
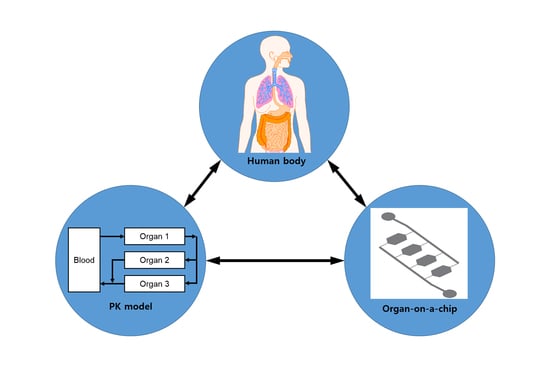
Microtechnology-Based Multi-Organ Models

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
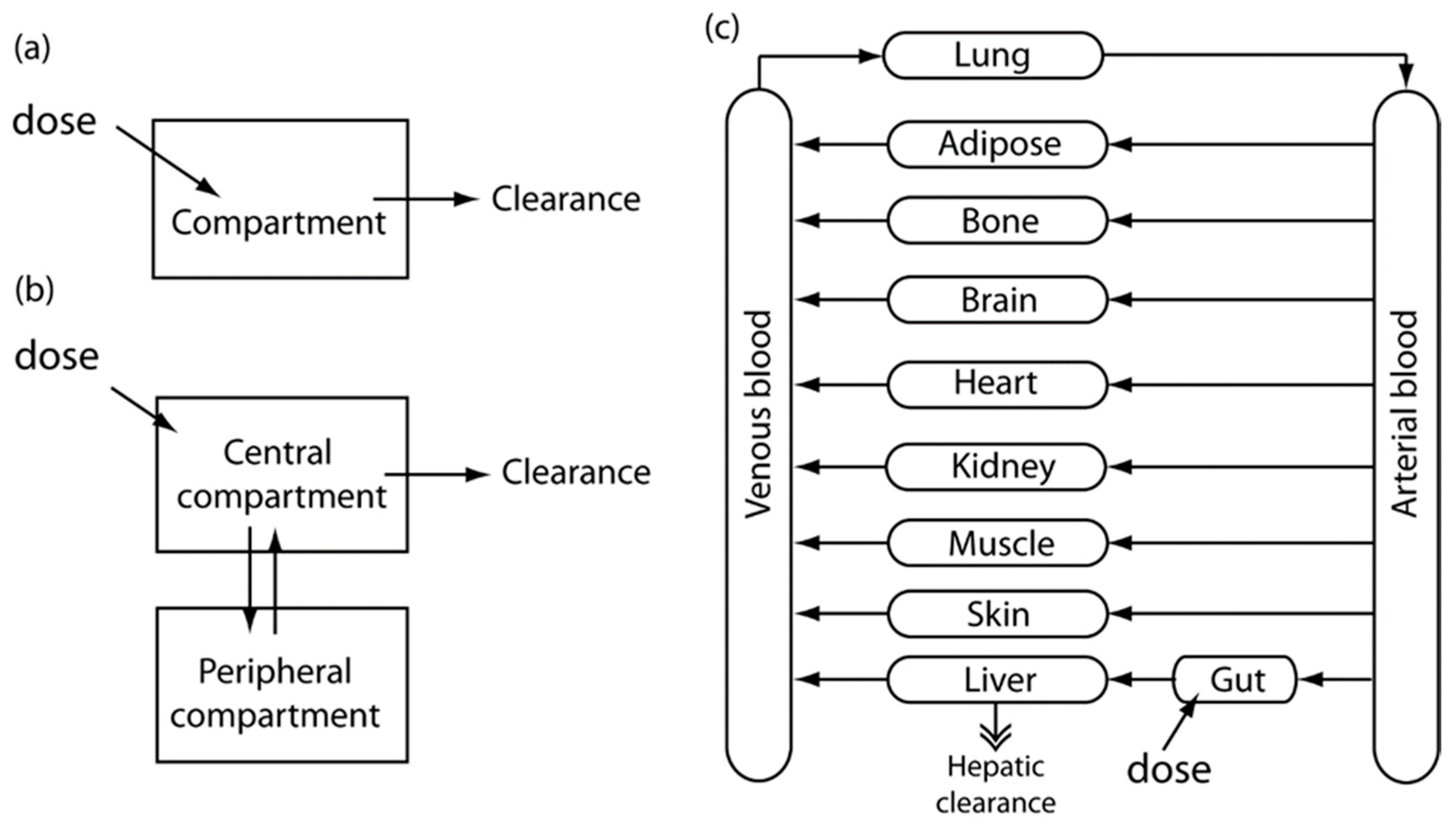
2. Pharmacokinetic-Pharmacodynamic (PK-PD) Modeling
2.1. PK Modeling
2.2. PD Modeling
3. Two-Organ-Based Organ Models
3.1. Cell Line-Based Two-Organ Models
3.2. Co-Culture of Organ Specific Cells
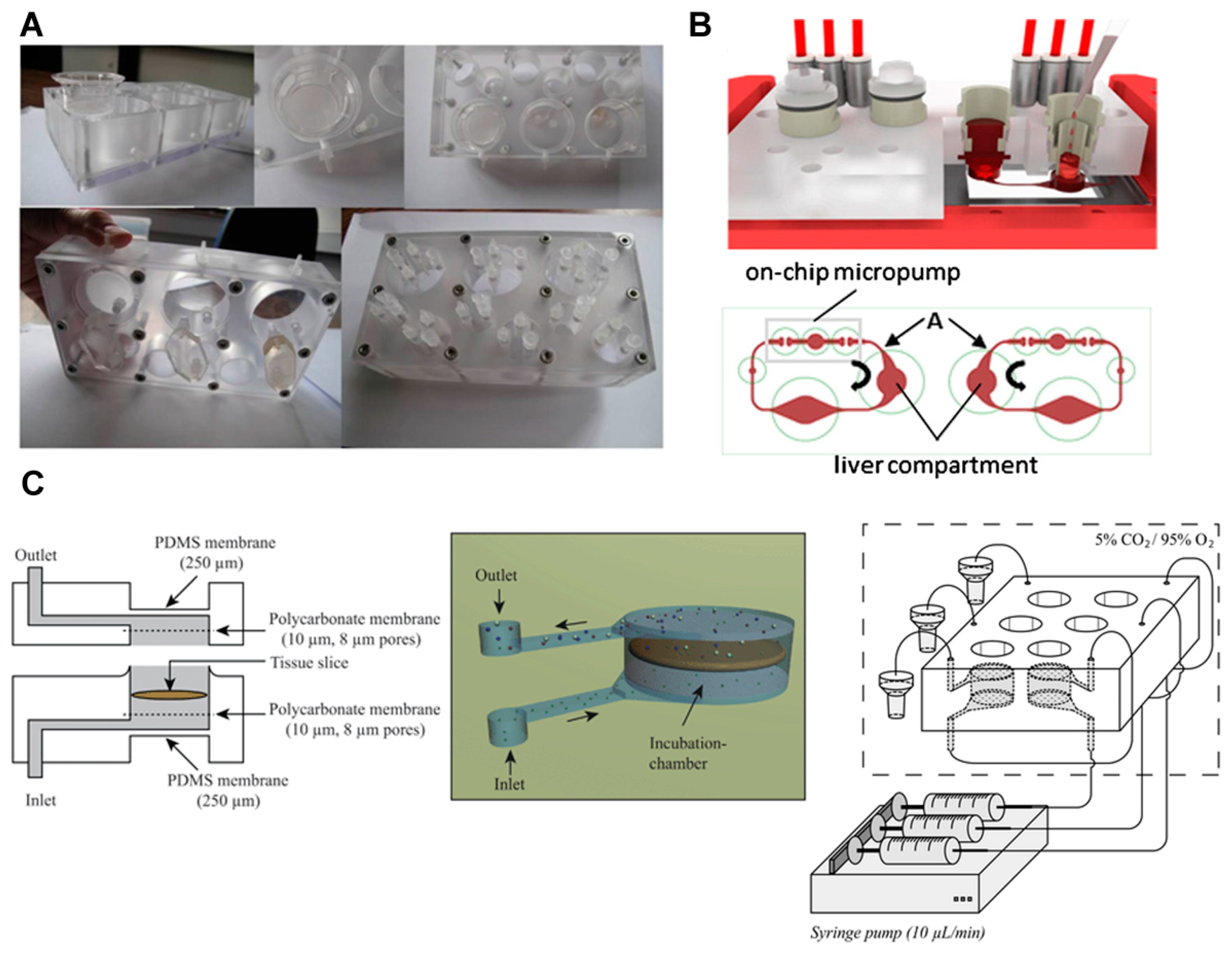
3.3. Primary Cells and Tissue Slices
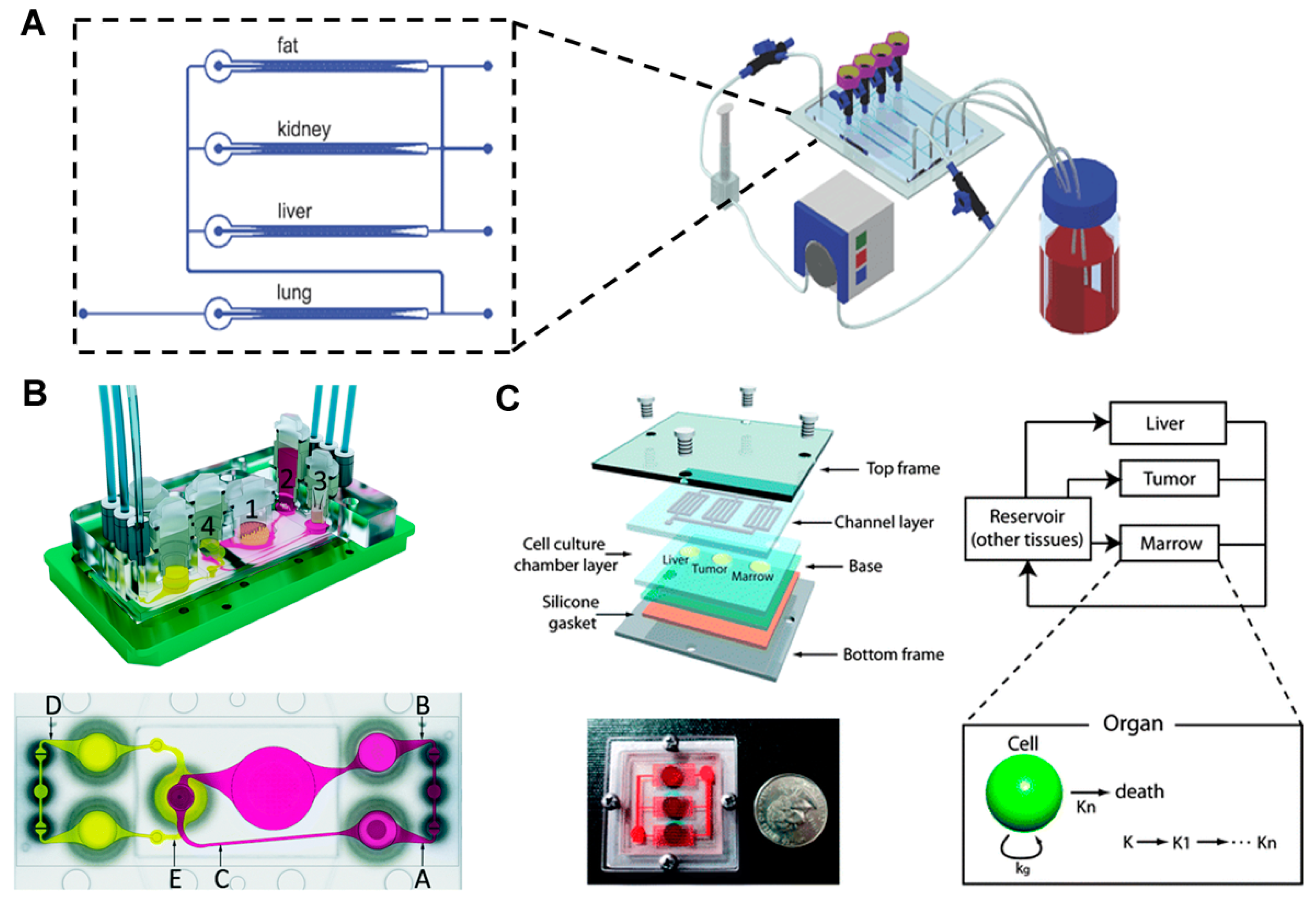
4. Multi-Organ Model with More than Three Organs
5. PK-PD Modeling-Based Multiple Organ Model
6. Remaining Challenges and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lahoz, A.; Gombau, L.; Donato, M.T.; V Castell, J.; Gomez-Lechon, M.J. In vitro ADME medium/high-throughput screening in drug preclinical development. Mini Rev. Med. Chem. 2006, 6, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Shuler, M.L. In vitro microscale systems for systematic drug toxicity study. Bioprocess Biosyst. Eng. 2010, 33, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Dingemanse, J.; Appel-Dingemanse, S. Integrated pharmacokinetics and pharmacodynamics in drug development. Clin. Pharmacokinet. 2007, 46, 713–737. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Sung, J.H. Organ-on-a-chip technology and microfluidic whole-body models for pharmacokinetic drug toxicity screening. Biotechnol. J. 2013, 8, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Bugrim, A.; Nikolskaya, T.; Nikolsky, Y. Early prediction of drug metabolism and toxicity: Systems biology approach and modeling. Drug Discov. Today 2004, 9, 127–135. [Google Scholar] [CrossRef]
- Dickson, M.; Gagnon, J.P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 2004, 3, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Agoram, B.M.; Martin, S.W.; van der Graaf, P.H. The role of mechanism-based pharmacokinetic–pharmacodynamic (PK–PD) modelling in translational research of biologics. Drug Discov. Today 2007, 12, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Esch, M.B.; Shuler, M.L. Integration of in silico and in vitro platforms for pharmacokinetic-pharmacodynamic modeling. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1063–1081. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Shuler, M.L. Microtechnology for mimicking in vivo tissue environment. Ann. Biomed. Eng. 2012, 40, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Rhee, H.-W.; van Noort, D.; Lee, H.J.; Park, H.H.; Shin, I.-S.; Hong, J.-I.; Park, T.H. Microfluidic bead-based sensing platform for monitoring kinase activity. Biosens. Bioelectron. 2014, 57, 1–9. [Google Scholar] [CrossRef] [PubMed]
- El-Ali, J.; Sorger, P.K.; Jensen, K.F. Cells on chips. Nature 2006, 442, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A. Cell culture: Biology’s new dimension. Nature 2003, 424, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Yum, K.; Hong, S.G.; Healy, K.E.; Lee, L.P. Physiologically relevant organs on chips. Biotechnol. J. 2014, 9, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Esch, M.B.; Prot, J.-M.; Long, C.J.; Smith, A.; Hickman, J.J.; Shuler, M.L. Microfabricated mammalian organ systems and their integration into models of whole animals and humans. Lab Chip 2013, 13, 1201–1212. [Google Scholar] [CrossRef]
- Choe, A.; Ha, S.K.; Choi, I.; Choi, N.; Sung, J.H. Microfluidic Gut-liver chip for reproducing the first pass metabolism. Biomed. Microdevices 2017, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Rostami-Hodjegan, A. Physiologically based pharmacokinetics joined with in vitro-in vivo extrapolation of ADME: A marriage under the arch of systems pharmacology. Clin. Pharmacol. Ther. 2012, 92, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Lu, C. PBPK modeling and simulation in drug research and development. Acta Pharm. Sin. B 2016, 6, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Bergen, P.J.; Lora-Tamayo, J.; Landersdorfer, C.B.; Li, J. PK/PD models in antibacterial development. Curr. Opin. Microbiol. 2013, 16, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.; Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aarons, L. Physiologically based pharmacokinetic modelling: A sound mechanistic basis is needed. Br. J. Clin. Pharmacol. 2005, 60, 581–583. [Google Scholar] [CrossRef]
- McCarley, K.D.; Bunge, A.L. Physiologically relevant two-compartment pharmacokinetic models for skin. J. Pharm. Sci. 2000, 89, 1212–1235. [Google Scholar] [CrossRef]
- Bois, F.Y.; Jamei, M.; Clewell, H.J. PBPK modelling of inter-individual variability in the pharmacokinetics of environmental chemicals. Toxicology 2010, 278, 256–267. [Google Scholar] [CrossRef]
- Kearns, C.M.; Gianni, L.; Egorin, M.J. Paclitaxel pharmacokinetics and pharmacodynamics. Semin. Oncol. 1995, 22, 16–23. [Google Scholar] [PubMed]
- Jusko, W.J. Pharmacodynamics of chemotherapeutic effects: Dose-time-response relationships for phase-nonspecific agents. J. Pharm. Sci. 1971, 60, 892–895. [Google Scholar] [CrossRef]
- Meibohm, B.; Derendorf, H. Pharmacokinetic/pharmacodynamic studies in drug product development. J. Pharm. Sci. 2002, 91, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Inamdar, N.K.; Borenstein, J.T. Microfluidic cell culture models for tissue engineering. Curr. Opin. Microbiol. 2011, 22, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, A.M.; Hancock, M.J.; Harrington, H.; Kaji, H.; Khademhosseini, A. Biomimetic tissues on a chip for drug discovery. Drug Discov. Today 2012, 17, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Mahler, G.J.; Esch, M.B.; Glahn, R.P.; Shuler, M.L. Characterization of a gastrointestinal tract microscale cell culture analog used to predict drug toxicity. Biotechnol. Bioeng. 2009, 104, 193–205. [Google Scholar] [CrossRef] [PubMed]
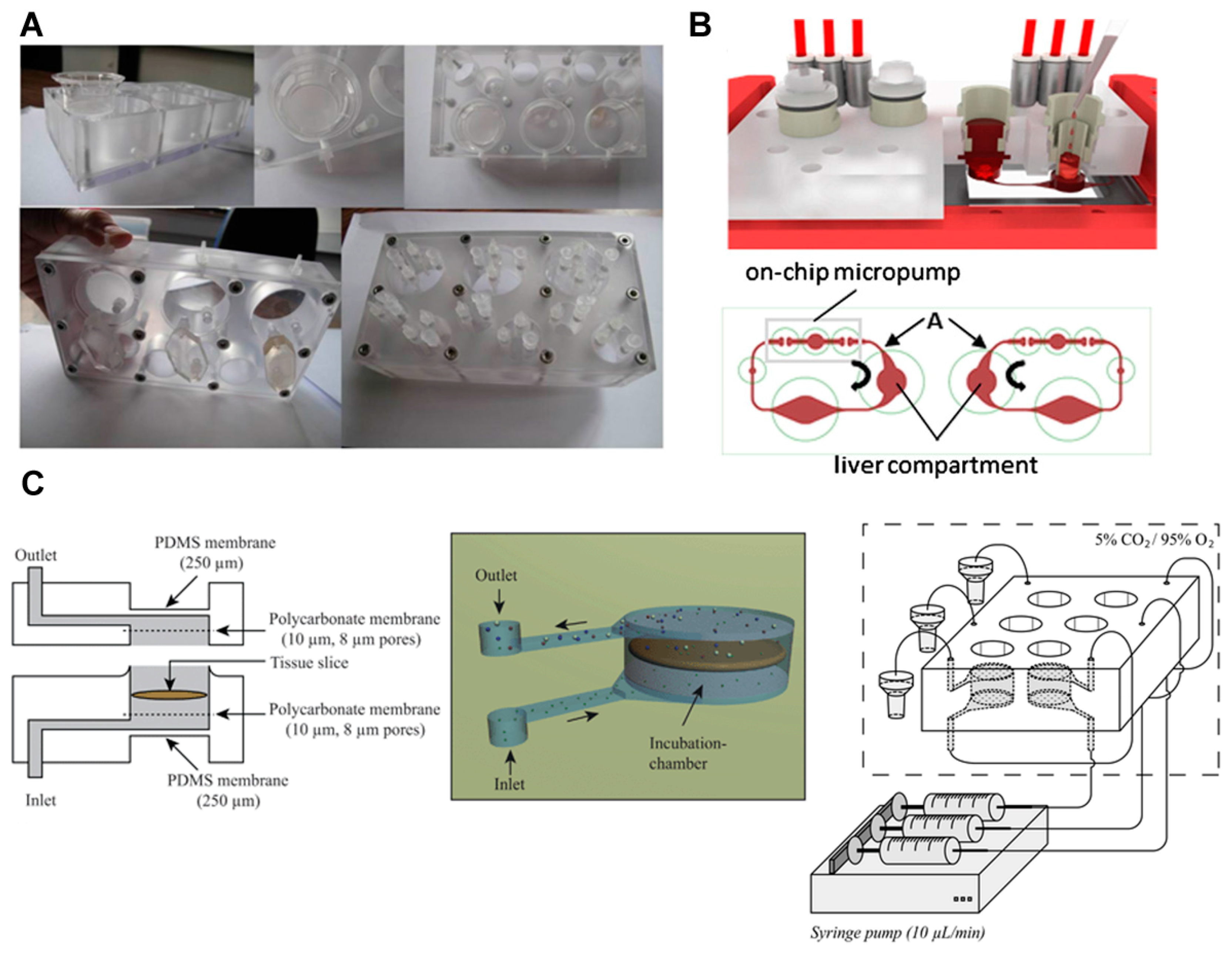
- Van Midwoud, P.M.; Merema, M.T.; Verpoorte, E.; Groothuis, G.M. A microfluidic approach for in vitro assessment of interorgan interactions in drug metabolism using intestinal and liver slices. Lab Chip 2010, 10, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
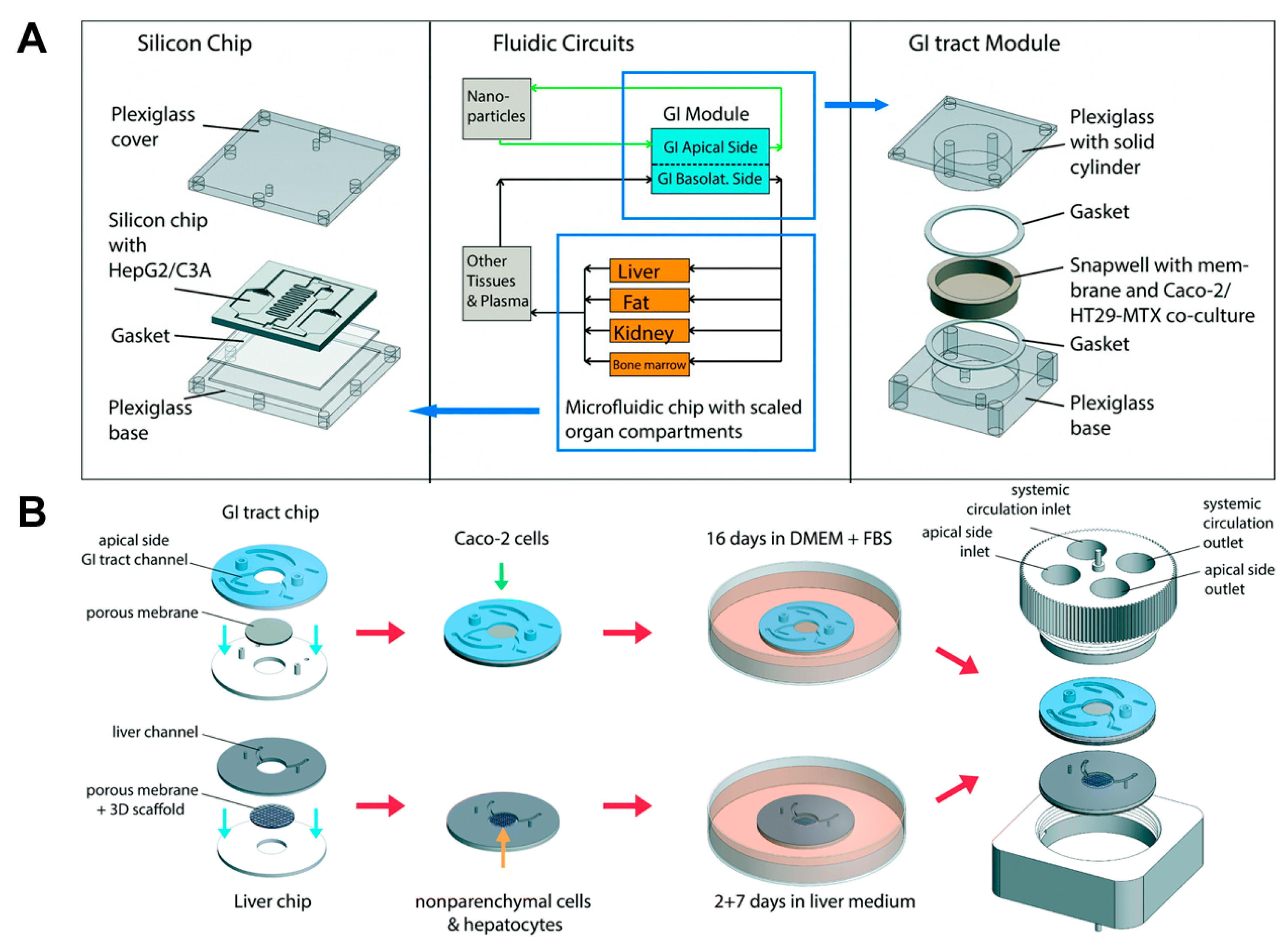
- Esch, M.B.; Mahler, G.J.; Stokol, T.; Shuler, M.L. Body-on-a-chip simulation with gastrointestinal tract and liver tissues suggests that ingested nanoparticles have the potential to cause liver injury. Lab Chip 2014, 14, 3081–3092. [Google Scholar] [CrossRef] [PubMed]
- Bricks, T.; Paullier, P.; Legendre, A.; Fleury, M.-J.; Zeller, P.; Merlier, F.; Anton, P.M.; Leclerc, E. Development of a new microfluidic platform integrating co-cultures of intestinal and liver cell lines. Toxicol. In Vitro 2014, 28, 885–895. [Google Scholar] [CrossRef]
- Kimura, H.; Ikeda, T.; Nakayama, H.; Sakai, Y.; Fujii, T. An On-Chip Small Intestine–Liver Model for Pharmacokinetic Studies. J. Lab. Autom. 2015, 20, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Mather, J.P.; Roberts, P.E. Introduction to Cell and Tissue Culture: Theory and Technique; Springer Science & Business Media: Berlin, Germany, 1998. [Google Scholar]
- McAuliffe, G.J.; Chang, J.Y.; Glahn, R.P.; Shuler, M.L. Development of a gastrointestinal tract microscale cell culture analog to predict drug transport. Mol. Cell. Biomech. 2008, 5, 119. [Google Scholar] [PubMed]
- Hilgendorf, C.; Spahn-Langguth, H.; Regårdh, C.G.; Lipka, E.; Amidon, G.L.; Langguth, P. Caco-2 versus Caco-2/HT29-MTX co-cultured cell lines: Permeabilities via diffusion, inside-and outside-directed carrier-mediated transport. J. Pharm. Sci. 2000, 89, 63–75. [Google Scholar] [CrossRef]
- Mahler, G.J.; Shuler, M.L.; Glahn, R.P. Characterization of Caco-2 and HT29-MTX cocultures in an in vitro digestion/cell culture model used to predict iron bioavailability. J. Nutr. Biochem. 2009, 20, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.; Janich, S.; Roessler, B.J.; Hilfinger, J.M.; Amidon, G.L. HT29-MTX/Caco-2 cocultures as an in vitro model for the intestinal epithelium: In vitro-in vivo correlation with permeability data from rats and humans. J. Pharm. Sci. 1996, 85, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; Ueno, H.; Applegate, D.R.; Shuler, M.L. Modular, pumpless body-on-a-chip platform for the co-culture of GI tract epithelium and 3D primary liver tissue. Lab Chip 2016, 16, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Prot, J.M.; Maciel, L.; Bricks, T.; Merlier, F.; Cotton, J.; Paullier, P.; Bois, F.Y.; Leclerc, E. First pass intestinal and liver metabolism of paracetamol in a microfluidic platform coupled with a mathematical modeling as a means of evaluating ADME processes in humans. Biotechnol. Bioeng. 2014, 111, 2027–2040. [Google Scholar] [CrossRef] [PubMed]
- Maschmeyer, I.; Hasenberg, T.; Jaenicke, A.; Lindner, M.; Lorenz, A.K.; Zech, J.; Garbe, L.-A.; Sonntag, F.; Hayden, P.; Ayehunie, S.; et al. Chip-based human liver–intestine and liver–skin co-cultures—A first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm. 2015, 95, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Grosberg, A.; Alford, P.W.; McCain, M.L.; Parker, K.K. Ensembles of engineered cardiac tissues for physiological and pharmacological study: Heart on a chip. Lab Chip 2011, 11, 4165–4173. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Z.; Rahim, N.A.A.; van Noort, D.; Yu, H. Towards a human-on-chip: Culturing multiple cell types on a chip with compartmentalized microenvironments. Lab Chip 2009, 9, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Wagner, I.; Materne, E.-M.; Brincker, S.; Süßbier, U.; Frädrich, C.; Busek, M.; Sonntag, F.; Sakharov, D.A.; Trushkin, E.V.; Tonevitsky, A.G.; et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 2013, 13, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Schimek, K.; Busek, M.; Brincker, S.; Groth, B.; Hoffmann, S.; Lauster, R.; Lindner, G.; Lorenz, A.; Menzel, U.; Sonntag, F.; et al. Integrating biological vasculature into a multi-organ-chip microsystem. Lab Chip 2013, 13, 3588–3598. [Google Scholar] [CrossRef] [PubMed]
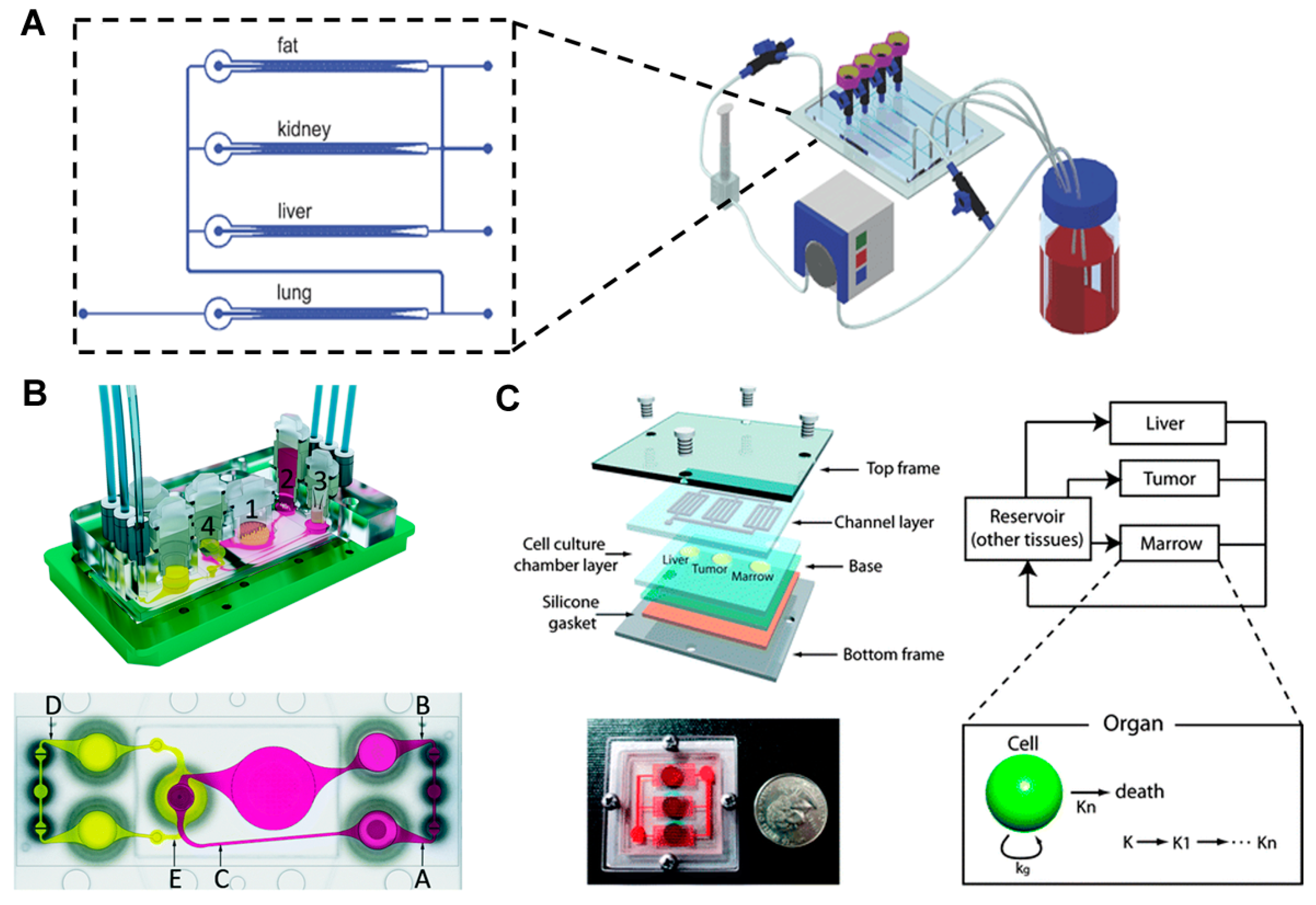
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Kam, C.; Shuler, M.L. A microfluidic device for a pharmacokinetic–pharmacodynamic (PK–PD) model on a chip. Lab Chip 2010, 10, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Shuler, M.; Ghanem, A.; Quick, D.; Wong, M.; Miller, P. A self-regulating cell culture analog device to mimic animal and human toxicological responses. Biotechnol. Bioeng. 1996, 52, 45–60. [Google Scholar] [CrossRef]
- Sin, A.; Chin, K.C.; Jamil, M.F.; Kostov, Y.; Rao, G.; Shuler, M.L. The design and fabrication of three-chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol. Prog. 2004, 20, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Viravaidya, K.; Sin, A.; Shuler, M.L. Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnol. Prog. 2004, 20, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Shuler, M.L. A micro cell culture analog (µCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab Chip 2009, 9, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Shuler, M.L. Prevention of air bubble formation in a microfluidic perfusion cell culture system using a microscale bubble trap. Biomed. Microdevices 2009, 11, 731–738. [Google Scholar] [CrossRef]
- Moraes, C.; Labuz, J.M.; Leung, B.M.; Inoue, M.; Chun, T.-H.; Takayama, S. On being the right size: Scaling effects in designing a human-on-a-chip. Integr. Biol. 2013, 5, 1149–1161. [Google Scholar] [CrossRef]
- Ucciferri, N.; Sbrana, T.; Ahluwalia, A. Allometric scaling and cell ratios in multi-organ in vitro models of human metabolism. Front. Bioeng. Biotechnol. 2014, 2, 74. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Yu, J.; Luo, D.; Shuler, M.L.; March, J.C. Microscale 3-D hydrogel scaffold for biomimetic gastrointestinal (GI) tract model. Lab Chip 2011, 11, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef]
- Du, Y.; Li, N.; Yang, H.; Luo, C.; Gong, Y.; Tong, C.; Gao, Y.; Lü, S.; Long, M. Mimicking Liver Sinusoidal Structures and Functions using a 3D-configured Microfluidic Chip. Lab Chip 2017, 17, 782–794. [Google Scholar] [CrossRef]
- Vernetti, L.A.; Senutovitch, N.; Boltz, R.; DeBiasio, R.; Ying Shun, T.; Gough, A.; Taylor, D.L. A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp. Biol. Med. 2016, 241, 101–114. [Google Scholar] [CrossRef]
- Prodanov, L.; Jindal, R.; Bale, S.S.; Hegde, M.; McCarty, W.J.; Golberg, I.; Bhushan, A.; Yarmush, M.L.; Usta, O.B. Long-term maintenance of a microfluidic 3D human liver sinusoid. Biotechnol. Bioeng. 2016, 113, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Luni, C.; Serena, E.; Elvassore, N. Human-on-chip for therapy development and fundamental science. Curr. Opin. Biotechnol. 2014, 25, 45–50. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Sung, J.H. Microtechnology-Based Multi-Organ Models. Bioengineering 2017, 4, 46. https://doi.org/10.3390/bioengineering4020046
Lee SH, Sung JH. Microtechnology-Based Multi-Organ Models. Bioengineering. 2017; 4(2):46. https://doi.org/10.3390/bioengineering4020046
Chicago/Turabian StyleLee, Seung Hwan, and Jong Hwan Sung. 2017. "Microtechnology-Based Multi-Organ Models" Bioengineering 4, no. 2: 46. https://doi.org/10.3390/bioengineering4020046
APA StyleLee, S. H., & Sung, J. H. (2017). Microtechnology-Based Multi-Organ Models. Bioengineering, 4(2), 46. https://doi.org/10.3390/bioengineering4020046