Decker, S.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; Hollingshead, M.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Kraljevic, S.; Stambrook, P.J.; Pavelic, K. Accelerating drug discovery. EMBO Rep. 2004, 5, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Samson, K. New Campaign Seeks to Stimulate Research on Pediatric Brain Cancers. Oncol. Times 2016, 38, 14–15. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Z.; Yu, L.; Zhang, Y.; Baxter, P.; Voicu, H.; Gurusiddappa, S.; Luan, J.; Su, J.M.; Leung, H.C.; et al. Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro Oncol. 2012, 14, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Bingel, C.; Koeneke, E.; Ridinger, J.; Bittmann, A.; Sill, M.; Peterziel, H.; Wrobel, J.K.; Rettig, I.; Milde, T.; Fernekorn, U.; et al. Three-dimensional tumor cell growth stimulates autophagic flux and recapitulates chemotherapy resistance. Cell. Death Dis. 2017, 8, e3013. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell. Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Sobo, M.; Lee, K.; Senthil, K.S.; White, A.R.; Mender, I.; Fuller, C.; Chow, L.M.L.; Fouladi, M.; Shay, J.W.; et al. Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Mol. Cancer Ther. 2018, 17, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Casey, M.J.; Modzelewska, K.; Anderson, D.; Goodman, J.; Boer, E.F.; Jimenez, L.; Grossman, D.; Stewart, R.A. Transplantation of Zebrafish Pediatric Brain Tumors into Immune-competent Hosts for Long-term Study of Tumor Cell Behavior and Drug Response. J. Vis. Exp. 2017, 123, e55712. [Google Scholar] [CrossRef] [PubMed]
- Kirchberger, S.; Sturtzel, C.; Pascoal, S.; Distel, M. Quo natas, Danio?-Recent Progress in Modeling Cancer in Zebrafish. Front. Oncol. 2017, 7, 186. [Google Scholar] [CrossRef] [PubMed]
- Eden, C.J.; Ju, B.; Murugesan, M.; Phoenix, T.N.; Nimmervoll, B.; Tong, Y.; Ellison, D.W.; Finkelstein, D.; Wright, K.; Boulos, N.; et al. Orthotopic models of pediatric brain tumors in zebrafish. Oncogene 2015, 34, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Modzelewska, K.; Boer, E.F.; Mosbruger, T.L.; Picard, D.; Anderson, D.; Miles, R.R.; Kroll, M.; Oslund, W.; Pysher, T.J.; Schiffman, J.D.; Jensen, R.; et al. MEK Inhibitors Reverse Growth of Embryonal Brain Tumors Derived from Oligoneural Precursor Cells. Cell. Rep. 2016, 17, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Chen, Z.; Li, K.; Morais, G.R.; Yerneni, J.K.; Pisani, L.; Chin, F.T.; Mitra, S.; Cheshier, S.; Chang, E.; et al. A novel theranostic strategy for MMP-14-expressing glioblastomas impacts survival. Mol. Cancer Ther. 2017, 16, 1909–1921. [Google Scholar] [CrossRef] [PubMed]
- Welker, A.M.; Jaros, B.D.; Puduvalli, V.K.; Imitola, J.; Kaur, B.; Beattie, C.E. Standardized orthotopic xenografts in zebrafish reveal glioma cell-line-specific characteristics and tumor cell heterogeneity. Dis. Model. Mech. 2016, 9, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Chen, W.; Orr, B.A.; Spitsbergen, J.M.; Jia, S.; Eden, C.J.; Henson, H.E.; Taylor, M.R. Oncogenic KRAS promotes malignant brain tumors in zebrafish. Mol. Cancer 2015, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, M.; Gourain, V.; Reischl, M.; Affaticati, P.; Jenett, A.; Joly, J.S.; Benelli, M.; Demichelis, F.; Poliani, P.L.; Sieger, D.; et al. A novel brain tumour model in zebrafish reveals the role of YAP activation in MAPK- and PI3K-induced malignant growth. Dis. Model. Mech. 2017, 10, 15–28. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Benefits | Limitations | |||
|---|---|---|---|---|
| Translational relevance: similarities between humans and zebrafish | Largely conserved development and signaling pathways | Function of innate and adaptive immune cells is highly conserved | Over 80% of human disease-related genes present | Whole-genome duplication (more than one ortholog for some human genes) may interfere with genetic studies |
| Xenograft models [79,81,84] | Can be generated from human, mouse, or zebrafish tumors | No rejection due to immature adaptive immune system in larvae | Recapitulates parental tumor behaviors including proliferation, survival, invasion, and dissemination | Molecular interactions between transplanted human or mouse tumors and zebrafish cells unclear |
| Genetically engineered zebrafish models (GEZMs) [82,85,86] | Easy genetic manipulation—injection into one-cell-stage larvae possible | Fast development | Comparable histology to human cancers | Tumor initiation and progression studies hindered by lack of a functional adaptive immune system in early-stage models |
| Drug studies | Easy, cost-effective, and high scalability | Ease of imaging and high-throughput screening with transparent larvae | High degree of conservation of metabolic enzymes between human and zebrafish larvae | Pharmacokinetic processes still unclear |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobson, T.H.W.; Gopalakrishnan, V. Preclinical Models of Pediatric Brain Tumors—Forging Ahead. Bioengineering 2018, 5, 81. https://doi.org/10.3390/bioengineering5040081
Dobson THW, Gopalakrishnan V. Preclinical Models of Pediatric Brain Tumors—Forging Ahead. Bioengineering. 2018; 5(4):81. https://doi.org/10.3390/bioengineering5040081
Chicago/Turabian StyleDobson, Tara H.W., and Vidya Gopalakrishnan. 2018. "Preclinical Models of Pediatric Brain Tumors—Forging Ahead" Bioengineering 5, no. 4: 81. https://doi.org/10.3390/bioengineering5040081
APA StyleDobson, T. H. W., & Gopalakrishnan, V. (2018). Preclinical Models of Pediatric Brain Tumors—Forging Ahead. Bioengineering, 5(4), 81. https://doi.org/10.3390/bioengineering5040081