
Comparative N-Glycosylation Analysis of the Fc Portions of a Chimeric Human Coagulation Factor VIII and Immunoglobulin G1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
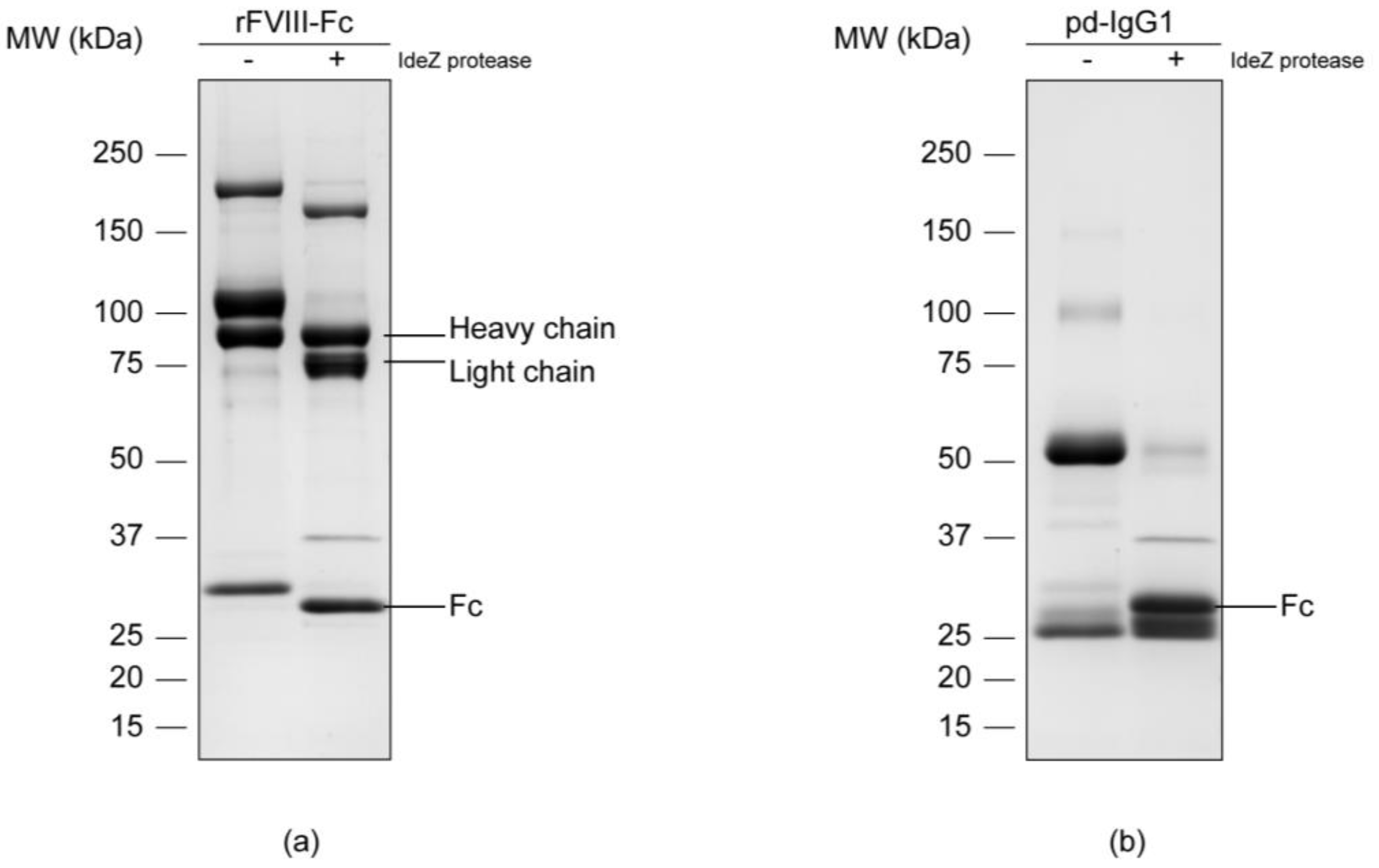
2.2. Enzymatic Cleavage and Separation of the Fc Portion
2.3. In-Gel Deglycosylation
2.4. Neuraminidase Digestion
2.5. N-Glycan Derivatization
2.5.1. Permethylation
2.5.2. Fluorescent Labeling with 2-Amino Benzamide (2-AB)
2.6. Matrix-Assisted Laser Desorption/Ionization Time-Of-Flight (MALDI-TOF) MS
2.7. Ultra High-Performance Liquid Chromatography (UHPLC)
2.7.1. Hydrophilic Interaction Liquid Chromatography (HILIC)
2.7.2. Mixed-Mode N-Glycan Separation
3. Results
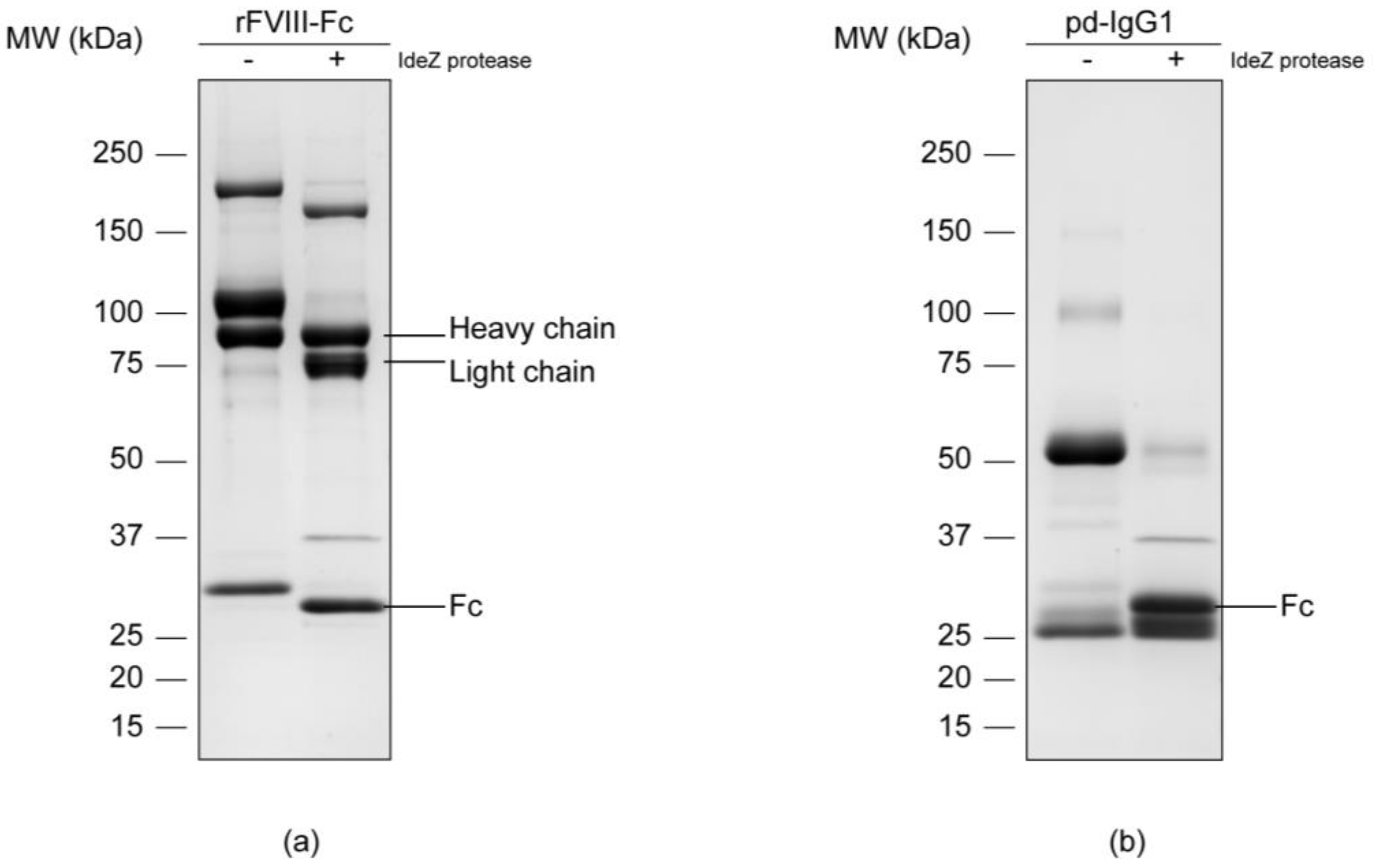
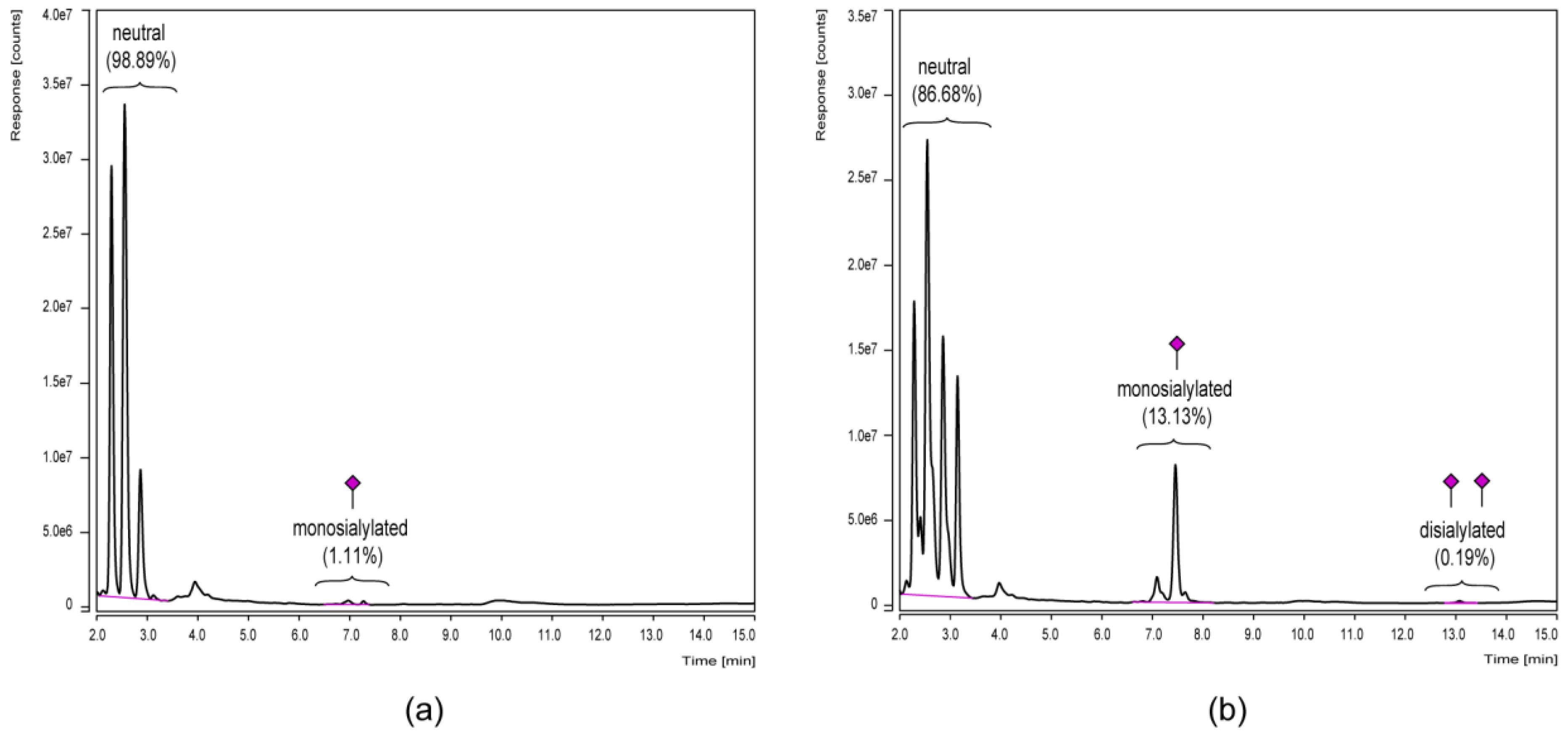
3.1. Separation of the Fc Parts and N-Glycan Release
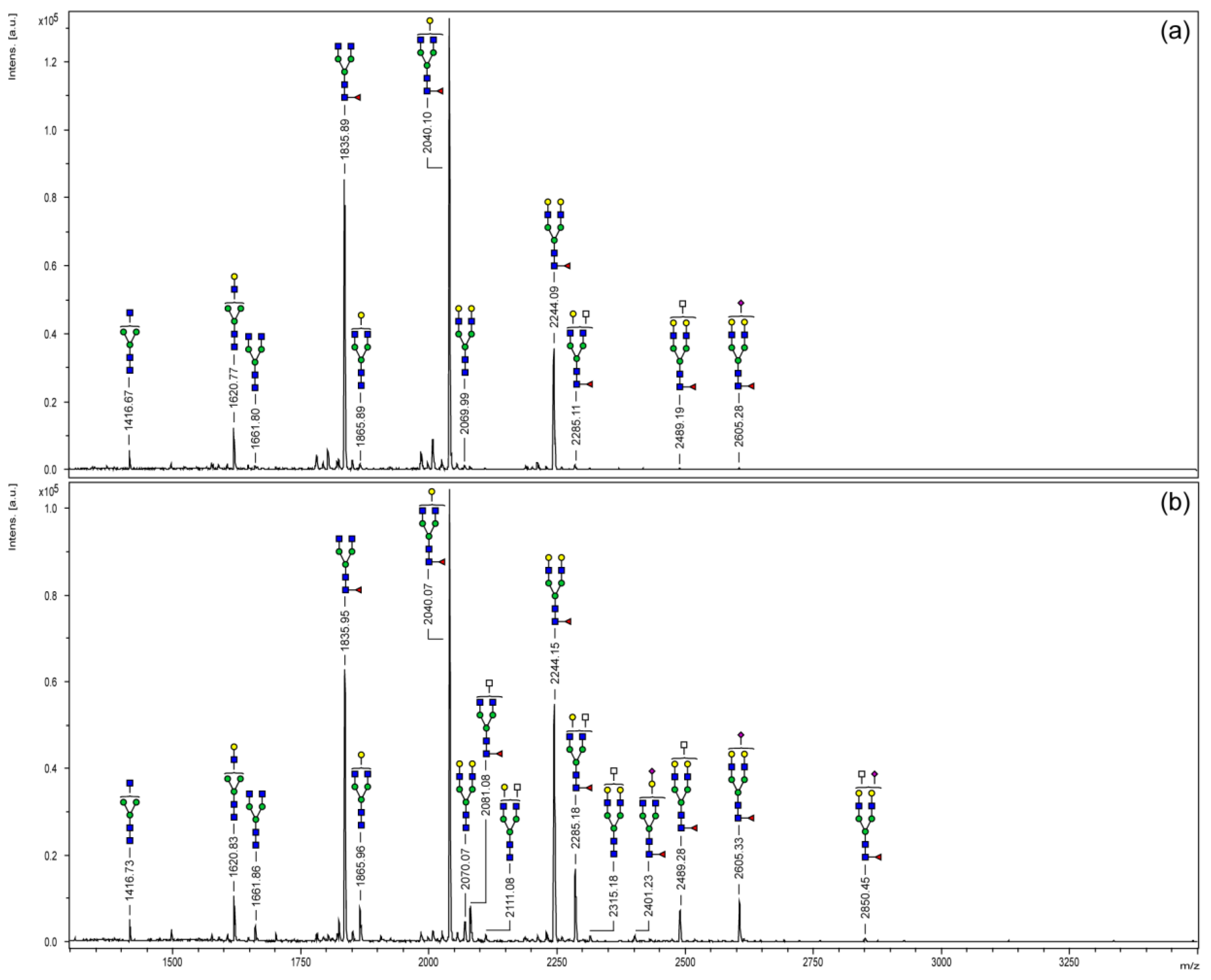
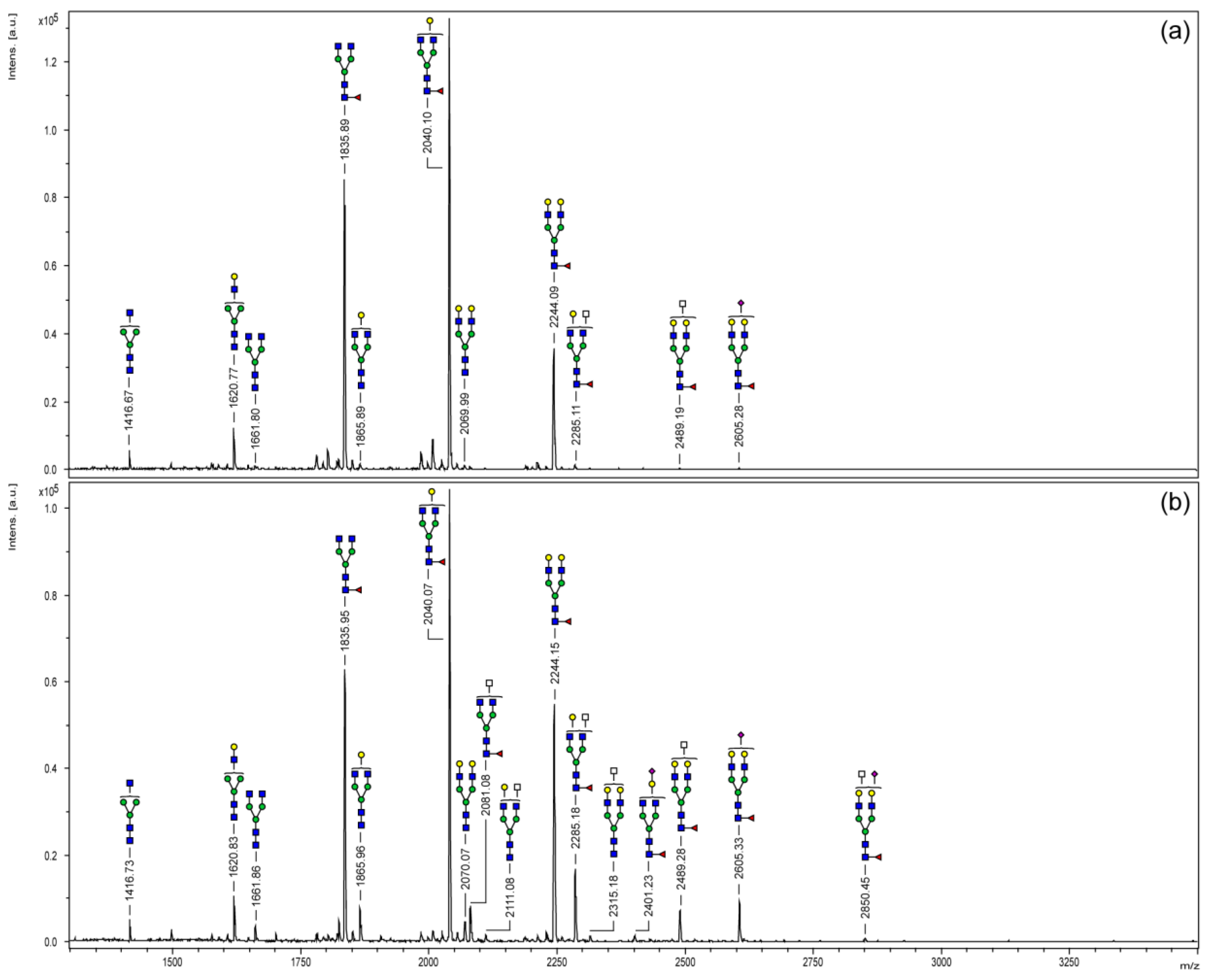
3.2. MALDI-TOF Mass Spectrometry
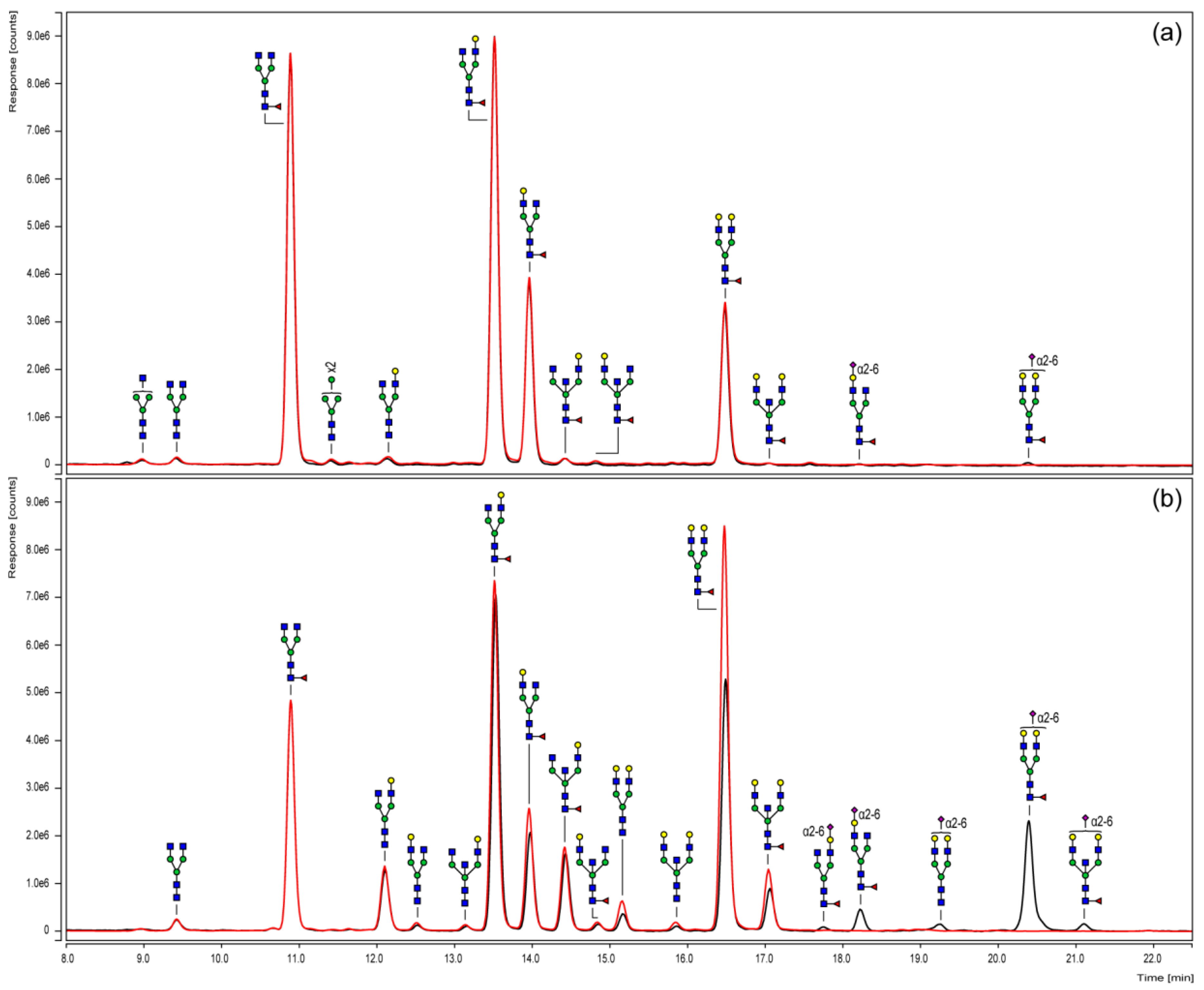
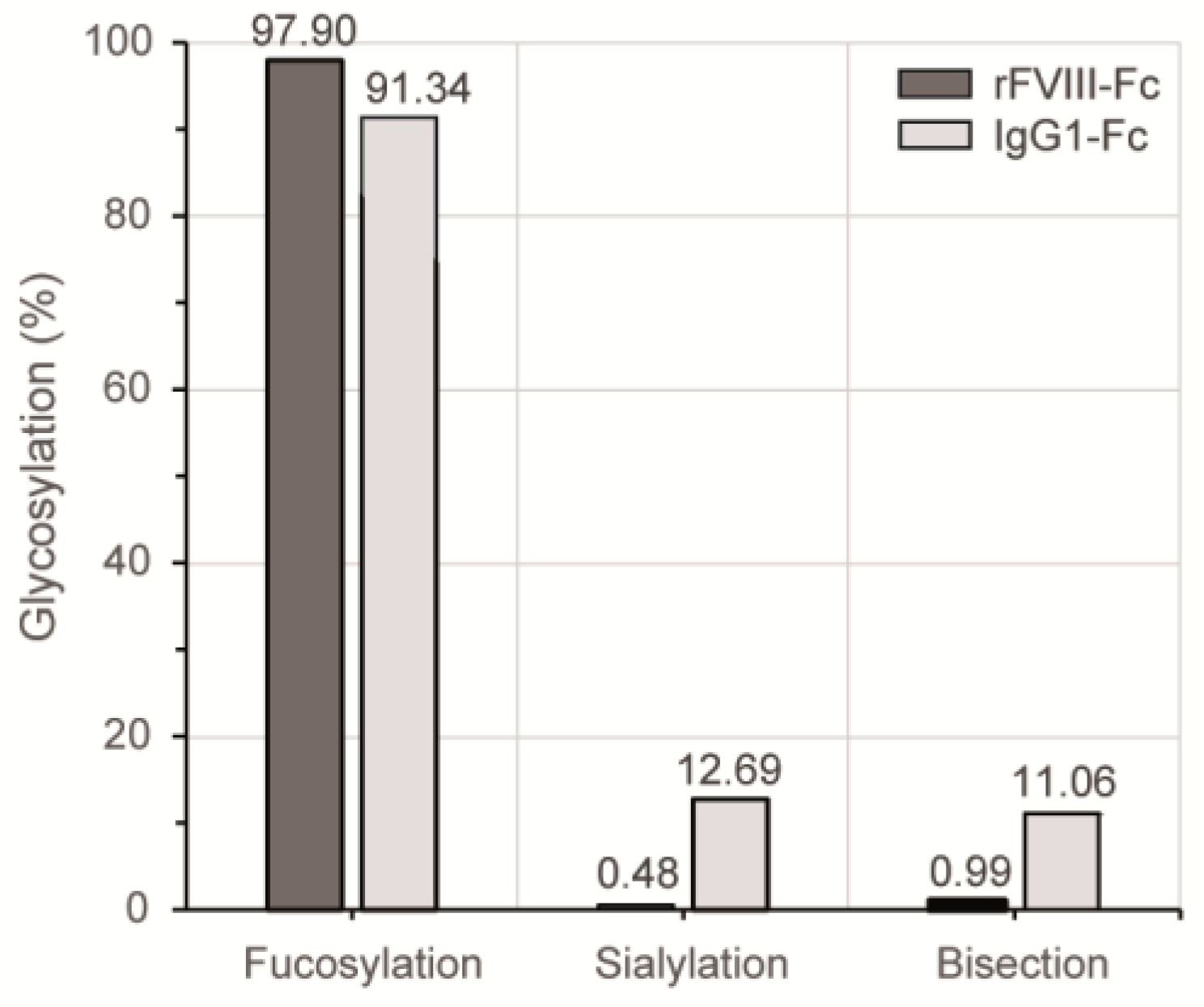
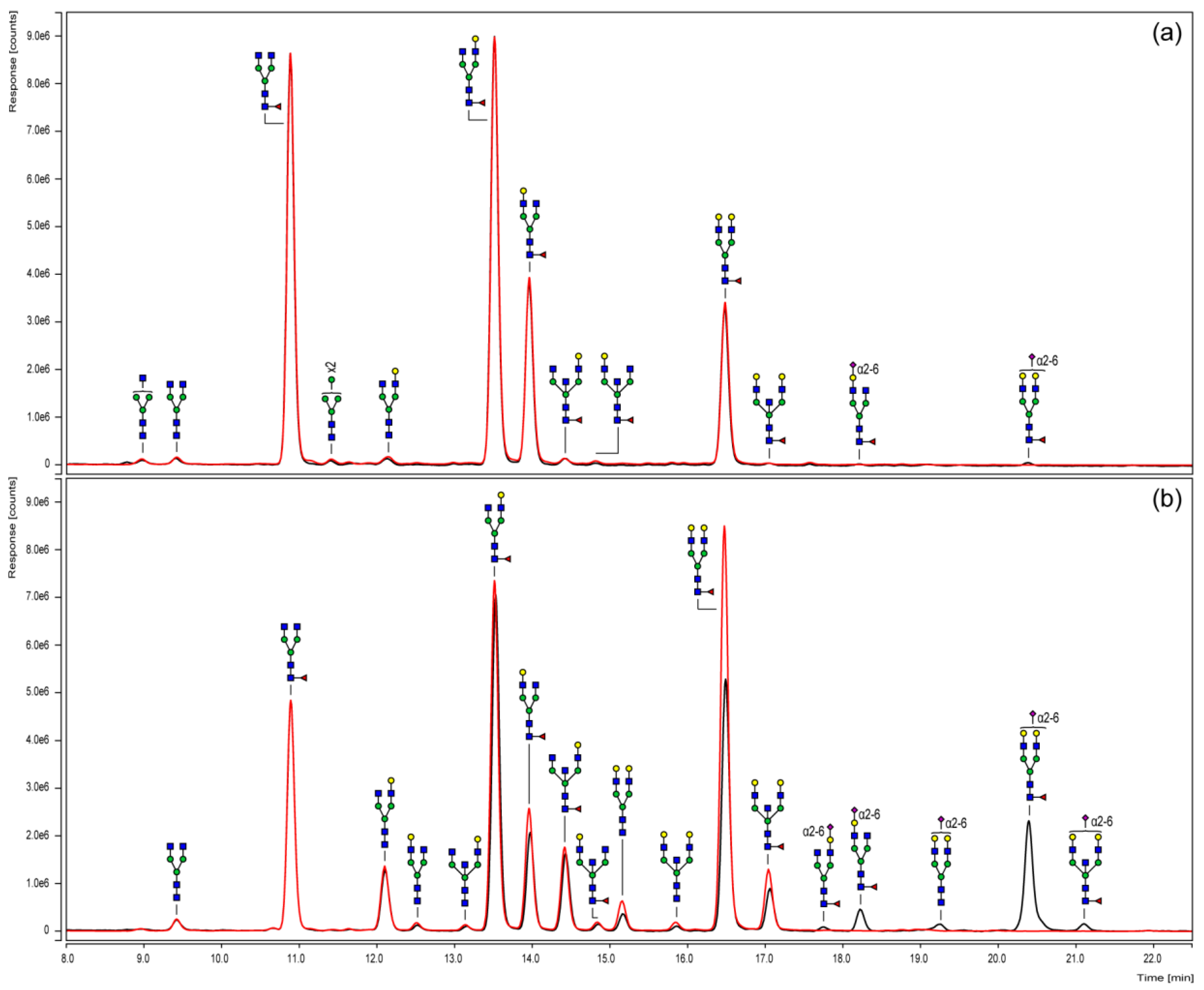
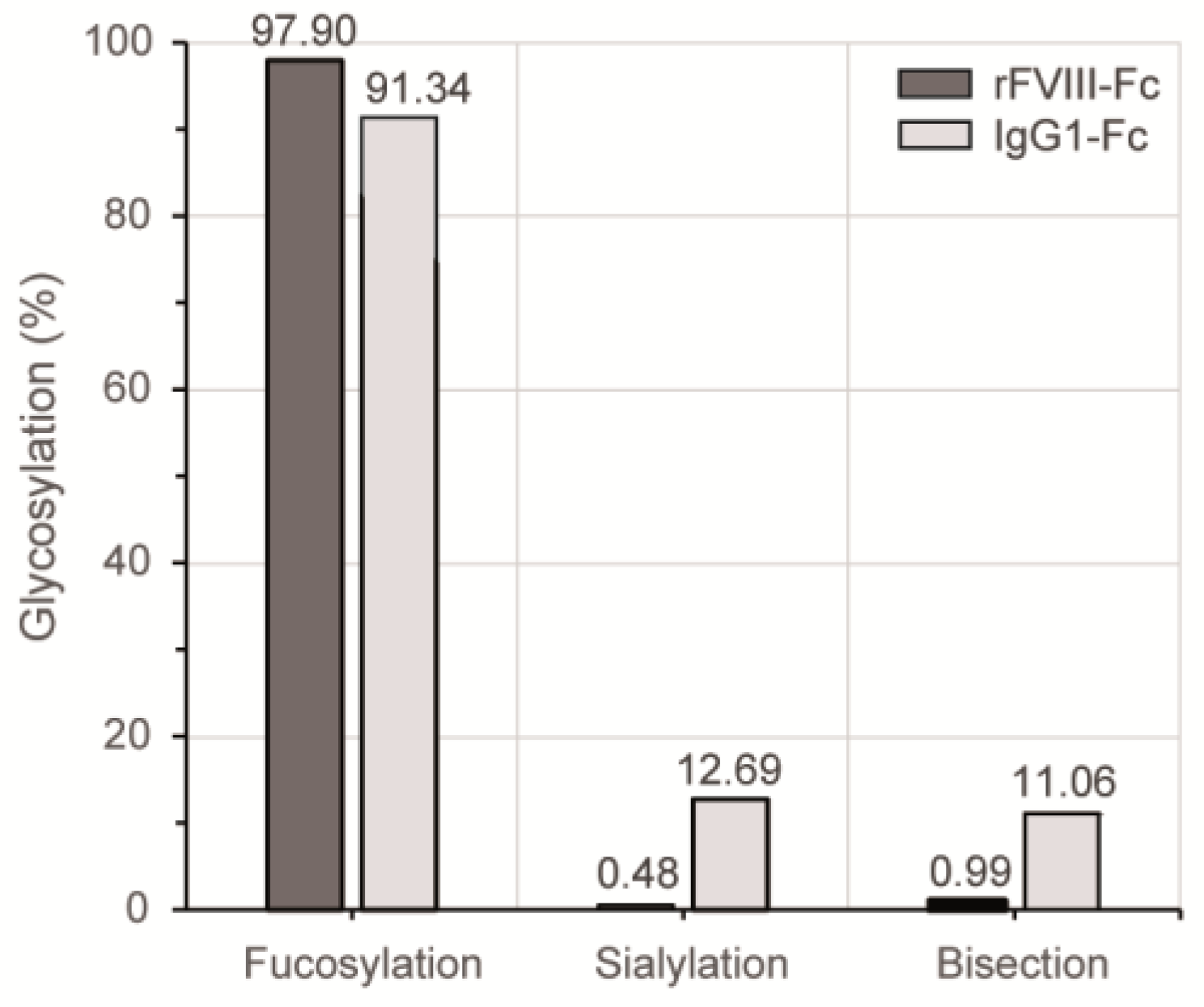
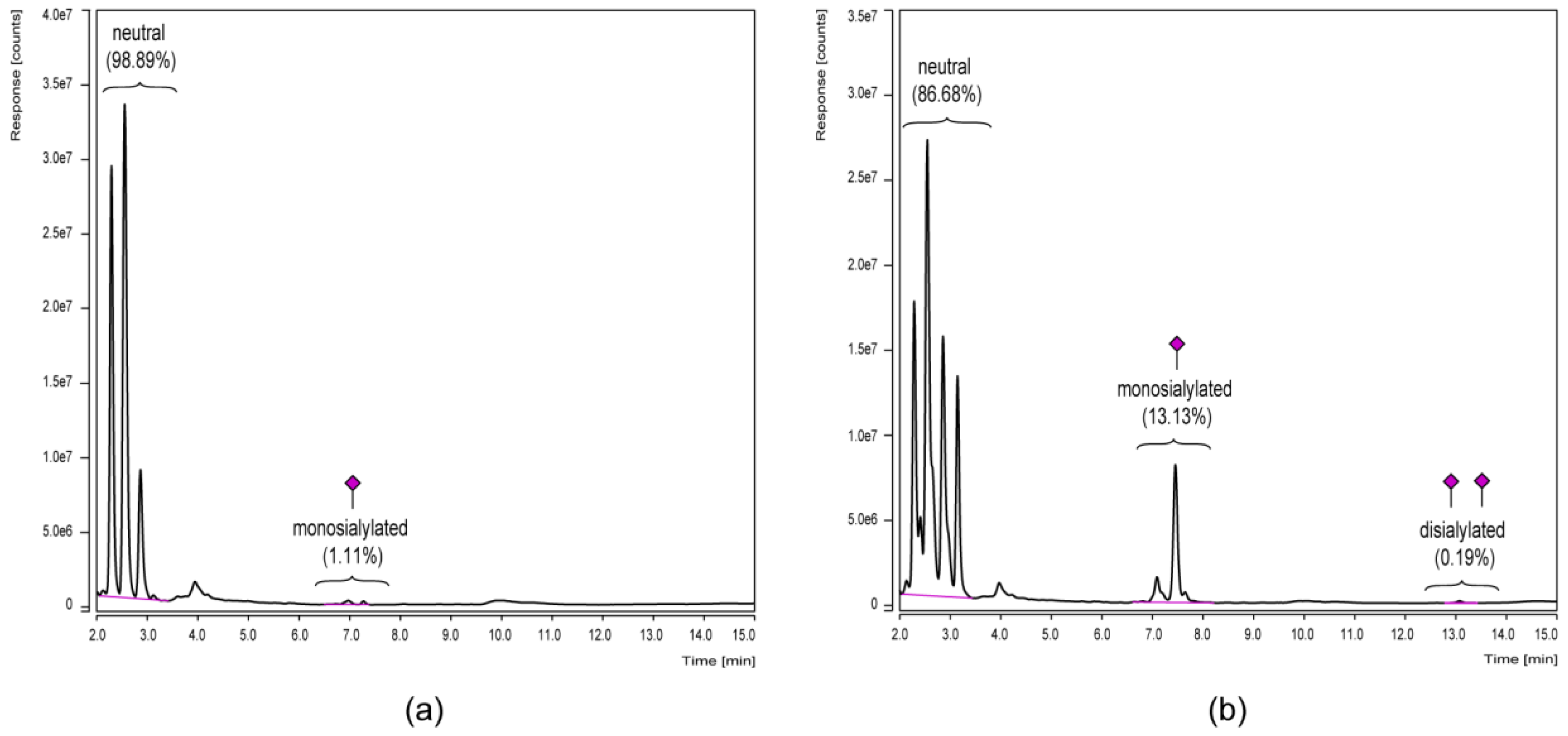
3.3. Chromatographic N-Glycan Profiling
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aledort, L.; Ljung, R.; Mann, K.; Pipe, S. Factor VIII therapy for hemophilia A: Current and future issues. Expert Rev. Hematol. 2014, 7, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Valentino, L.A.; Kawji, M.; Grygotis, M. Venous access in the management of hemophilia. Blood Rev. 2011, 25, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Ing, M.; Gupta, N.; Teyssandier, M.; Maillere, B.; Pallardy, M.; Delignat, S.; Lacroix-Desmazes, S. Immunogenicity of long-lasting recombinant factor VIII products. Cell. Immunol. 2015, 301, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Kessler, C.; Oldenburg, J.; Ettingshausen, C.E.; Tiede, A.; Khair, K.; Negrier, C.; Klamroth, R. Spotlight on the human factor: Building a foundation for the future of haemophilia A management: Report from a symposium on human recombinant FVIII at the World Federation of Hemophilia World Congress, Melbourne, Australia on 12 May 2014. Haemophilia 2015, 21 (Suppl. S1), 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.M.; Ravetch, J.V. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.; Golding, B.; Strome, S.E.; Sauna, Z.E. Fc fusion as a platform technology: Potential for modulating immunogenicity. Trends Biotechnol. 2015, 33, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.; Lagasse, H.A.; Burch, E.; Strome, S.; Tan, S.; Jiang, H.; Sauna, Z.E.; Golding, B. Modulating immunogenicity of factor IX by fusion to an immunoglobulin Fc domain: A study using a hemophilia B mouse model. J. Thromb. Haemost. 2017, 15, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Raju, T.S. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr. Opin. Immunol. 2008, 20, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.T.; Toby, G.; Lu, Q.; Liu, T.; Kulman, J.D.; Low, S.C.; Bitonti, A.J.; Pierce, G.F. Biochemical and functional characterization of a recombinant monomeric Factor VIII-Fc fusion protein. J. Thromb. Haemost. 2013, 11, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Küster, B.; Wheeler, S.F.; Hunter, A.P.; Dwek, R.A.; Harvey, D.J. Sequencing of N-linked oligosaccharides directly from protein gels: In-gel deglycosylation followed by matrix-assisted laser desorption/ionization mass spectrometry and normal-phase high-performance liquid chromatography. Anal. Biochem. 1997, 250, 82–101. [Google Scholar] [CrossRef] [PubMed]
- Weiz, S.; Kamalakumar, A.; Biskup, K.; Blanchard, V. Enhanced detection of in-gel released N-glycans by MALDI-TOF-MS. Proteomics 2015, 15, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.O.; Bayer, M.; Berger, M.; Blanchard, V.; Hinderlich, S. Analysis of Cell Surface N-glycosylation of the Human Embryonic Kidney 293T Cell Line. J. Carbohydr. Chem. 2011, 30, 218–232. [Google Scholar] [CrossRef]
- Wada, Y.; Azadi, P.; Costello, C.E.; Dell, A.; Dwek, R.A.; Geyer, H.; Geyer, R.; Kakehi, K.; Karlsson, N.G.; Kato, K.; et al. Comparison of the methods for profiling glycoprotein glycans--HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 2007, 17, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Rosenlöcher, J.; Böhrsch, V.; Sacharjat, M.; Blanchard, V.; Giese, C.; Sandig, V.; Hackenberger, C.P.R.; Hinderlich, S. Applying Acylated Fucose Analogues to Metabolic Glycoengineering. Bioengineering 2015, 2, 213–234. [Google Scholar] [CrossRef]
- Ceroni, A.; Maass, K.; Geyer, H.; Geyer, R.; Dell, A.; Haslam, S.M. GlycoWorkbench: A tool for the computer-assisted annotation of mass spectra of glycans. J. Proteome Res. 2008, 7, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Aebi, M.; Packer, N.H.; Seeberger, P.H.; Esko, J.D.; Stanley, P.; Hart, G.; Darvill, A.; Kinoshita, T.; et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.C.; Chen, X.; Liu, Y.D.; Shah, B.; Zhang, Z. Naturally occurring glycan forms of human immunoglobulins G1 and G2. Mol. Immunol. 2010, 47, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Krapp, S.; Mimura, Y.; Jefferis, R.; Huber, R.; Sondermann, P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 2003, 325, 979–989. [Google Scholar] [CrossRef]
- Dunn, A. The long and short of it: Using the new factor products. Hematol. Am. Soc. Hematol. Educ. Program 2015, 2015, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C.; Uribe-Querol, E. Fc receptors: Cell activators of antibody functions. Adv. Biosci. Biotechnol. 2013, 4, 21–33. [Google Scholar] [CrossRef]
- Kannicht, C.; Ramstrom, M.; Kohla, G.; Tiemeyer, M.; Casademunt, E.; Walter, O.; Sandberg, H. Characterisation of the post-translational modifications of a novel, human cell line-derived recombinant human factor VIII. Thromb. Res. 2013, 131, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Kosloski, M.P.; Miclea, R.D.; Balu-Iyer, S.V. Role of glycosylation in conformational stability, activity, macromolecular interaction and immunogenicity of recombinant human factor VIII. AAPS J. 2009, 11, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Sommer, J.M.; Moore, N.; Guffie-Valentine, B.; Bardan, S.; Buyue, Y.; Kamphaus, G.D.; Konkle, B.A.; Pierce, G.F. Comparative field study evaluating the activity of recombinant factor VIII Fc fusion protein in plasma samples at clinical haemostasis laboratories. Haemophilia 2014, 20, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.M.; Cosgrave, E.F.; Struwe, W.B.; Wormald, M.; Davey, G.P.; Jefferis, R.; Rudd, P.M. Glycosylation and Fc receptors. Curr. Top. Microbiol. Immunol. 2014, 382, 165–199. [Google Scholar] [CrossRef] [PubMed]
- Reusch, D.; Haberger, M.; Maier, B.; Maier, M.; Kloseck, R.; Zimmermann, B.; Hook, M.; Szabo, Z.; Tep, S.; Wegstein, J.; et al. Comparison of methods for the analysis of therapeutic immunoglobulin G Fc-glycosylation profiles-Part 1: Separation-based methods. MAbs 2015, 7, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.; Ochiai, H.; Huang, W.; Yang, Q.; Li, C.; Wang, L.X. Chemoenzymatic synthesis and Fcgamma receptor binding of homogeneous glycoforms of antibody Fc domain. Presence of a bisecting sugar moiety enhances the affinity of Fc to FcgammaIIIa receptor. J. Am. Chem. Soc. 2011, 133, 18975–18991. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Jiang, L.; Pan, L.Z.; LaBarre, M.J.; Anderson, D.; Reff, M. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. Biotechnol. Bioeng. 2001, 74, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, G.; Plomp, R.; Koeleman, C.A.; Visser, R.; von Horsten, H.H.; Sandig, V.; Rispens, T.; Wuhrer, M.; Vidarsson, G. Multi-level glyco-engineering techniques to generate IgG with defined Fc-glycans. Sci. Rep. 2016, 6, 36964. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Kobayashi, T.; Wermeling, F.; Ravetch, J.V. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature 2011, 475, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Quast, I.; Peschke, B.; Lunemann, J.D. Regulation of antibody effector functions through IgG Fc N-glycosylation. Cell. Mol. Life Sci. 2017, 74, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Liu, T.; Drager, D.; Patarroyo-White, S.; Chhabra, E.S.; Peters, R.; Josephson, N.; Lillicrap, D.; Blumberg, R.S.; Pierce, G.F.; et al. Recombinant factor VIII Fc (rFVIIIFc) fusion protein reduces immunogenicity and induces tolerance in hemophilia A mice. Cell. Immunol. 2016, 301, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lenting, P.J.; van Mourik, J.A.; Mertens, K. The life cycle of coagulation factor VIII in view of its structure and function. Blood 1998, 92, 3983–3996. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kannicht, C.; Kröning, M.; Solecka-Witulska, B.A.; Kohla, G.; Rosenlöcher, J. Comparative N-Glycosylation Analysis of the Fc Portions of a Chimeric Human Coagulation Factor VIII and Immunoglobulin G1. Bioengineering 2017, 4, 44. https://doi.org/10.3390/bioengineering4020044
Kannicht C, Kröning M, Solecka-Witulska BA, Kohla G, Rosenlöcher J. Comparative N-Glycosylation Analysis of the Fc Portions of a Chimeric Human Coagulation Factor VIII and Immunoglobulin G1. Bioengineering. 2017; 4(2):44. https://doi.org/10.3390/bioengineering4020044
Chicago/Turabian StyleKannicht, Christoph, Mario Kröning, Barbara A. Solecka-Witulska, Guido Kohla, and Julia Rosenlöcher. 2017. "Comparative N-Glycosylation Analysis of the Fc Portions of a Chimeric Human Coagulation Factor VIII and Immunoglobulin G1" Bioengineering 4, no. 2: 44. https://doi.org/10.3390/bioengineering4020044
APA StyleKannicht, C., Kröning, M., Solecka-Witulska, B. A., Kohla, G., & Rosenlöcher, J. (2017). Comparative N-Glycosylation Analysis of the Fc Portions of a Chimeric Human Coagulation Factor VIII and Immunoglobulin G1. Bioengineering, 4(2), 44. https://doi.org/10.3390/bioengineering4020044