
Analysis of Microbiome for AP and CRC Discrimination

 , , ,
, , ,  ,
,  ,
,  and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Origin of the Data
2.2. Data Synthesis
2.3. Data Quality Metrics
2.3.1. Support Vector Classifier Optimisation
2.3.2. Methods for Data Classification
2.4. Statistical Analysis
2.5. Feature Selection
2.6. Beta-Diversity Analysis
2.7. Classification and SHAP Analysis
3. Results
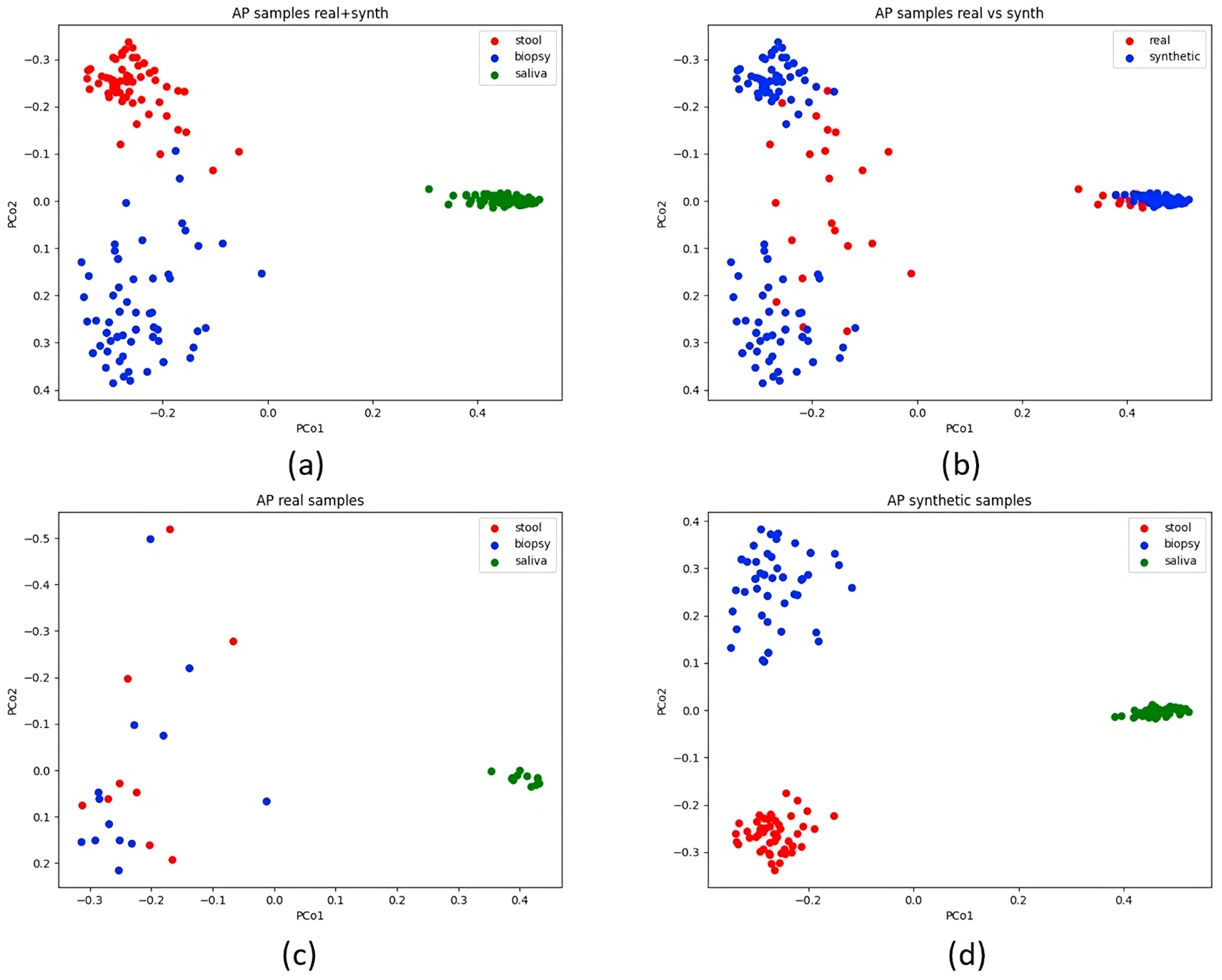
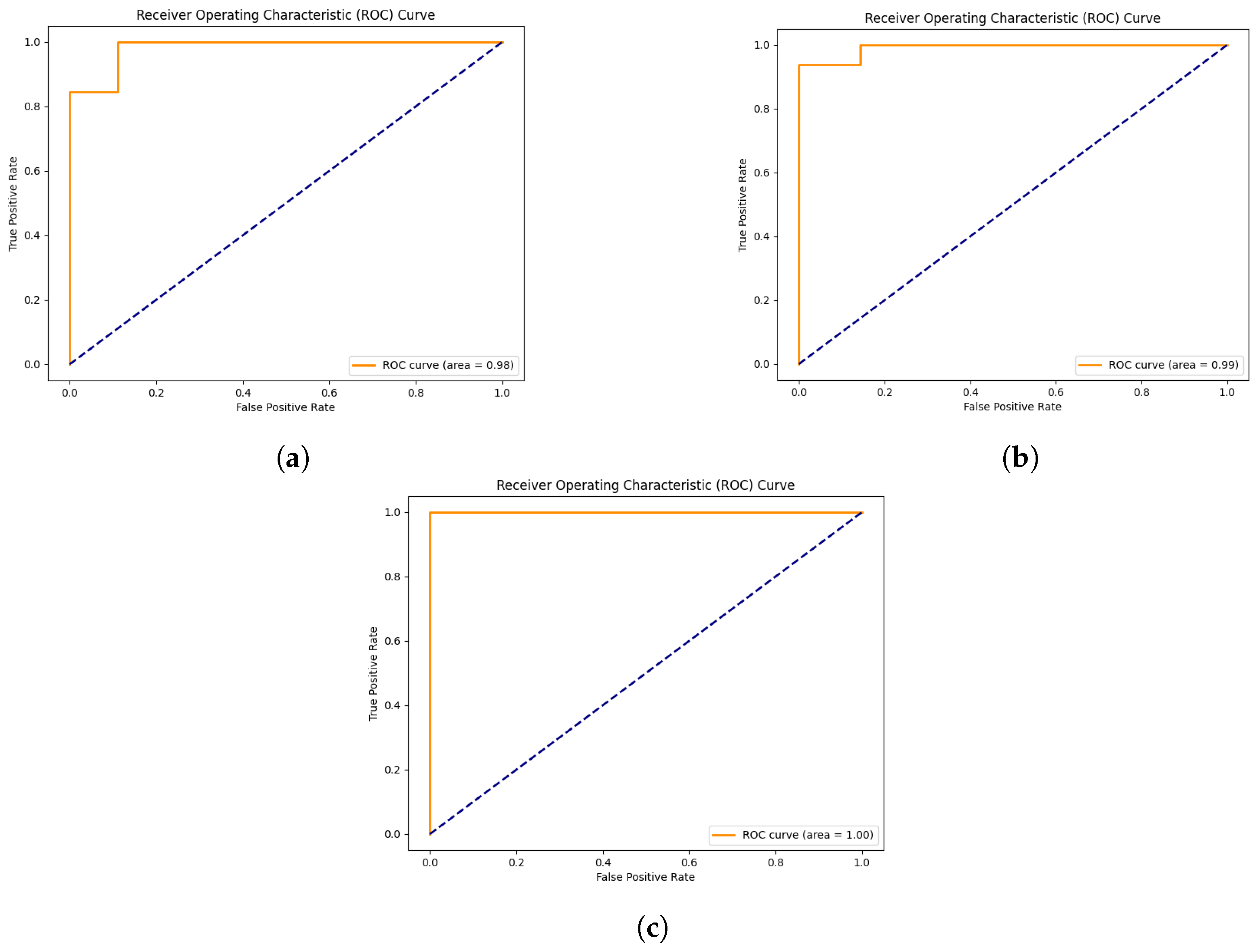
3.1. Synthetic Data Evaluation
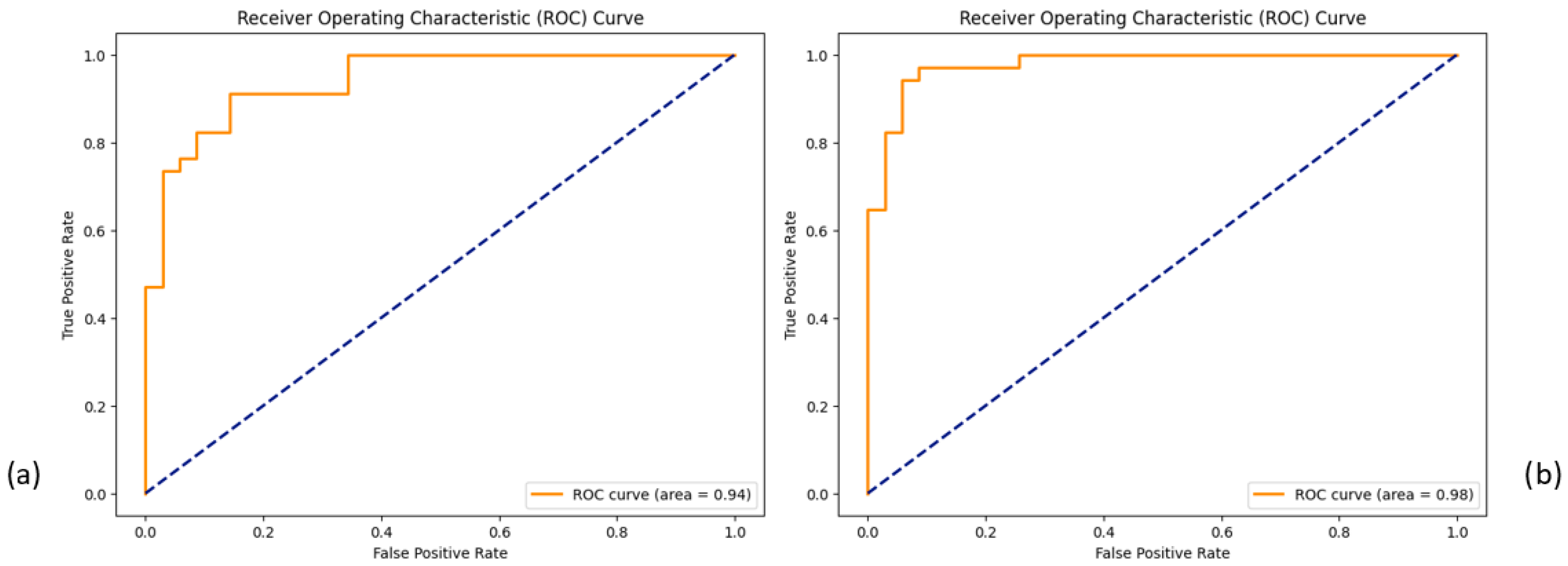
3.1.1. Data Classification
3.1.2. Statistical and Quality Assessment
3.2. Feature Extraction
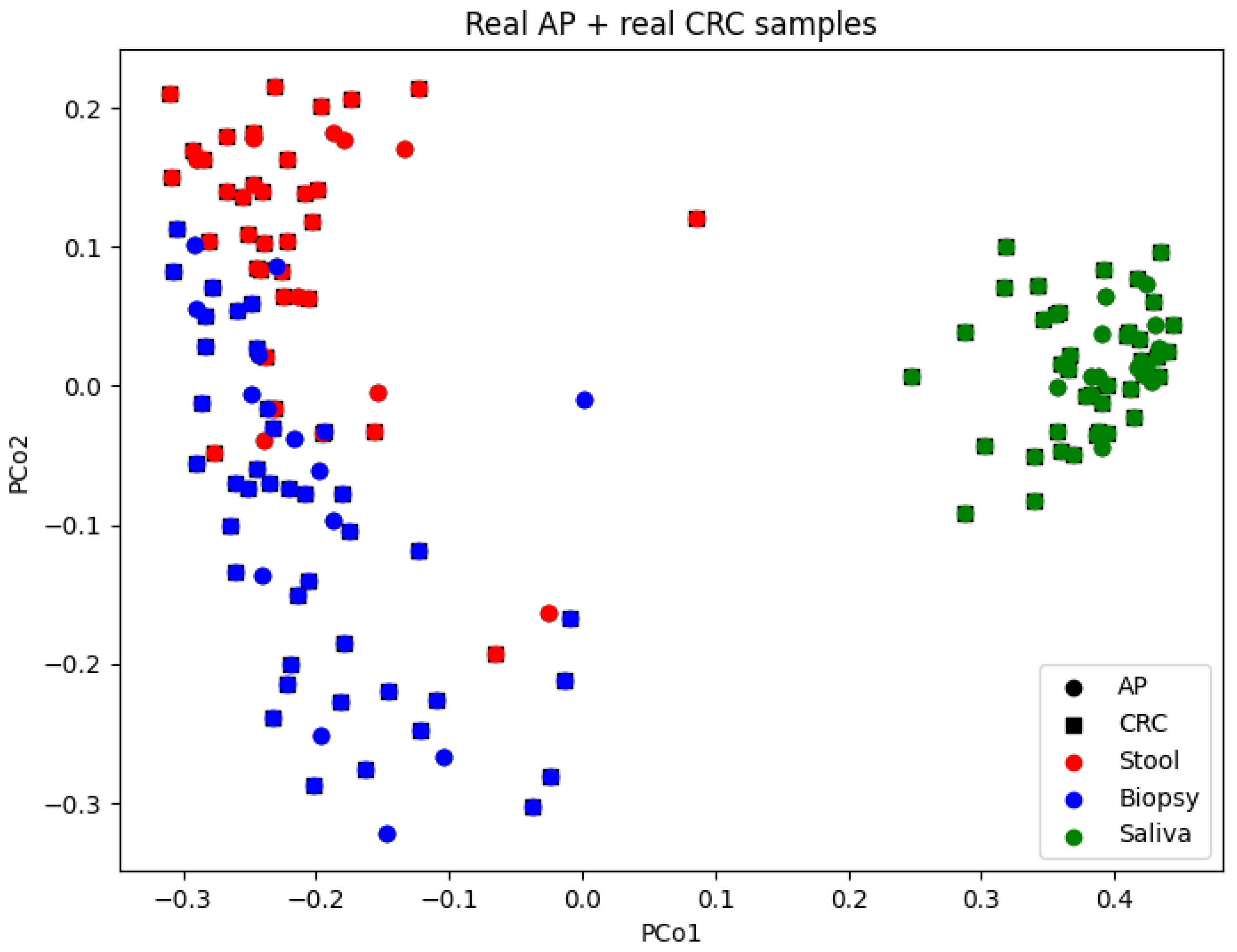
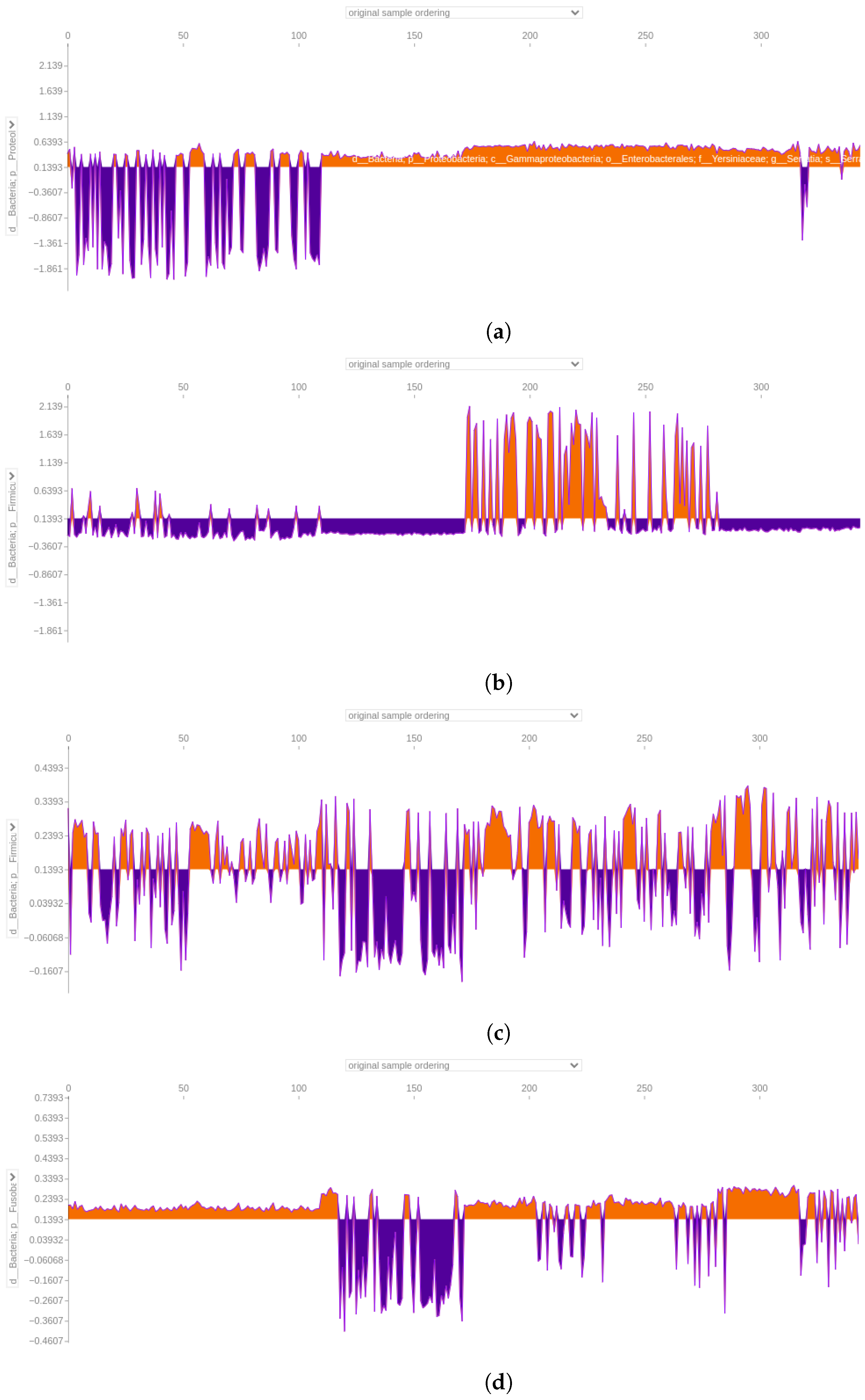
3.3. Beta-Diversity Insights
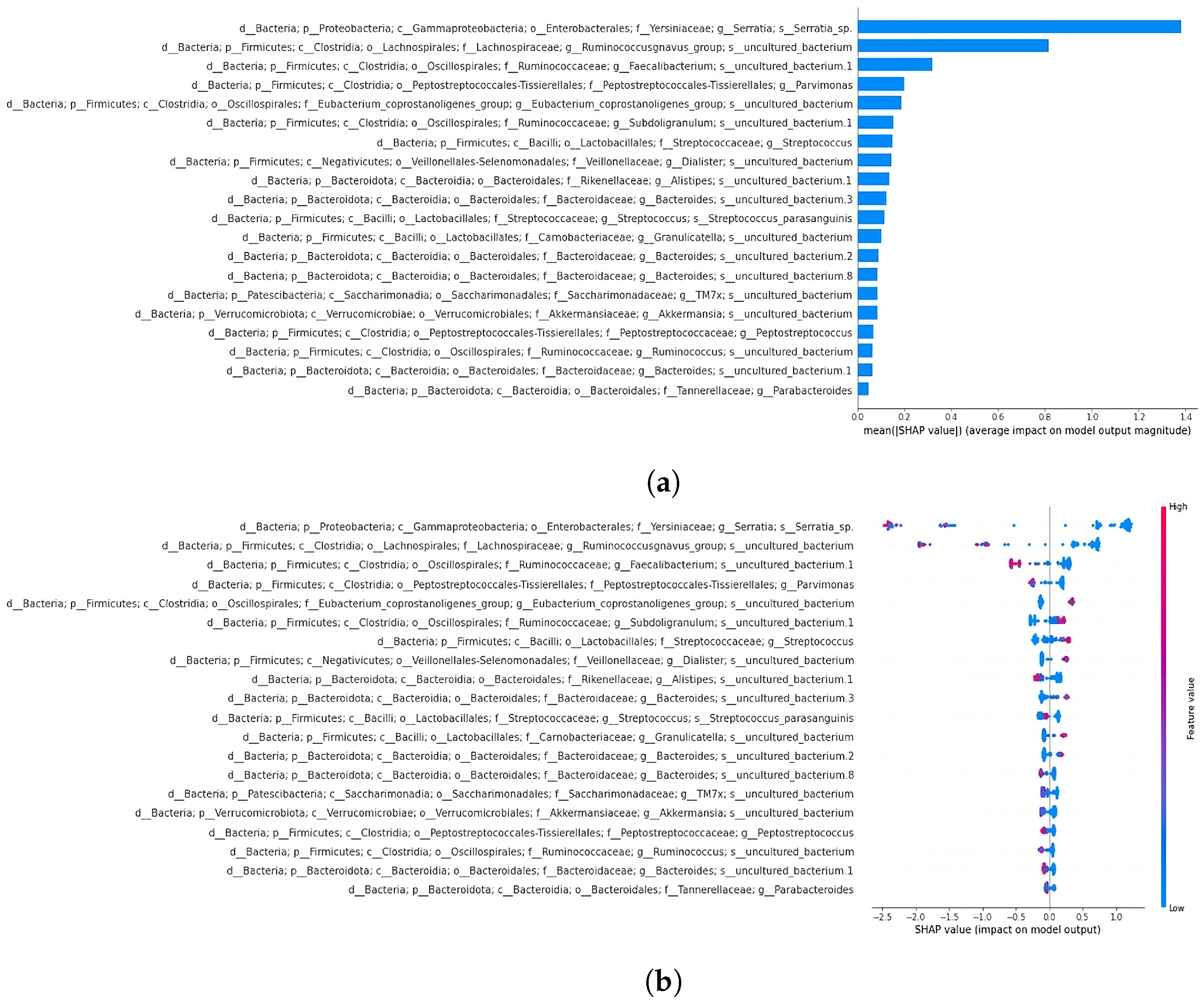
3.4. Classification and SHAP Analysis Outcomes
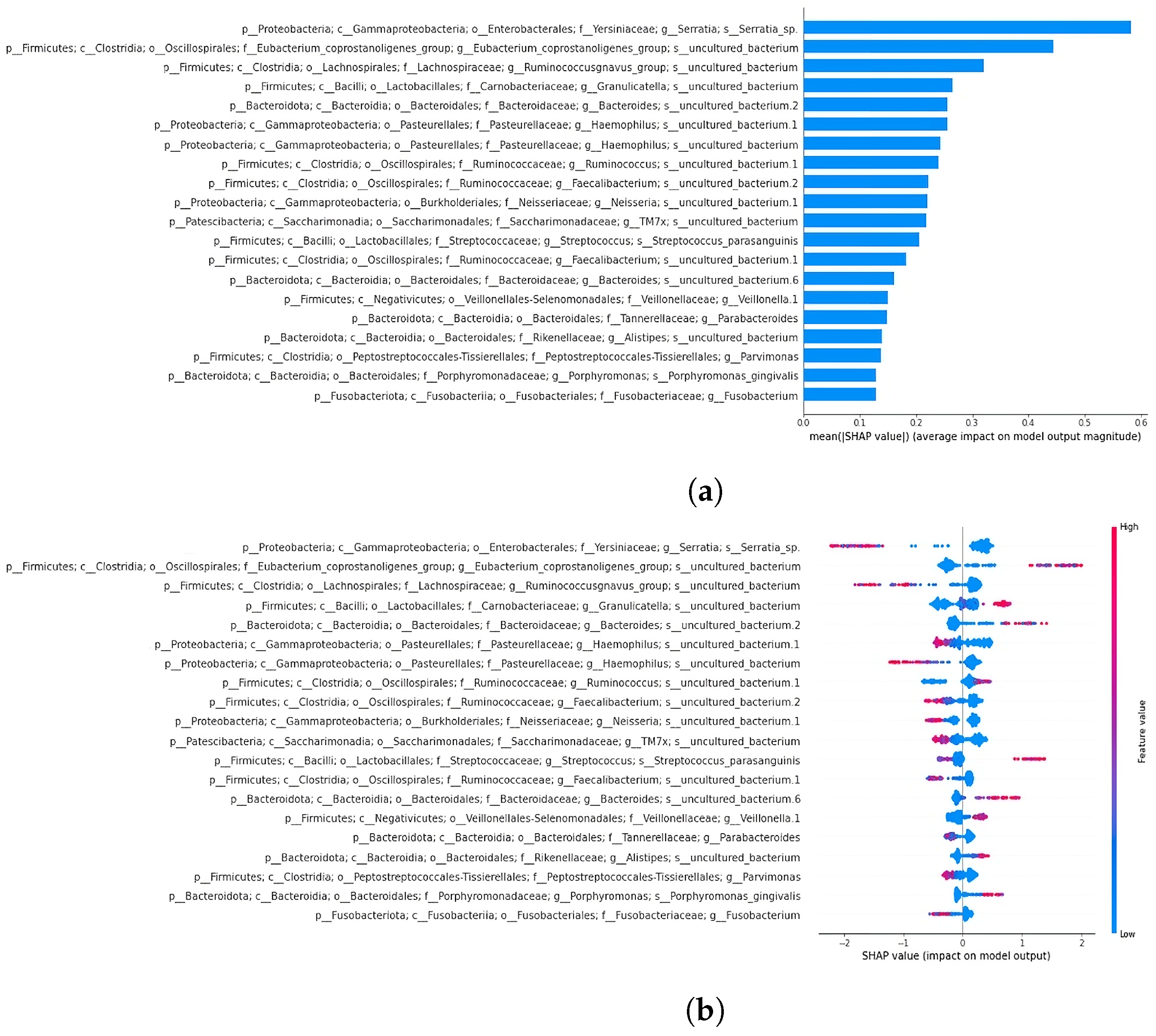
3.4.1. SHAP Analysis Overview
3.4.2. Classification and SHAP Analysis of the 19-Feature Dataset
3.4.3. Classification and SHAP Analysis of the 64-Feature Dataset
3.4.4. Force Plot of the 64-Feature Dataset
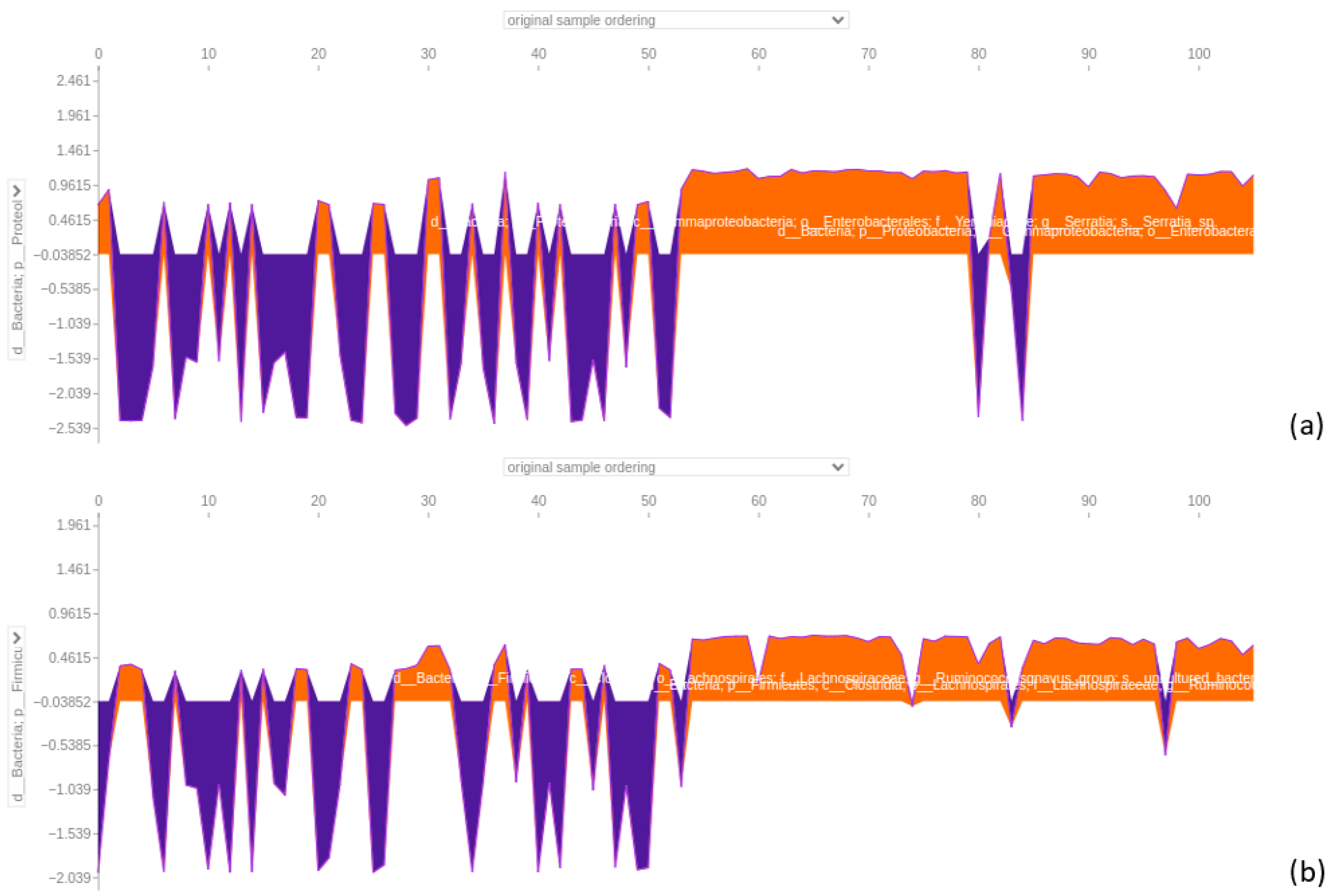
3.4.5. Classification and SHAP for Individual Datasets
3.4.6. Stool Dataset
3.4.7. Biopsy Dataset
3.4.8. Saliva Dataset
4. Discussion
4.1. Evaluation of Classifier Performance and Additional Metrics for Synthetic Data
4.2. Feature Extraction and Identification of Relevant Bacterial Taxa
4.3. Discrepancies in PCoA Patterns: Real vs. Synthetic AP Samples
4.4. XGBoost Classifier Dominance in Sample Segregation and Feature Distinction
4.5. SHAP Analysis Reveals Discrepancies in Feature Importance: A Closer Look at XGBoost Classification of the 64-Feature Dataset
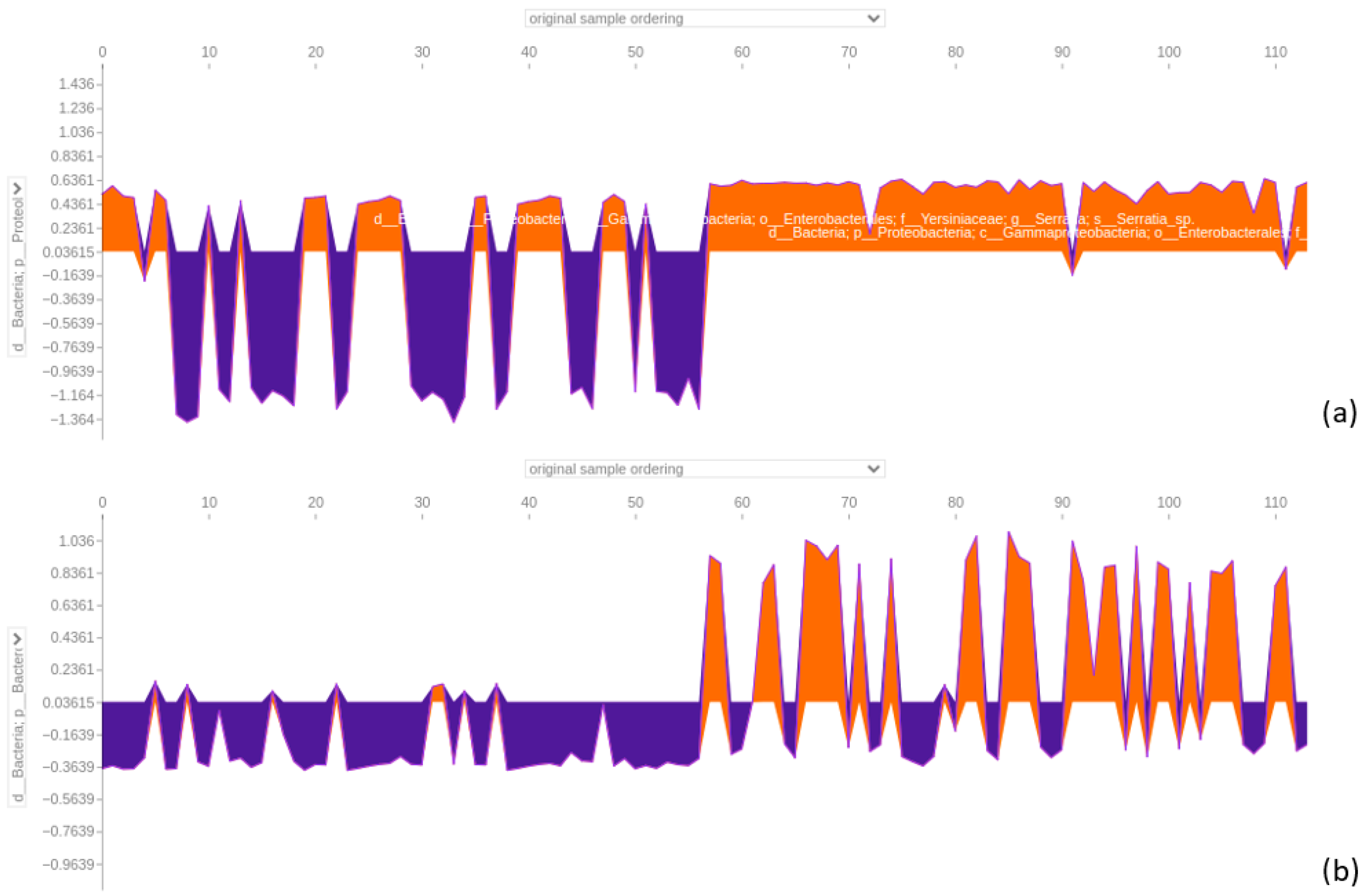
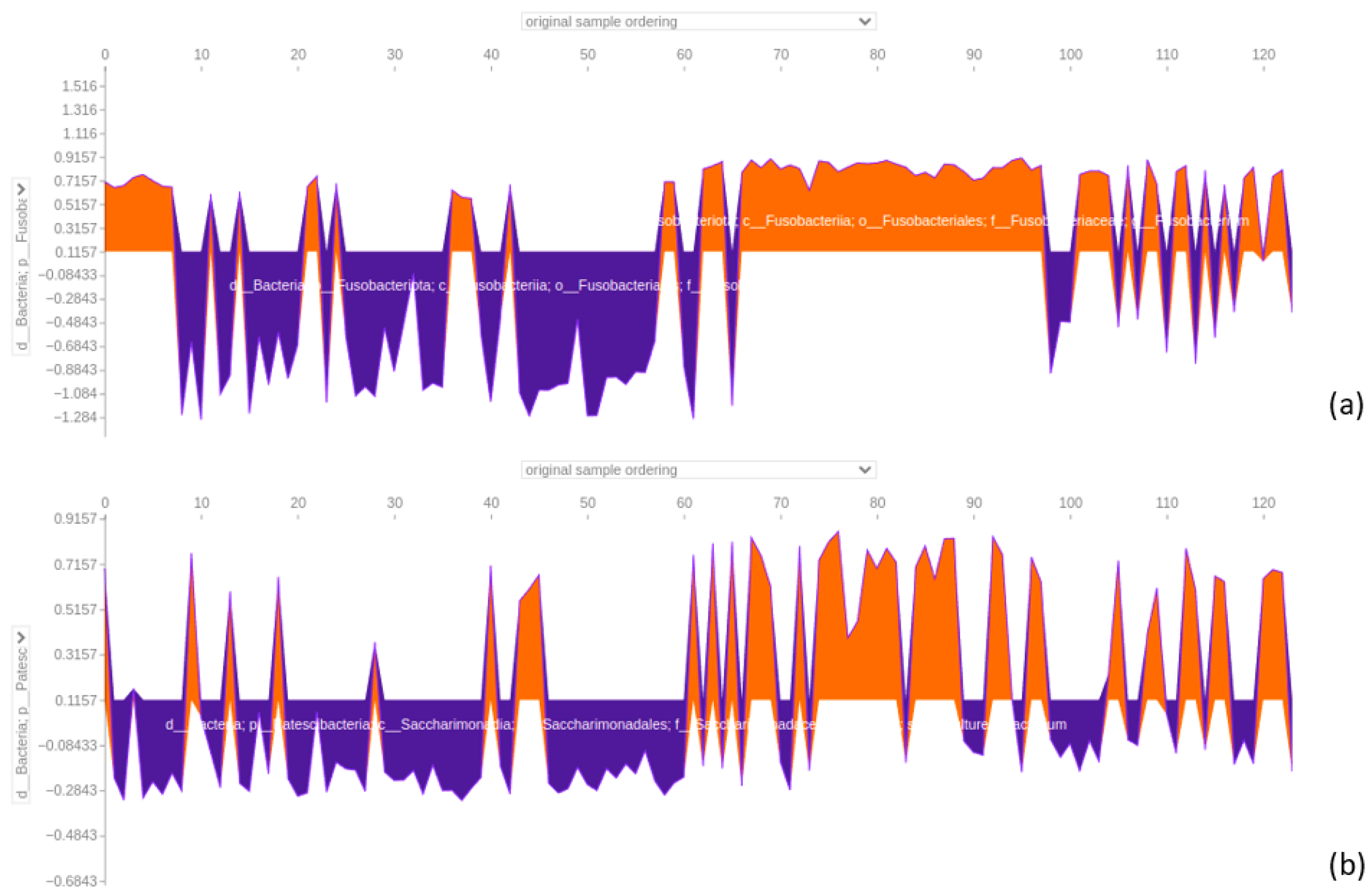
4.6. Significance of Fusobacterium and Parvimonas Genus
4.7. Comparisons of Taxa with Previous Findings
4.7.1. Similarities in Faecal Microbiota
4.7.2. Similarities in Biopsy Microbiota
4.7.3. Insights from Key Genera in Stool and Biopsy Samples
4.7.4. Similarities in Saliva Microbiota
4.7.5. Insights from Key Genera in Saliva Samples
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Adenomatous Polyps |
| AUC | Area Under the Curve |
| BCD | Bray–Curtis Dissimilarity |
| CRC | Colorectal Cancer |
| DL | Deep Learning |
| GeLU | Gaussian Error Linear Unit |
| KDE | Kernel Density Estimation |
| KS | Kolmogorov–Smirnov (test) |
| LeakyReLU | Leaky Rectified Linear Unit |
| LR | Logistic Regression |
| LRP | Layer-Wise Relevance Propagation |
| ML | Machine Learning |
| MSE | Mean Squared Error |
| OTU | Operational Taxonomic Unit |
| PCoA | Principal Coordinates Analysis |
| ReLU | Rectified Linear Unit |
| RF | Random Forest |
| ROC | Receiver Operating Characteristic |
| SDV | Synthetic Data Vault |
| SHAP | SHapley Additive exPlanations |
| STD | Standard Deviation |
| SVC | Support Vector Classifier |
| XAI | eXplainable Artificial Intelligence |
References
- Mármol, I.; Sánchez-de Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Douaiher, J.; Ravipati, A.; Grams, B.; Chowdhury, S.; Alatise, O.; Are, C. Colorectal cancer—Global burden, trends, and geographical variations. J. Surg. Oncol. 2017, 115, 619–630. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Di Gloria, L.; Nannini, G.; Meoni, G.; Niccolai, E.; Ringressi, M.N.; Baldi, S.; Fani, R.; Tenori, L.; Taddei, A.; et al. From adenoma to CRC stages: The oral-gut microbiome axis as a source of potential microbial and metabolic biomarkers of malignancy. Neoplasia 2023, 40, 100901. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Goel, A.; Chung, D.C. Pathways of colorectal carcinogenesis. Gastroenterology 2020, 158, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The gut microbiota and inflammation: An overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Alhinai, E.A.; Walton, G.E.; Commane, D.M. The role of the gut microbiota in colorectal cancer causation. Int. J. Mol. Sci. 2019, 20, 5295. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.K. Potential role of the gut microbiome in colorectal cancer progression. Front. Immunol. 2022, 12, 807648. [Google Scholar] [CrossRef]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, W.D.; Wang, Y.D. The relationship between gut microbiota and inflammatory diseases: The role of macrophages. Front. Microbiol. 2020, 11, 1065. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Iadanza, E.; Fabbri, R.; Bašić-ČiČak, D.; Amedei, A.; Telalovic, J.H. Gut microbiota and artificial intelligence approaches: A scoping review. Health Technol. 2020, 10, 1343–1358. [Google Scholar] [CrossRef]
- Johnson, J.M.; Khoshgoftaar, T.M. Survey on deep learning with class imbalance. J. Big Data 2019, 6, 1–54. [Google Scholar] [CrossRef]
- Hittmeir, M.; Mayer, R.; Ekelhart, A. Utility and privacy assessment of synthetic microbiome data. In Proceedings of the IFIP Annual Conference on Data and Applications Security and Privacy, Newark, NJ, USA, 18–20 July 2022; Springer: Cham, Switzerland, 2022; pp. 15–27. [Google Scholar]
- Patki, N.; Wedge, R.; Veeramachaneni, K. The synthetic data vault. In Proceedings of the 2016 IEEE International Conference on Data Science and Advanced Analytics (DSAA), Montreal, QC, Canada, 17–19 October 2016; pp. 399–410. [Google Scholar]
- Karthikeyan, R.; Alli, P. Feature selection and parameters optimization of support vector machines based on hybrid glowworm swarm optimization for classification of diabetic retinopathy. J. Med. Syst. 2018, 42, 195. [Google Scholar] [CrossRef]
- Chen, R.J.; Lu, M.Y.; Chen, T.Y.; Williamson, D.F.; Mahmood, F. Synthetic data in machine learning for medicine and healthcare. Nat. Biomed. Eng. 2021, 5, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Montavon, G.; Binder, A.; Lapuschkin, S.; Samek, W.; Müller, K.R. Layer-wise relevance propagation: An overview. In Explainable AI: Interpreting, Explaining and Visualizing Deep Learning; Springer: Cham, Switzerland, 2019; pp. 193–209. [Google Scholar]
- Nanni, L.; Brahnam, S.; Paci, M.; Ghidoni, S. Comparison of different convolutional neural network activation functions and methods for building ensembles for small to midsize medical data sets. Sensors 2022, 22, 6129. [Google Scholar] [CrossRef]
- Peng, X.; Cheng, L.; You, Y.; Tang, C.; Ren, B.; Li, Y.; Xu, X.; Zhou, X. Oral microbiota in human systematic diseases. Int. J. Oral Sci. 2022, 14, 14. [Google Scholar] [CrossRef]
- Giuffrè, M.; Shung, D.L. Harnessing the power of synthetic data in healthcare: Innovation, application, and privacy. Npj Digit. Med. 2023, 6, 186. [Google Scholar] [CrossRef]
- Liu, J.; Huang, X.; Chen, C.; Wang, Z.; Huang, Z.; Qin, M.; He, F.; Tang, B.; Long, C.; Hu, H.; et al. Identification of colorectal cancer progression-associated intestinal microbiome and predictive signature construction. J. Transl. Med. 2023, 21, 373. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Huttenhower, C. Dysbiosis of the fecal microbiota in patients with colorectal cancer. Genome Res. 2012, 22, 442–448. [Google Scholar]
- Parker, B.J.; Wearsch, P.A.; Velasquez, M.T. The role of the gut microbiome in colorectal cancer risk: Alcohol, diet and inflammation. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 345–356. [Google Scholar]
- Chen, J.; Vitetta, L. Gut microbiota and colorectal cancer: From pathogenesis to therapy. Cell Death Dis. 2021, 12, 1–15. [Google Scholar]
- Löwenmark, T.; Löfgren-Burström, A.; Zingmark, C.; Eklöf, V.; Dahlberg, M.; Wai, S.N.; Larsson, P.; Ljuslinder, I.; Edin, S.; Palmqvist, R. Parvimonas micra as a putative non-invasive faecal biomarker for colorectal cancer. Sci. Rep. 2020, 10, 15250. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Bohörquez, L.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Mitsuhashi, K.; Nosho, K.; Sukawa, Y.; Matsunaga, Y.; Ito, M.; Kurihara, H.; Kanno, S.; Igarashi, H.; Naito, T.; Adachi, Y.; et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget 2015, 6, 7209–7220. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Gui, X.; Zhang, Y.; Zhang, Z.; Chen, W.; Zhang, X.; Wang, Y.; Zhang, M.; Shang, Z.; et al. Salivary Fusobacterium nucleatum serves as a potential biomarker for colorectal cancer. iScience 2022, 25, 104203. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, Y.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Wong, S.Y.; Yu, J. Quantification of Fusobacterium nucleatum in colorectal cancer tissues reveals a link with epithelial-mesenchymal transition. Gut Pathog. 2017, 9, 1–7. [Google Scholar]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hyperparameter | Tested Values |
|---|---|
| Model Architecture | Dense (64) → ReLU/LeakyReLU ( = 0.01)/Softmax/GeLU Dense (32) → ReLU/LeakyReLU ( = 0.01)/Softmax/GeLU Dense(1,Sigmoid) |
| Optimizer | Adam |
| Loss Score | Binary Crossentropy |
| Batch Size | 32 |
| Epochs | 45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotelli, A.; Salman, A.; Di Gloria, L.; Nannini, G.; Niccolai, E.; Luschi, A.; Amedei, A.; Iadanza, E. Analysis of Microbiome for AP and CRC Discrimination. Bioengineering 2025, 12, 713. https://doi.org/10.3390/bioengineering12070713
Rotelli A, Salman A, Di Gloria L, Nannini G, Niccolai E, Luschi A, Amedei A, Iadanza E. Analysis of Microbiome for AP and CRC Discrimination. Bioengineering. 2025; 12(7):713. https://doi.org/10.3390/bioengineering12070713
Chicago/Turabian StyleRotelli, Alessio, Ali Salman, Leandro Di Gloria, Giulia Nannini, Elena Niccolai, Alessio Luschi, Amedeo Amedei, and Ernesto Iadanza. 2025. "Analysis of Microbiome for AP and CRC Discrimination" Bioengineering 12, no. 7: 713. https://doi.org/10.3390/bioengineering12070713
APA StyleRotelli, A., Salman, A., Di Gloria, L., Nannini, G., Niccolai, E., Luschi, A., Amedei, A., & Iadanza, E. (2025). Analysis of Microbiome for AP and CRC Discrimination. Bioengineering, 12(7), 713. https://doi.org/10.3390/bioengineering12070713