
New Insights into the Role of Cellular Senescence and Its Therapeutic Implications in Ocular Diseases


Abstract

1. Introduction
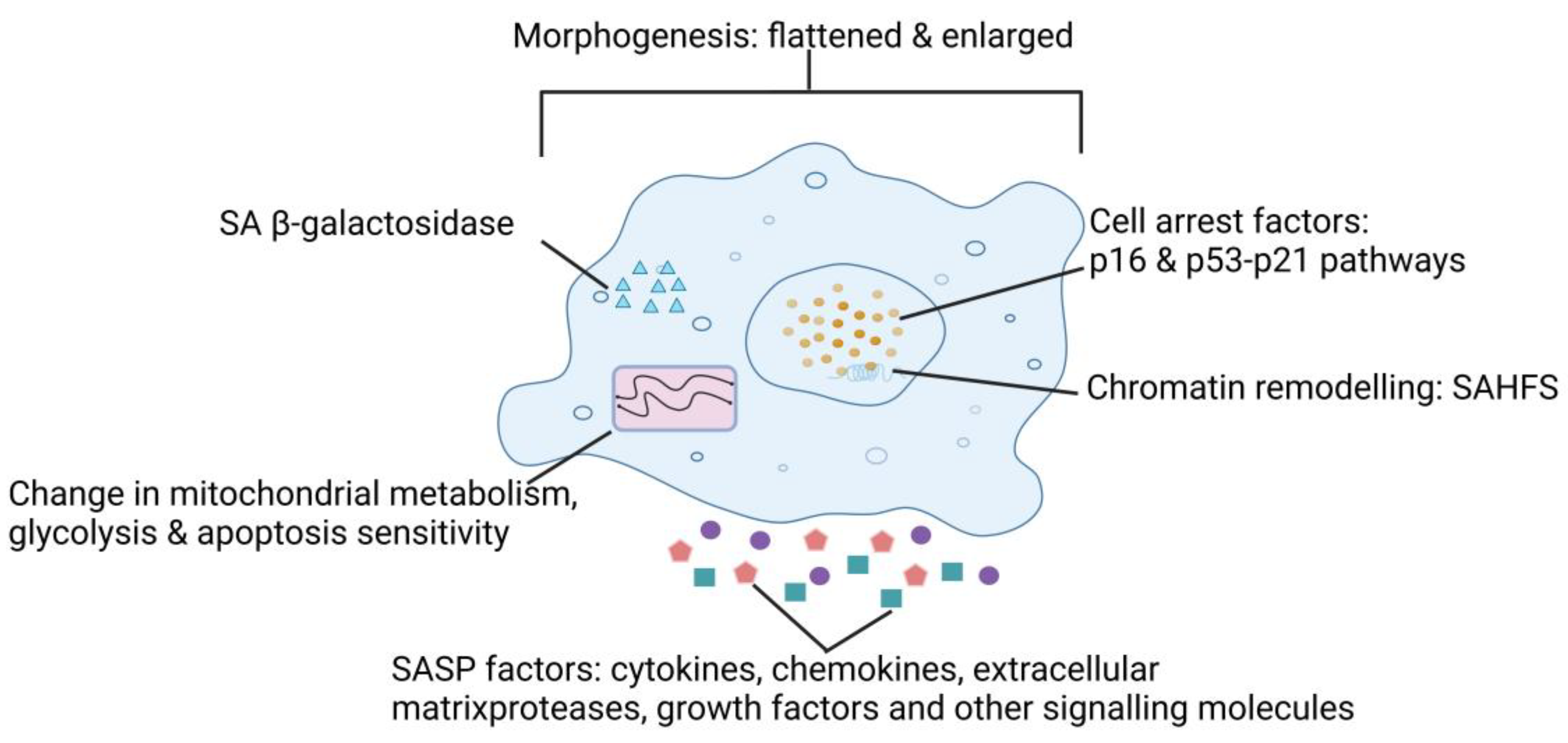
2. Characteristics of SnCs
3. Brief Introduction and Classification of SASP
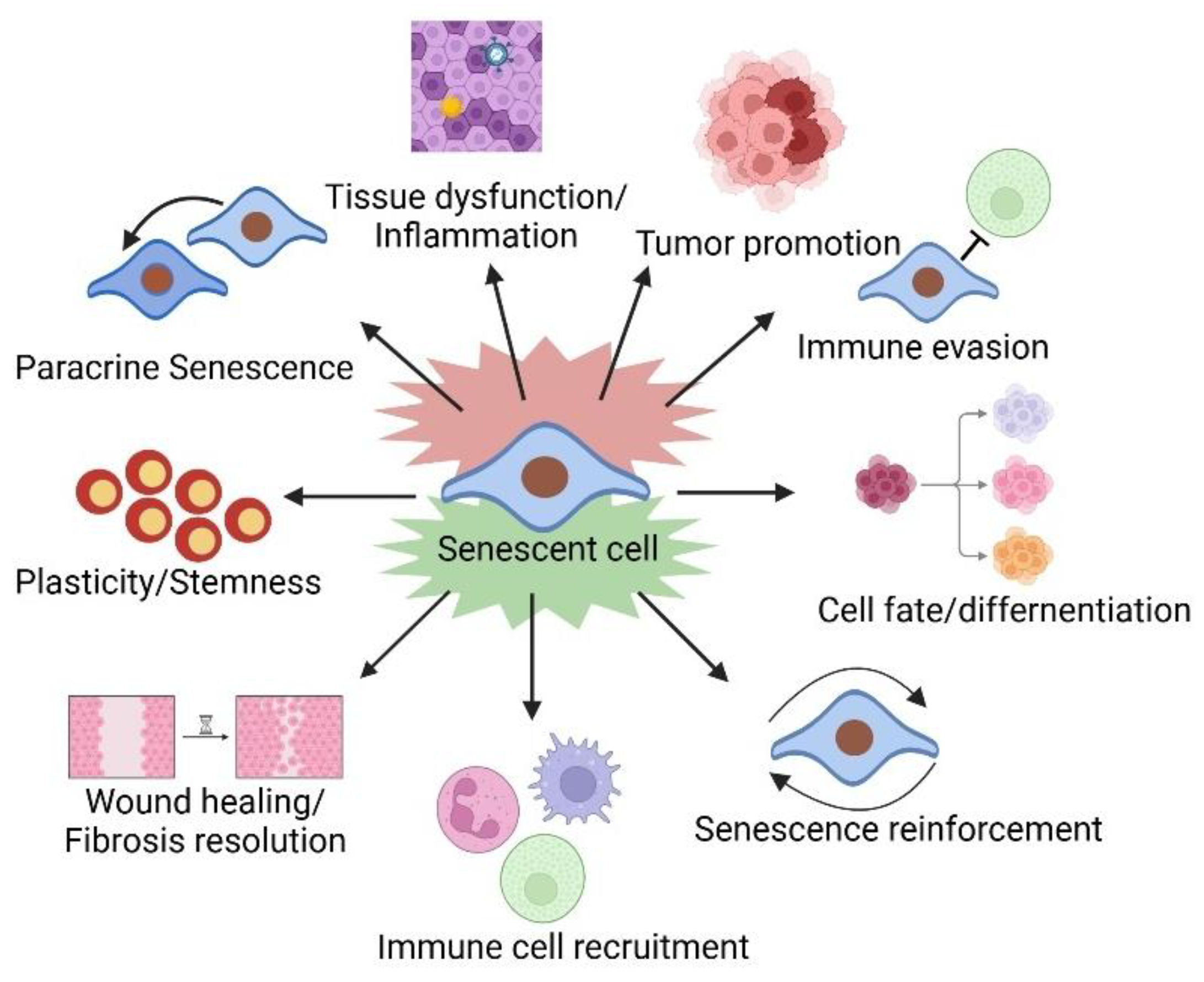
4. The Pleiotropic Effects of the SASP
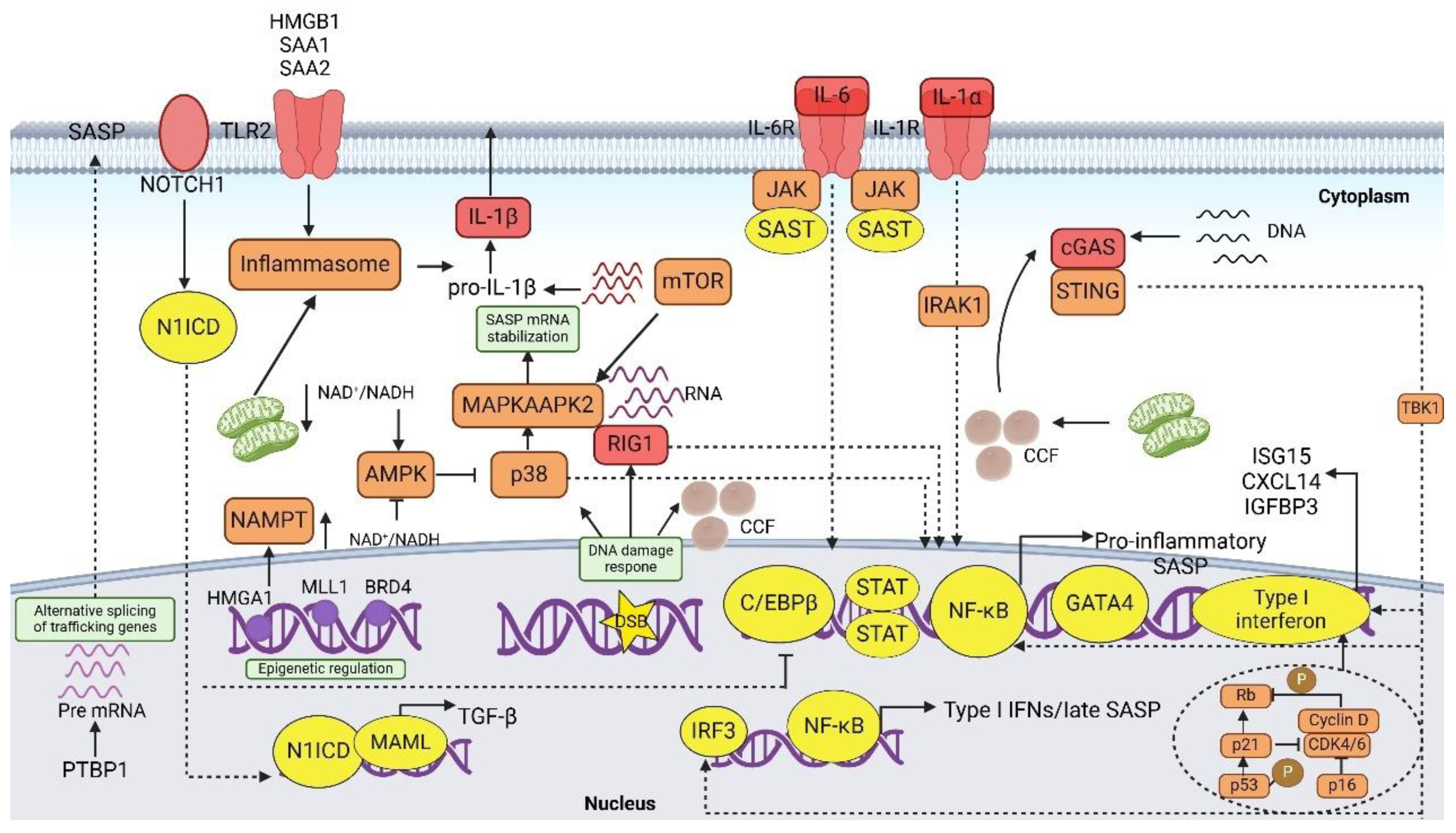
5. Regulation of the SASP
5.1. SASP Induction
5.2. Signaling Pathway
6. Physiological Features of SnCs in the Eye

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Senescent Cell Type/Mechanism | Key Senescence Markers | Representative SASP Components | Emerging Therapeutic Implications | References |
|---|---|---|---|---|---|
| DR | Endothelial cell senescence in retinal microvasculature; SASP-driven vascular dysfunction | p16INK4a, p21, p53, IGFBP3 | IL-6, IL-1β, VEGF, MCP-1 | Senolytics (UBX1325), cGAS–STING inhibitors, antioxidants, IL-6 trans-signaling blockade | [138,139] |
| AMD | Senescence of RPE cells; SASP-driven inflammation and drusen accumulation | p16, p21, p53, BMP4 | IL-6, IL-8, MMPs, VEGF, complement proteins | Senolytics, autophagy enhancers, Piezo1-BMP4 inhibitors, PGAM5 modulators | [64,140] |
| Glaucoma | Senescence in trabecular meshwork and RGCs; increased outflow resistance leads to elevated intraocular pressure | p53, p21, p16, TGF-β1 | IL-6, IL-8, MMP-9, TNF-α | Senolytics (Dasatinib, Quercetin), mTOR inhibitors, mitochondrial antioxidants, OSK factors | [65,141] |
| Cataracts | Senescence in lens epithelial cells; autophagy dysfunction and oxidative stress-induced protein aggregation | p21, LC3, p62 | IL-6, ROS | HO-1/TFEB axis activators, autophagy modulators, antioxidant therapy | [142] |
| DED | Senescence-associated inflammation in lacrimal and meibomian glands; SASP-driven immune dysregulation | p16, 8-OHdG, SASP cytokines | IL-1β, MMP-3, MMP-9, IL-6 | Senomorphic eye drops, mesenchymal stem cell therapy, mTOR inhibitors, mitochondrial transfer | [66] |
| Autoimmune/Inflammatory Eye Diseases | Senescence-associated immune dysfunction contributes to chronic inflammation in autoimmune uveitis | p16, p21, IFN-γ, IL-17A | GM-CSF, IL-17, IFN-γ, TNF-α | JAK-STAT inhibitors, immunosenescence modulators, Foxp3+ Treg reprogramming | [143,144] |
7. Mechanistic Insights into Cellular Senescence in Ocular Diseases
7.1. Diabetic Retinal Disease
7.2. Age-Related Macular Degeneration (AMD)
7.3. Glaucoma
7.4. Age-Related Cataract
7.5. Dry Eye Disease
7.6. Autoimmune and Inflammatory Eye Diseases
8. Clinical Applications of Senescence-Targeting Strategies in Ocular Diseases
8.1. Senolytics
8.2. Senomorphics
8.3. Treatment Strategies for Diabetes-Related Retinopathy
8.4. Treatment Strategies for AMD
8.5. Treatment Strategies for Glaucoma
8.6. Treatment Strategies for Cataract
8.7. Treatment Strategies for Ocular Surface Diseases
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMD | Age-related macular degeneration |
| AMPK | AMP-activated protein kinase |
| ARNC | Age-related nuclear cataract |
| AU | Autoimmune uveitis |
| BCL-xL | B cell lymphoma-extra large |
| BET | Bromodomain and extraterminal |
| BETd | BET family protein degraders |
| C/EBP | CCAAT/enhancer-binding protein |
| CCFs | Cytoplasmic chromatin fragments |
| CDKs | Cyclin-dependent kinases |
| cGAS–STING | Cyclic GMP-AMP synthase–stimulator of interferon genes |
| CNS | Central nervous system |
| DAMPs | Damage-associated molecular patterns |
| DBD | DNA-binding domain |
| DDR | DNA damage response |
| DED | Dry eye disease |
| DME | Diabetic macular edema |
| DPS | Developmentally programmed senescence |
| DR | Diabetic retinopathy |
| DSBs | Double-strand breaks |
| EAU | Experimental autoimmune uveitis |
| EMT | Epithelial-to-mesenchymal transition |
| EVs | Extracellular vesicles |
| FED | Fuchs’ endothelial dystrophy |
| GWAS | Genome-wide association studies |
| HCC | Hepatocellular carcinoma |
| HMGB1 | High mobility group box 1 protein |
| HUVECs | Human umbilical vein endothelial cells |
| IGFBP | Insulin-like growth factor-binding proteins |
| INL | Inner nuclear layer |
| IOP | Intraocular pressure |
| LECs | Lens epithelial cells |
| LNs | Lymph nodes |
| MCJ | mucocutaneous junction |
| NHEJ | Non-homologous end joining |
| NK | Natural killer |
| OIS | Oncogene-induced senescence |
| PAI | Plasminogen activator inhibitor |
| PARP1 | Polymerase 1 |
| PCC1 | Procyanidin C1 |
| PDR | Proliferative diabetic retinopathy |
| PKD | Protein kinase D |
| PML | Promyelocytic leukemia protein |
| POAG | Primary open-angle glaucoma |
| RECs | Retinal endothelial cells |
| RGCs | Retinal ganglion cells |
| RONS | Reactive oxygen and nitrogen species |
| ROS | Reactive oxygen species |
| RPE | Retinal pigment epithelium |
| RS | Replicative senescence |
| SAHF | Senescence-associated heterochromatic foci |
| SASP | Senescence-associated secretory phenotype |
| SIPS | Stress-induced premature senescence |
| SnCs | Senescent cells |
| STING | Stimulator of interferon genes |
| TAD | Transactivation domain |
| TFEB | Transcription factor EB |
| TIMP | Tissue inhibitors of metalloproteases |
| TIS | Therapy-induced senescence |
| TOP1cc | Topoisomerase 1 cleavage complexes |
| TSPCs | Tendon stem/progenitor cells |
| UPS | Ubiquitin-proteasome system |
| VEGF | Vascular endothelial growth factor |
References
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes. Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M.; Cheraqpour, K.; Koganti, R.; Djalilian, A.R. Cellular senescence and ophthalmic diseases: Narrative review. Graefes Arch. Clin. Exp. Ophthalmol. 2023, 261, 3067–3082. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Luo, L.J.; Yang, C.J.; Lai, J.Y. Highly Retina-Permeating and Long-Acting Resveratrol/Metformin Nanotherapeutics for Enhanced Treatment of Macular Degeneration. ACS Nano 2023, 17, 168–183. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Liao, Z.; Yeo, H.L.; Wong, S.W.; Zhao, Y. Cellular Senescence: Mechanisms and Therapeutic Potential. Biomedicines 2021, 9, 1769. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Ramos, I.; Stamatakis, K.; Oeste, C.L.; Pérez-Sala, D. Vimentin as a Multifaceted Player and Potential Therapeutic Target in Viral Infections. Int. J. Mol. Sci. 2020, 21, 4675. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Yun, M.H.; Davaapil, H.; Brockes, J.P. Recurrent turnover of senescent cells during regeneration of a complex structure. Elife 2015, 4, e05505. [Google Scholar] [CrossRef]
- Ramakrishna, G.; Anwar, T.; Angara, R.K.; Chatterjee, N.; Kiran, S.; Singh, S. Role of cellular senescence in hepatic wound healing and carcinogenesis. Eur. J. Cell Biol. 2012, 91, 739–747. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Hoare, M.; Narita, M. Transmitting senescence to the cell neighbourhood. Nat. Cell Biol. 2013, 15, 887–889. [Google Scholar] [CrossRef]
- Byun, H.O.; Lee, Y.K.; Kim, J.M.; Yoon, G. From cell senescence to age-related diseases: Differential mechanisms of action of senescence-associated secretory phenotypes. BMB Rep. 2015, 48, 549–558. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Hinton, D.R.; Kannan, R. The Emerging Role of Senescence in Ocular Disease. Oxid. Med. Cell Longev. 2020, 2020, 2583601. [Google Scholar] [CrossRef]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes. Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef]
- Sheekey, E.; Narita, M. p53 in senescence—it’s a marathon, not a sprint. FEBS J. 2023, 290, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Kupnicka, P.; Chlubek, M.; Gorący, J.; Gutowska, I.; Baranowska-Bosiacka, I. CXCR2 Receptor: Regulation of Expression, Signal Transduction, and Involvement in Cancer. Int. J. Mol. Sci. 2022, 23, 2168. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Development 2019, 146, dev151837. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes. Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Soni, S.; Rosas, L.; Rojas, M.; Horowitz, J.C.; Nho, R. The aged extracellular matrix and the profibrotic role of senescence-associated secretory phenotype. Am. J. Physiol. Cell Physiol. 2023, 325, C565–C579. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Abbas, M.; Jesel, L.; Auger, C.; Amoura, L.; Messas, N.; Manin, G.; Rumig, C.; León-González, A.J.; Ribeiro, T.P.; Silva, G.C.; et al. Endothelial Microparticles From Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the Ang II/AT1 Receptor/NADPH Oxidase-Mediated Activation of MAPKs and PI3-Kinase Pathways. Circulation 2017, 135, 280–296. [Google Scholar] [CrossRef]
- Davis, C.; Dukes, A.; Drewry, M.; Helwa, I.; Johnson, M.H.; Isales, C.M.; Hill, W.D.; Liu, Y.; Shi, X.; Fulzele, S.; et al. MicroRNA-183-5p Increases with Age in Bone-Derived Extracellular Vesicles, Suppresses Bone Marrow Stromal (Stem) Cell Proliferation, and Induces Stem Cell Senescence. Tissue Eng. Part A 2017, 23, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef] [PubMed]
- Weilner, S.; Schraml, E.; Wieser, M.; Messner, P.; Schneider, K.; Wassermann, K.; Micutkova, L.; Fortschegger, K.; Maier, A.B.; Westendorp, R.; et al. Secreted microvesicular miR-31 inhibits osteogenic differentiation of mesenchymal stem cells. Aging Cell 2016, 15, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Borras, C.; Mas-Bargues, C.; Sanz-Ros, J.; Román-Domínguez, A.; Gimeno-Mallench, L.; Inglés, M.; Gambini, J.; Viña, J. Extracellular vesicles and redox modulation in aging. Free Radic. Biol. Med. 2020, 149, 44–50. [Google Scholar] [CrossRef]
- O’Loghlen, A. Role for extracellular vesicles in the tumour microenvironment. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160488. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Würdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes. Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Liu, S.; Limbad, C.; Zawadzka, A.M.; Beck, J.; Demaria, M.; Artwood, R.; Alimirah, F.; Lopez-Dominguez, J.A.; Kuehnemann, C.; et al. SILAC Analysis Reveals Increased Secretion of Hemostasis-Related Factors by Senescent Cells. Cell Rep. 2019, 28, 3329–3337.e3325. [Google Scholar] [CrossRef]
- Ohtani, N. The roles and mechanisms of senescence-associated secretory phenotype (SASP): Can it be controlled by senolysis? Inflamm. Regen. 2022, 42, 11. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, X.; Le, W.; Xie, L.; Li, H.; Wen, W.; Wang, S.; Ma, S.; Huang, Z.; Ye, J.; et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell 2020, 11, 740–770. [Google Scholar] [CrossRef]
- Hadziahmetovic, M.; Malek, G. Age-Related Macular Degeneration Revisited: From Pathology and Cellular Stress to Potential Therapies. Front. Cell Dev. Biol. 2020, 8, 612812. [Google Scholar] [CrossRef]
- Guo, L.; Wang, N.; Chen, J.; Zhang, R.; Li, D.; Yang, L. Cellular senescence and glaucoma. Exp. Gerontol. 2025, 202, 112718. [Google Scholar] [CrossRef]
- Bu, J.; Liu, Y.; Zhang, R.; Lin, S.; Zhuang, J.; Sun, L.; Zhang, L.; He, H.; Zong, R.; Wu, Y.; et al. Potential New Target for Dry Eye Disease-Oxidative Stress. Antioxidants 2024, 13, 422. [Google Scholar] [CrossRef]
- Wang, B.; Varela-Eirin, M.; Brandenburg, S.M.; Hernandez-Segura, A.; van Vliet, T.; Jongbloed, E.M.; Wilting, S.M.; Ohtani, N.; Jager, A.; Demaria, M. Pharmacological CDK4/6 inhibition reveals a p53-dependent senescent state with restricted toxicity. EMBO J. 2022, 41, e108946. [Google Scholar] [CrossRef]
- Wiley, C.D.; Sharma, R.; Davis, S.S.; Lopez-Dominguez, J.A.; Mitchell, K.P.; Wiley, S.; Alimirah, F.; Kim, D.E.; Payne, T.; Rosko, A.; et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell Metab. 2021, 33, 1124–1136.e1125. [Google Scholar] [CrossRef]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef]
- Borghesan, M.; Fafián-Labora, J.; Eleftheriadou, O.; Carpintero-Fernández, P.; Paez-Ribes, M.; Vizcay-Barrena, G.; Swisa, A.; Kolodkin-Gal, D.; Ximénez-Embún, P.; Lowe, R.; et al. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM3. Cell Rep. 2019, 27, 3956–3971.e3956. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Crescenzi, E.; Pacifico, F.; Lavorgna, A.; De Palma, R.; D’Aiuto, E.; Palumbo, G.; Formisano, S.; Leonardi, A. NF-κB-dependent cytokine secretion controls Fas expression on chemotherapy-induced premature senescent tumor cells. Oncogene 2011, 30, 2707–2717. [Google Scholar] [CrossRef]
- Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene 2011, 30, 2356–2366. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Kolesnichenko, M.; Mikuda, N.; Höpken, U.E.; Kärgel, E.; Uyar, B.; Tufan, A.B.; Milanovic, M.; Sun, W.; Krahn, I.; Schleich, K.; et al. Transcriptional repression of NFKBIA triggers constitutive IKK- and proteasome-independent p65/RelA activation in senescence. EMBO J. 2021, 40, e104296. [Google Scholar] [CrossRef]
- Alexander, E.; Hildebrand, D.G.; Kriebs, A.; Obermayer, K.; Manz, M.; Rothfuss, O.; Schulze-Osthoff, K.; Essmann, F. IκBζ is a regulator of the senescence-associated secretory phenotype in DNA damage- and oncogene-induced senescence. J. Cell Sci. 2013, 126, 3738–3745. [Google Scholar] [CrossRef]
- Salotti, J.; Johnson, P.F. Regulation of senescence and the SASP by the transcription factor C/EBPβ. Exp. Gerontol. 2019, 128, 110752. [Google Scholar] [CrossRef]
- Wang, P.; Han, L.; Shen, H.; Wang, P.; Lv, C.; Zhao, G.; Niu, J.; Xue, L.; Wang, Q.J.; Tong, T.; et al. Protein kinase D1 is essential for Ras-induced senescence and tumor suppression by regulating senescence-associated inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, 7683–7688. [Google Scholar] [CrossRef] [PubMed]
- Chong, M.; Yin, T.; Chen, R.; Xiang, H.; Yuan, L.; Ding, Y.; Pan, C.C.; Tang, Z.; Alexander, P.B.; Li, Q.J.; et al. CD36 initiates the secretory phenotype during the establishment of cellular senescence. EMBO Rep. 2018, 19, e45274. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, Y.; Yuan, W.W.; Yin, Y.; Song, W.W.; Wang, Y.; Huang, Q.Q.; Zhao, W.H.; Wu, J.Q. Membrane-Bound CD40L Promotes Senescence and Initiates Senescence-Associated Secretory Phenotype via NF-κB Activation in Lung Adenocarcinoma. Cell Physiol. Biochem. 2018, 48, 1793–1803. [Google Scholar] [CrossRef]
- Cinat, D.; Coppes, R.P.; Barazzuol, L. DNA Damage-Induced Inflammatory Microenvironment and Adult Stem Cell Response. Front. Cell Dev. Biol. 2021, 9, 729136. [Google Scholar] [CrossRef]
- Wang, B.; Han, J.; Elisseeff, J.H.; Demaria, M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell Biol. 2024, 25, 958–978. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci. Rep. 2018, 8, 2410. [Google Scholar] [CrossRef]
- Efeyan, A.; Ortega-Molina, A.; Velasco-Miguel, S.; Herranz, D.; Vassilev, L.T.; Serrano, M. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 2007, 67, 7350–7357. [Google Scholar] [CrossRef]
- Coppé, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Zhang, C.; Sine, C.C.; van Deursen, E.J.; Jeganathan, K.B.; Hamada, N.; Grasic, J.; Friedman, D.; Stutchman, J.T.; Can, I.; et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 2021, 374, eabb3420. [Google Scholar] [CrossRef]
- Buj, R.; Leon, K.E.; Anguelov, M.A.; Aird, K.M. Suppression of p16 alleviates the senescence-associated secretory phenotype. Aging 2021, 13, 3290–3312. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Flanagan, K.C.; Luo, X.; Ruhland, M.K.; Huang, H.; Pazolli, E.; Donlin, M.J.; Marsh, T.; Piwnica-Worms, D.; Monahan, J.; et al. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 2014, 4, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Fu, D.; Xu, Q.; Cong, X.; Wu, C.; Zhong, X.; Ma, Y.; Lv, Z.; Chen, F.; Han, L.; et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat. Commun. 2018, 9, 1723. [Google Scholar] [CrossRef]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol. Cell 2021, 81, 2041–2052.e2046. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef]
- Habibi-Kavashkohie, M.R.; Scorza, T.; Oubaha, M. Senescent Cells: Dual Implications on the Retinal Vascular System. Cells 2023, 12, 2341. [Google Scholar] [CrossRef]
- Wang, W.; Xia, X.; Mao, L.; Wang, S. The CCAAT/Enhancer-Binding Protein Family: Its Roles in MDSC Expansion and Function. Front. Immunol. 2019, 10, 1804. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Huggins, C.J.; Malik, R.; Lee, S.; Salotti, J.; Thomas, S.; Martin, N.; Quiñones, O.A.; Alvord, W.G.; Olanich, M.E.; Keller, J.R.; et al. C/EBPγ suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPβ. Mol. Cell Biol. 2013, 33, 3242–3258. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and Temporal Control of Senescence. Trends Cell Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Kirkland, J.L. Perspective: Targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol. Res. 2016, 111, 152–154. [Google Scholar] [CrossRef]
- Chen, M.; Xiao, L.; Dai, G.; Lu, P.; Zhang, Y.; Li, Y.; Ni, M.; Rui, Y. Inhibition of JAK-STAT Signaling Pathway Alleviates Age-Related Phenotypes in Tendon Stem/Progenitor Cells. Front. Cell Dev. Biol. 2021, 9, 650250. [Google Scholar] [CrossRef]
- Luo, W.; Li, Y.X.; Jiang, L.J.; Chen, Q.; Wang, T.; Ye, D.W. Targeting JAK-STAT Signaling to Control Cytokine Release Syndrome in COVID-19. Trends Pharmacol. Sci. 2020, 41, 531–543. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 2018, 9, 1249. [Google Scholar] [CrossRef]
- Zhao, B.; Liu, P.; Fukumoto, T.; Nacarelli, T.; Fatkhutdinov, N.; Wu, S.; Lin, J.; Aird, K.M.; Tang, H.Y.; Liu, Q.; et al. Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nat. Commun. 2020, 11, 908. [Google Scholar] [CrossRef]
- Omer, A.; Barrera, M.C.; Moran, J.L.; Lian, X.J.; Di Marco, S.; Beausejour, C.; Gallouzi, I.E. G3BP1 controls the senescence-associated secretome and its impact on cancer progression. Nat. Commun. 2020, 11, 4979. [Google Scholar] [CrossRef] [PubMed]
- Hari, P.; Millar, F.R.; Tarrats, N.; Birch, J.; Quintanilla, A.; Rink, C.J.; Fernández-Duran, I.; Muir, M.; Finch, A.J.; Brunton, V.G.; et al. The innate immune sensor Toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci. Adv. 2019, 5, eaaw0254. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Li, R.; Liang, Y.; Lin, B. Accumulation of systematic TPM1 mediates inflammation and neuronal remodeling by phosphorylating PKA and regulating the FABP5/NF-κB signaling pathway in the retina of aged mice. Aging Cell 2022, 21, e13566. [Google Scholar] [CrossRef]
- Samuel, M.A.; Voinescu, P.E.; Lilley, B.N.; de Cabo, R.; Foretz, M.; Viollet, B.; Pawlyk, B.; Sandberg, M.A.; Vavvas, D.G.; Sanes, J.R. LKB1 and AMPK regulate synaptic remodeling in old age. Nat. Neurosci. 2014, 17, 1190–1197. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Chen, X.; Yang, Z.; Chen, J.; Zhu, W.; Li, Y.; Wen, Y.; Deng, C.; Gu, C.; et al. Senolytic and senomorphic agent procyanidin C1 alleviates structural and functional decline in the aged retina. Proc. Natl. Acad. Sci. USA 2024, 121, e2311028121. [Google Scholar] [CrossRef]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990-2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef]
- Liu, H.; Prokosch, V. Energy Metabolism in the Inner Retina in Health and Glaucoma. Int. J. Mol. Sci. 2021, 22, 3689. [Google Scholar] [CrossRef] [PubMed]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018, 28, 595–607. [Google Scholar] [CrossRef]
- Caprioli, J. Glaucoma: A disease of early cellular senescence. Investig. Ophthalmol. Vis. Sci. 2013, 54, Orsf60–Orsf67. [Google Scholar] [CrossRef]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef]
- Yazdankhah, M.; Shang, P.; Ghosh, S.; Hose, S.; Liu, H.; Weiss, J.; Fitting, C.S.; Bhutto, I.A.; Zigler, J.S., Jr.; Qian, J.; et al. Role of glia in optic nerve. Prog. Retin. Eye Res. 2021, 81, 100886. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Ashok, A.; Pooranawattanakul, S.; Tai, W.L.; Cho, K.S.; Utheim, T.P.; Cestari, D.M.; Chen, D.F. Epigenetic Regulation of Optic Nerve Development, Protection, and Repair. Int. J. Mol. Sci. 2022, 23, 8927. [Google Scholar] [CrossRef]
- Cooper, M.L.; Pasini, S.; Lambert, W.S.; D’Alessandro, K.B.; Yao, V.; Risner, M.L.; Calkins, D.J. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 18810–18821. [Google Scholar] [CrossRef] [PubMed]
- García-Bermúdez, M.Y.; Freude, K.K.; Mouhammad, Z.A.; van Wijngaarden, P.; Martin, K.K.; Kolko, M. Glial Cells in Glaucoma: Friends, Foes, and Potential Therapeutic Targets. Front. Neurol. 2021, 12, 624983. [Google Scholar] [CrossRef] [PubMed]
- Fernandez de Castro, J.P.; Mullins, R.F.; Manea, A.M.; Hernandez, J.; Wallen, T.; Kuehn, M.H. Lipofuscin in human glaucomatous optic nerves. Exp. Eye Res. 2013, 111, 61–66. [Google Scholar] [CrossRef]
- Ma, W.; Wong, W.T. Aging Changes in Retinal Microglia and their Relevance to Age-related Retinal Disease. Adv. Exp. Med. Biol. 2016, 854, 73–78. [Google Scholar] [CrossRef]
- Zhu, J.D.; Tarachand, S.P.; Abdulwahab, Q.; Samuel, M.A. Structure, Function, and Molecular Landscapes of the Aging Retina. Annu. Rev. Vis. Sci. 2023, 9, 177–199. [Google Scholar] [CrossRef]
- Chaum, E.; Winborn, C.S.; Bhattacharya, S. Genomic regulation of senescence and innate immunity signaling in the retinal pigment epithelium. Mamm. Genome 2015, 26, 210–221. [Google Scholar] [CrossRef]
- Yan, Q.; Ding, Y.; Weeks, D.E.; Chen, W. AMD Genetics: Methods and Analyses for Association, Progression, and Prediction. Adv. Exp. Med. Biol. 2021, 1256, 191–200. [Google Scholar] [CrossRef]
- Liu, H.; Ghosh, S.; Vaidya, T.; Bammidi, S.; Huang, C.; Shang, P.; Nair, A.P.; Chowdhury, O.; Stepicheva, N.A.; Strizhakova, A.; et al. Activated cGAS/STING signaling elicits endothelial cell senescence in early diabetic retinopathy. JCI Insight 2023, 8, e168945. [Google Scholar] [CrossRef]
- Iwasaki, K.; Abarca, C.; Aguayo-Mazzucato, C. Regulation of Cellular Senescence in Type 2 Diabetes Mellitus: From Mechanisms to Clinical Applications. Diabetes Metab. J. 2023, 47, 441–453. [Google Scholar] [CrossRef]
- Blasiak, J.; Sobczuk, P.; Pawlowska, E.; Kaarniranta, K. Interplay between aging and other factors of the pathogenesis of age-related macular degeneration. Ageing Res. Rev. 2022, 81, 101735. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiong, Y.; Li, R.; Wang, X.; Lin, X.; Tong, Y. Targeting cGAS-STING signaling protects retinal ganglion cells from DNA damage-induced cell loss and promotes visual recovery in glaucoma. Aging 2024, 16, 9813–9823. [Google Scholar] [CrossRef]
- Wishart, T.F.L.; Flokis, M.; Shu, D.Y.; Das, S.J.; Lovicu, F.J. Hallmarks of lens aging and cataractogenesis. Exp. Eye Res. 2021, 210, 108709. [Google Scholar] [CrossRef]
- Li, Z.; Li, Z.; Hu, Y.; Xie, Y.; Shi, Y.; Chen, G.; Huang, J.; Xiao, Z.; Zhu, W.; Huang, H.; et al. Neutrophil extracellular traps potentiate effector T cells via endothelial senescence in uveitis. JCI Insight 2025, 10, e180248. [Google Scholar] [CrossRef] [PubMed]
- Geertsema-Hoeve, B.C.; van Laar, J.A.M.; Raaphorst, J.; Tas, S.W.; Welsing, P.M.J.; Goekoop, R.J.; Checa, C.M.; Thurlings, R.M.; Rekers, N.H.; Present, E.; et al. Multicentre, 26-week, open-label phase 2 trial of the JAK inhibitor filgotinib in Behçet’s disease, idiopathic inflammatory myopathies and IgG4-related disease: DRIMID study protocol. BMJ Open 2025, 15, e089827. [Google Scholar] [CrossRef]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Silva, P.S.; Stitt, A.W. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat. Rev. Endocrinol. 2021, 17, 195–206. [Google Scholar] [CrossRef]
- Levine, S.R.; Sapieha, P.; Dutta, S.; Sun, J.K.; Gardner, T.W. It is time for a moonshot to find “Cures” for diabetic retinal disease. Prog. Retin. Eye Res. 2022, 90, 101051. [Google Scholar] [CrossRef]
- Yu, D.Y.; Cringle, S.J.; Yu, P.K.; Balaratnasingam, C.; Mehnert, A.; Sarunic, M.V.; An, D.; Su, E.N. Retinal capillary perfusion: Spatial and temporal heterogeneity. Prog. Retin. Eye Res. 2019, 70, 23–54. [Google Scholar] [CrossRef]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060.e1010. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e124. [Google Scholar] [CrossRef]
- Su, L.; Kong, X.; Loo, S.J.; Gao, Y.; Kovalik, J.P.; Su, X.; Ma, J.; Ye, L. Diabetic Endothelial Cells Differentiated From Patient iPSCs Show Dysregulated Glycine Homeostasis and Senescence Associated Phenotypes. Front. Cell Dev. Biol. 2021, 9, 667252. [Google Scholar] [CrossRef] [PubMed]
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, eaay5356. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 2021, 33, 818–832.e817. [Google Scholar] [CrossRef]
- Shosha, E.; Xu, Z.; Narayanan, S.P.; Lemtalsi, T.; Fouda, A.Y.; Rojas, M.; Xing, J.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. Int. J. Mol. Sci. 2018, 19, 1215. [Google Scholar] [CrossRef]
- Frisch, S.M.; MacFawn, I.P. Type I interferons and related pathways in cell senescence. Aging Cell 2020, 19, e13234. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Lettice, L.; Tarnauskaitė, Ž.; Reddy, K.; Dix, F.; Revuelta, A.; Abbondati, E.; Rigby, R.E.; Rabe, B.; et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 2016, 35, 831–844. [Google Scholar] [CrossRef]
- Gentili, M.; Lahaye, X.; Nadalin, F.; Nader, G.P.F.; Puig Lombardi, E.; Herve, S.; De Silva, N.S.; Rookhuizen, D.C.; Zueva, E.; Goudot, C.; et al. The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep. 2019, 26, 2377–2393.e2313. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Jonas, J.B.; Cheung, C.M.G.; Panda-Jonas, S. Updates on the Epidemiology of Age-Related Macular Degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 493–497. [Google Scholar] [CrossRef]
- Thomas, C.J.; Mirza, R.G.; Gill, M.K. Age-Related Macular Degeneration. Med. Clin. N. Am. 2021, 105, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Chirco, K.R.; Sohn, E.H.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Structural and molecular changes in the aging choroid: Implications for age-related macular degeneration. Eye 2017, 31, 10–25. [Google Scholar] [CrossRef]
- Yeo, N.J.Y.; Chan, E.J.J.; Cheung, C. Choroidal Neovascularization: Mechanisms of Endothelial Dysfunction. Front. Pharmacol. 2019, 10, 1363. [Google Scholar] [CrossRef]
- Nesper, P.L.; Ong, J.X.; Fawzi, A.A. Exploring the Relationship Between Multilayered Choroidal Neovascularization and Choriocapillaris Flow Deficits in AMD. Investig. Ophthalmol. Vis. Sci. 2021, 62, 12. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Reddy, S.T.; Hinton, D.R.; Kannan, R. Mechanisms of RPE senescence and potential role of αB crystallin peptide as a senolytic agent in experimental AMD. Exp. Eye Res. 2022, 215, 108918. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Ach, T.; Tolstik, E.; Messinger, J.D.; Zarubina, A.V.; Heintzmann, R.; Curcio, C.A. Lipofuscin redistribution and loss accompanied by cytoskeletal stress in retinal pigment epithelium of eyes with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3242–3252. [Google Scholar] [CrossRef]
- Malek, G.; Campisi, J.; Kitazawa, K.; Webster, C.; Lakkaraju, A.; Skowronska-Krawczyk, D. Does senescence play a role in age-related macular degeneration? Exp. Eye Res. 2022, 225, 109254. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Kenney, M.C.; Atilano, S.R.; Hurley, J.B.; Brown, E.E.; Ash, J.D. Mitochondria: The Retina’s Achilles’ Heel in AMD. Adv. Exp. Med. Biol. 2021, 1256, 237–264. [Google Scholar] [CrossRef]
- Cheng, M.; Lin, N.; Dong, D.; Ma, J.; Su, J.; Sun, L. PGAM5: A crucial role in mitochondrial dynamics and programmed cell death. Eur. J. Cell Biol. 2021, 100, 151144. [Google Scholar] [CrossRef]
- Yu, B.; Ma, J.; Li, J.; Wang, D.; Wang, Z.; Wang, S. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat. Commun. 2020, 11, 2549. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.M.; Tanna, A.P. Glaucoma. Med. Clin. N. Am. 2021, 105, 493–510. [Google Scholar] [CrossRef]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef]
- Han, X.; Gharahkhani, P.; Hamel, A.R.; Ong, J.S.; Rentería, M.E.; Mehta, P.; Dong, X.; Pasutto, F.; Hammond, C.; Young, T.L.; et al. Large-scale multitrait genome-wide association analyses identify hundreds of glaucoma risk loci. Nat. Genet. 2023, 55, 1116–1125. [Google Scholar] [CrossRef]
- Wang, Z.; Wiggs, J.L.; Aung, T.; Khawaja, A.P.; Khor, C.C. The genetic basis for adult onset glaucoma: Recent advances and future directions. Prog. Retin. Eye Res. 2022, 90, 101066. [Google Scholar] [CrossRef] [PubMed]
- Traoré, L.; Sanou, J.; Bakyono, B.S.; Zoure, A.A.; Zohoncon, T.M.; Sombié, H.K.; Yonli, A.T.; Meda-Hien, G.; Tibiri, E.B.; Djigma, F.W.; et al. Prevalence of Glu323Lys Mutation of the TIGR/MYOC Gene and Risk Factors amongst Primary Open-angle Glaucoma Patients in Ouagadougou, Burkina Faso. J. Curr. Glaucoma Pract. 2023, 17, 79–84. [Google Scholar] [CrossRef]
- Huang, C.; Xie, L.; Wu, Z.; Cao, Y.; Zheng, Y.; Pang, C.P.; Zhang, M. Detection of mutations in MYOC, OPTN, NTF4, WDR36 and CYP1B1 in Chinese juvenile onset open-angle glaucoma using exome sequencing. Sci. Rep. 2018, 8, 4498. [Google Scholar] [CrossRef]
- Kasetti, R.B.; Maddineni, P.; Kiehlbauch, C.; Patil, S.; Searby, C.C.; Levine, B.; Sheffield, V.C.; Zode, G.S. Autophagy stimulation reduces ocular hypertension in a murine glaucoma model via autophagic degradation of mutant myocilin. JCI Insight 2021, 6, e143359. [Google Scholar] [CrossRef]
- Want, A.; Gillespie, S.R.; Wang, Z.; Gordon, R.; Iomini, C.; Ritch, R.; Wolosin, J.M.; Bernstein, A.M. Autophagy and Mitochondrial Dysfunction in Tenon Fibroblasts from Exfoliation Glaucoma Patients. PLoS ONE 2016, 11, e0157404. [Google Scholar] [CrossRef]
- Bernstein, A.M.; Ritch, R.; Wolosin, J.M. LOXL1 folding in exfoliation glaucoma. Adv. Protein Chem. Struct. Biol. 2019, 118, 273–288. [Google Scholar] [CrossRef]
- Nivison, M.P.; Ericson, N.G.; Green, V.M.; Bielas, J.H.; Campbell, J.S.; Horner, P.J. Age-related accumulation of phosphorylated mitofusin 2 protein in retinal ganglion cells correlates with glaucoma progression. Exp. Neurol. 2017, 296, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Wang, W.; Wu, S.; Zhu, X.; Zheng, T.; Chen, X.; Ren, J.; Gong, Y.; Ke, M. Mitochondria and Autophagy Dysfunction in Glucocorticoid-Induced Ocular Hypertension/Glaucoma Mice Model. Curr. Eye Res. 2020, 45, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Park, H.L.; Kim, J.H.; Park, C.K. Different contributions of autophagy to retinal ganglion cell death in the diabetic and glaucomatous retinas. Sci. Rep. 2018, 8, 13321. [Google Scholar] [CrossRef]
- Bell, K.; Rosignol, I.; Sierra-Filardi, E.; Rodriguez-Muela, N.; Schmelter, C.; Cecconi, F.; Grus, F.; Boya, P. Age related retinal Ganglion cell susceptibility in context of autophagy deficiency. Cell Death Discov. 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Awadalla, M.S.; Fingert, J.H.; Roos, B.E.; Chen, S.; Holmes, R.; Graham, S.L.; Chehade, M.; Galanopolous, A.; Ridge, B.; Souzeau, E.; et al. Copy number variations of TBK1 in Australian patients with primary open-angle glaucoma. Am. J. Ophthalmol. 2015, 159, 124–130.e121. [Google Scholar] [CrossRef]
- Sirohi, K.; Kumari, A.; Radha, V.; Swarup, G. A Glaucoma-Associated Variant of Optineurin, M98K, Activates Tbk1 to Enhance Autophagosome Formation and Retinal Cell Death Dependent on Ser177 Phosphorylation of Optineurin. PLoS ONE 2015, 10, e0138289. [Google Scholar] [CrossRef]
- Hayat, B.; Padhy, B.; Mohanty, P.P.; Alone, D.P. Altered unfolded protein response and proteasome impairment in pseudoexfoliation pathogenesis. Exp. Eye Res. 2019, 181, 197–207. [Google Scholar] [CrossRef]
- Hirt, J.; Porter, K.; Dixon, A.; McKinnon, S.; Liton, P.B. Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discov. 2018, 4, 14. [Google Scholar] [CrossRef]
- Thompson, J.; Lakhani, N. Cataracts. Prim. Care 2015, 42, 409–423. [Google Scholar] [CrossRef]
- Galichanin, K. Exposure to subthreshold dose of UVR-B induces apoptosis in the lens epithelial cells and does not in the lens cortical fibre cells. Acta Ophthalmol. 2017, 95, 834–838. [Google Scholar] [CrossRef]
- Wu, J.J.; Wu, W.; Tholozan, F.M.; Saunter, C.D.; Girkin, J.M.; Quinlan, R.A. A dimensionless ordered pull-through model of the mammalian lens epithelium evidences scaling across species and explains the age-dependent changes in cell density in the human lens. J. R. Soc. Interface 2015, 12, 20150391. [Google Scholar] [CrossRef]
- Ji, Y.; Cai, L.; Zheng, T.; Ye, H.; Rong, X.; Rao, J.; Lu, Y. The mechanism of UVB irradiation induced-apoptosis in cataract. Mol. Cell Biochem. 2015, 401, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, W.; He, Q.; He, X.; Yang, M.; Chen, W.; Han, W. Autophagy facilitates age-related cell apoptosis-a new insight from senile cataract. Cell Death Dis. 2022, 13, 37. [Google Scholar] [CrossRef]
- Cui, X.; Feng, R.; Wang, J.; Du, C.; Pi, X.; Chen, D.; Li, J.; Li, H.; Zhang, J.; Zhang, J.; et al. Heat shock factor 4 regulates lysosome activity by modulating the αB-crystallin-ATP6V1A-mTOR complex in ocular lens. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129496. [Google Scholar] [CrossRef]
- Ping, X.; Liang, J.; Shi, K.; Bao, J.; Wu, J.; Yu, X.; Tang, X.; Zou, J.; Shentu, X. Rapamycin relieves the cataract caused by ablation of Gja8b through stimulating autophagy in zebrafish. Autophagy 2021, 17, 3323–3337. [Google Scholar] [CrossRef]
- Quinlan, R.A.; Clark, J.I. Insights into the biochemical and biophysical mechanisms mediating the longevity of the transparent optics of the eye lens. J. Biol. Chem. 2022, 298, 102537. [Google Scholar] [CrossRef]
- Hazen, P.; Trossi-Torres, G.; Timsina, R.; Khadka, N.K.; Mainali, L. Association of Alpha-Crystallin with Human Cortical and Nuclear Lens Lipid Membrane Increases with the Grade of Cortical and Nuclear Cataract. Int. J. Mol. Sci. 2024, 25, 1936. [Google Scholar] [CrossRef]
- Clayton, J.A. Dry Eye. N. Engl. J. Med. 2018, 378, 2212–2223. [Google Scholar] [CrossRef]
- Craig, J.P.; Nelson, J.D.; Azar, D.T.; Belmonte, C.; Bron, A.J.; Chauhan, S.K.; de Paiva, C.S.; Gomes, J.A.P.; Hammitt, K.M.; Jones, L.; et al. TFOS DEWS II Report Executive Summary. Ocul. Surf. 2017, 15, 802–812. [Google Scholar] [CrossRef]
- Di Zazzo, A.; Micera, A.; Coassin, M.; Varacalli, G.; Foulsham, W.; De Piano, M.; Bonini, S. InflammAging at Ocular Surface: Clinical and Biomolecular Analyses in Healthy Volunteers. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1769–1775. [Google Scholar] [CrossRef]
- Barabino, S. Is dry eye disease the same in young and old patients? A narrative review of the literature. BMC Ophthalmol. 2022, 22, 85. [Google Scholar] [CrossRef] [PubMed]
- Yeotikar, N.S.; Zhu, H.; Markoulli, M.; Nichols, K.K.; Naduvilath, T.; Papas, E.B. Functional and Morphologic Changes of Meibomian Glands in an Asymptomatic Adult Population. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3996–4007. [Google Scholar] [CrossRef] [PubMed]
- Azcarate, P.M.; Venincasa, V.D.; Feuer, W.; Stanczyk, F.; Schally, A.V.; Galor, A. Androgen deficiency and dry eye syndrome in the aging male. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5046–5053. [Google Scholar] [CrossRef]
- Li, J.; Ma, J.; Hu, M.; Yu, J.; Zhao, Y. Assessment of tear film lipid layer thickness in patients with Meibomian gland dysfunction at different ages. BMC Ophthalmol. 2020, 20, 394. [Google Scholar] [CrossRef]
- Guillon, M.; Maïssa, C. Tear film evaporation--effect of age and gender. Cont. Lens Anterior Eye 2010, 33, 171–175. [Google Scholar] [CrossRef]
- Ozdemir, M.; Temizdemir, H. Age- and gender-related tear function changes in normal population. Eye 2010, 24, 79–83. [Google Scholar] [CrossRef]
- Mashaghi, A.; Hong, J.; Chauhan, S.K.; Dana, R. Ageing and ocular surface immunity. Br. J. Ophthalmol. 2017, 101, 1–5. [Google Scholar] [CrossRef]
- Galletti, J.G.; de Paiva, C.S. The ocular surface immune system through the eyes of aging. Ocul. Surf. 2021, 20, 139–162. [Google Scholar] [CrossRef]
- de Souza, R.G.; Yu, Z.; Hernandez, H.; Trujillo-Vargas, C.M.; Lee, A.; Mauk, K.E.; Cai, J.; Alves, M.R.; de Paiva, C.S. Modulation of Oxidative Stress and Inflammation in the Aged Lacrimal Gland. Am. J. Pathol. 2021, 191, 294–308. [Google Scholar] [CrossRef]
- Yoon, C.H.; Ryu, J.S.; Hwang, H.S.; Kim, M.K. Comparative Analysis of Age-Related Changes in Lacrimal Glands and Meibomian Glands of a C57BL/6 Male Mouse Model. Int. J. Mol. Sci. 2020, 21, 4169. [Google Scholar] [CrossRef]
- Tahir, S.; Fukushima, Y.; Sakamoto, K.; Sato, K.; Fujita, H.; Inoue, J.; Uede, T.; Hamazaki, Y.; Hattori, M.; Minato, N. A CD153+CD4+ T follicular cell population with cell-senescence features plays a crucial role in lupus pathogenesis via osteopontin production. J. Immunol. 2015, 194, 5725–5735. [Google Scholar] [CrossRef] [PubMed]
- Moreno, I.; Verma, S.; Gesteira, T.F.; Coulson-Thomas, V.J. Recent advances in age-related meibomian gland dysfunction (ARMGD). Ocul. Surf. 2023, 30, 298–306. [Google Scholar] [CrossRef]
- Parfitt, G.J.; Brown, D.J.; Jester, J.V. Transcriptome analysis of aging mouse meibomian glands. Mol. Vis. 2016, 22, 518–527. [Google Scholar]
- Sledge, S.; Henry, C.; Borchman, D.; Yappert, M.C.; Bhola, R.; Ramasubramanian, A.; Blackburn, R.; Austin, J.; Massey, K.; Sayied, S.; et al. Human Meibum Age, Lipid-Lipid Interactions and Lipid Saturation in Meibum from Infants. Int. J. Mol. Sci. 2017, 18, 1862. [Google Scholar] [CrossRef]
- Suzuki, T.; Kitazawa, K.; Cho, Y.; Yoshida, M.; Okumura, T.; Sato, A.; Kinoshita, S. Alteration in meibum lipid composition and subjective symptoms due to aging and meibomian gland dysfunction. Ocul. Surf. 2022, 26, 310–317. [Google Scholar] [CrossRef]
- Parfitt, G.J.; Xie, Y.; Geyfman, M.; Brown, D.J.; Jester, J.V. Absence of ductal hyper-keratinization in mouse age-related meibomian gland dysfunction (ARMGD). Aging 2013, 5, 825–834. [Google Scholar] [CrossRef]
- Wei, A.; Hong, J.; Sun, X.; Xu, J. Evaluation of age-related changes in human palpebral conjunctiva and meibomian glands by in vivo confocal microscopy. Cornea 2011, 30, 1007–1012. [Google Scholar] [CrossRef]
- Alghamdi, Y.A.; Mercado, C.; McClellan, A.L.; Batawi, H.; Karp, C.L.; Galor, A. Epidemiology of Meibomian Gland Dysfunction in an Elderly Population. Cornea 2016, 35, 731–735. [Google Scholar] [CrossRef]
- Peng, X.; Li, H.; Zhu, L.; Zhao, S.; Li, Z.; Li, S.; DongtingWu; Chen, J.; Zheng, S.; Su, W. Single-cell sequencing of the retina shows that LDHA regulates pathogenesis of autoimmune uveitis. J. Autoimmun. 2024, 143, 103160. [Google Scholar] [CrossRef]
- Thorne, J.E.; Suhler, E.; Skup, M.; Tari, S.; Macaulay, D.; Chao, J.; Ganguli, A. Prevalence of Noninfectious Uveitis in the United States: A Claims-Based Analysis. JAMA Ophthalmol. 2016, 134, 1237–1245. [Google Scholar] [CrossRef]
- Pennesi, G.; Mattapallil, M.J.; Sun, S.H.; Avichezer, D.; Silver, P.B.; Karabekian, Z.; David, C.S.; Hargrave, P.A.; McDowell, J.H.; Smith, W.C.; et al. A humanized model of experimental autoimmune uveitis in HLA class II transgenic mice. J. Clin. Investig. 2003, 111, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Ataide, M.; Kastenmüller, W. Lymph node—An organ for T-cell activation and pathogen defense. Immunol. Rev. 2016, 271, 200–220. [Google Scholar] [CrossRef]
- Richner, J.M.; Gmyrek, G.B.; Govero, J.; Tu, Y.; van der Windt, G.J.; Metcalf, T.U.; Haddad, E.K.; Textor, J.; Miller, M.J.; Diamond, M.S. Age-Dependent Cell Trafficking Defects in Draining Lymph Nodes Impair Adaptive Immunity and Control of West Nile Virus Infection. PLoS Pathog. 2015, 11, e1005027. [Google Scholar] [CrossRef]
- Turner, V.M.; Mabbott, N.A. Structural and functional changes to lymph nodes in ageing mice. Immunology 2017, 151, 239–247. [Google Scholar] [CrossRef]
- Li, H.; Zhu, L.; Wang, R.; Xie, L.; Ren, J.; Ma, S.; Zhang, W.; Liu, X.; Huang, Z.; Chen, B.; et al. Aging weakens Th17 cell pathogenicity and ameliorates experimental autoimmune uveitis in mice. Protein Cell 2022, 13, 422–445. [Google Scholar] [CrossRef]
- Coursey, T.G.; Bian, F.; Zaheer, M.; Pflugfelder, S.C.; Volpe, E.A.; de Paiva, C.S. Age-related spontaneous lacrimal keratoconjunctivitis is accompanied by dysfunctional T regulatory cells. Mucosal Immunol. 2017, 10, 743–756. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Passos, J.F.; Khosla, S.; Tchkonia, T.; Kirkland, J.L. Reducing Senescent Cell Burden in Aging and Disease. Trends Mol. Med. 2020, 26, 630–638. [Google Scholar] [CrossRef]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; DePinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006, 443, 421–426. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, P.; Liu, X.; Pan, C.; Shi, G.; Ye, P.; Zou, X.; Li, X.; Zheng, X.; Liu, Y.; et al. Obesity modulates hematopoietic stem cell fate decision via IL-1β induced p38/MAPK signaling pathway. Stem Cell Res. Ther. 2024, 15, 336. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Coppé, J.P.; Lam, E.W. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Knoppert, S.N.; Valentijn, F.A.; Nguyen, T.Q.; Goldschmeding, R.; Falke, L.L. Cellular Senescence and the Kidney: Potential Therapeutic Targets and Tools. Front. Pharmacol. 2019, 10, 770. [Google Scholar] [CrossRef]
- Cooley, J.C.; Javkhlan, N.; Wilson, J.A.; Foster, D.G.; Edelman, B.L.; Ortiz, L.A.; Schwartz, D.A.; Riches, D.W.; Redente, E.F. Inhibition of antiapoptotic BCL-2 proteins with ABT-263 induces fibroblast apoptosis, reversing persistent pulmonary fibrosis. JCI Insight 2023, 8, e163762. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Liu, X.; Wang, Y.; Chang, J.; Zhang, X.; Mackintosh, S.G.; Tackett, A.J.; He, Y.; Lv, D.; et al. Oxidation resistance 1 is a novel senolytic target. Aging Cell 2018, 17, e12780. [Google Scholar] [CrossRef]
- Yin, K.; Patten, D.; Gough, S.; de Barros Gonçalves, S.; Chan, A.; Olan, I.; Cassidy, L.; Poblocka, M.; Zhu, H.; Lun, A.; et al. Senescence-induced endothelial phenotypes underpin immune-mediated senescence surveillance. Genes. Dev. 2022, 36, 533–549. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Wang, T.W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Borghesan, M.; Hoogaars, W.M.H.; Varela-Eirin, M.; Talma, N.; Demaria, M. A Senescence-Centric View of Aging: Implications for Longevity and Disease. Trends Cell Biol. 2020, 30, 777–791. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Zhang, X.; Zhang, R.; Lao, K.; Mi, Y.; Gou, X. AMPK alleviates oxidative stress-induced premature senescence via inhibition of NF-κB/STAT3 axis-mediated positive feedback loop. Mech. Ageing Dev. 2020, 191, 111347. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Lin, S.; Copland, D.A.; Dick, A.D.; Liu, J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflamm. 2021, 18, 32. [Google Scholar] [CrossRef]
- van Deursen, J.M. Senolytic therapies for healthy longevity. Science 2019, 364, 636–637. [Google Scholar] [CrossRef]
- Crespo-Garcia, S.; Fournier, F.; Diaz-Marin, R.; Klier, S.; Ragusa, D.; Masaki, L.; Cagnone, G.; Blot, G.; Hafiane, I.; Dejda, A.; et al. Therapeutic targeting of cellular senescence in diabetic macular edema: Preclinical and phase 1 trial results. Nat. Med. 2024, 30, 443–454. [Google Scholar] [CrossRef]
- Robinson, R.; Brown, D.; Churchwell, L.; Lee, T.J.; Kodeboyina, S.K.; Bloom, J.; Sharma, A.; Sharma, S. RNA-Seq analysis reveals gene expression changes induced by IL-6 trans-signaling activation in retinal endothelial cells. Cytokine 2021, 139, 155375. [Google Scholar] [CrossRef]
- Robinson, R.; Srinivasan, M.; Shanmugam, A.; Ward, A.; Ganapathy, V.; Bloom, J.; Sharma, A.; Sharma, S. Interleukin-6 trans-signaling inhibition prevents oxidative stress in a mouse model of early diabetic retinopathy. Redox Biol. 2020, 34, 101574. [Google Scholar] [CrossRef]
- Valle, M.L.; Dworshak, J.; Sharma, A.; Ibrahim, A.S.; Al-Shabrawey, M.; Sharma, S. Inhibition of interleukin-6 trans-signaling prevents inflammation and endothelial barrier disruption in retinal endothelial cells. Exp. Eye Res. 2019, 178, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.M.; Robinson, R.; Greenway, G.; Glass, J.; Budkin, S.; Sharma, S. Blockade of interleukin-6 trans-signaling prevents mitochondrial dysfunction and cellular senescence in retinal endothelial cells. Exp. Eye Res. 2023, 237, 109721. [Google Scholar] [CrossRef] [PubMed]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef]
- Li, J.; Zhao, L.; Urabe, G.; Fu, Y.; Guo, L.W. Epigenetic intervention with a BET inhibitor ameliorates acute retinal ganglion cell death in mice. Mol. Vis. 2017, 23, 149–159. [Google Scholar]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Somasundaran, S.; Constable, I.J.; Mellough, C.B.; Carvalho, L.S. Retinal pigment epithelium and age-related macular degeneration: A review of major disease mechanisms. Clin. Exp. Ophthalmol. 2020, 48, 1043–1056. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kambhampati, S.P.; Bhutto, I.A.; McLeod, D.S.; Lutty, G.A.; Kannan, R.M. Evolution of oxidative stress, inflammation and neovascularization in the choroid and retina in a subretinal lipid induced age-related macular degeneration model. Exp. Eye Res. 2021, 203, 108391. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Qing, W.; Qi, R.; Zou, M.; Gong, L.; Liu, Y.; Li, D.W. Inhibition of Sumoylation Alleviates Oxidative Stress-induced Retinal Pigment Epithelial Cell Senescence and Represses Proinflammatory Gene Expression. Curr. Mol. Med. 2018, 18, 575–583. [Google Scholar] [CrossRef]
- Campello, L.; Esteve-Rudd, J.; Cuenca, N.; Martín-Nieto, J. The ubiquitin-proteasome system in retinal health and disease. Mol. Neurobiol. 2013, 47, 790–810. [Google Scholar] [CrossRef]
- Jun, B.; Mukherjee, P.K.; Asatryan, A.; Kautzmann, M.A.; Heap, J.; Gordon, W.C.; Bhattacharjee, S.; Yang, R.; Petasis, N.A.; Bazan, N.G. Elovanoids are novel cell-specific lipid mediators necessary for neuroprotective signaling for photoreceptor cell integrity. Sci. Rep. 2017, 7, 5279. [Google Scholar] [CrossRef]
- Do, K.V.; Kautzmann, M.I.; Jun, B.; Gordon, W.C.; Nshimiyimana, R.; Yang, R.; Petasis, N.A.; Bazan, N.G. Elovanoids counteract oligomeric β-amyloid-induced gene expression and protect photoreceptors. Proc. Natl. Acad. Sci. USA 2019, 116, 24317–24325. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, P.G.; Ishikawa, K.; Spee, C.; Mehta, H.H.; Wan, J.; Yen, K.; Cohen, P.; Kannan, R.; Hinton, D.R. The Mitochondrial-Derived Peptide Humanin Protects RPE Cells From Oxidative Stress, Senescence, and Mitochondrial Dysfunction. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1238–1253. [Google Scholar] [CrossRef] [PubMed]
- Handa, J.T.; Bowes Rickman, C.; Dick, A.D.; Gorin, M.B.; Miller, J.W.; Toth, C.A.; Ueffing, M.; Zarbin, M.; Farrer, L.A. A systems biology approach towards understanding and treating non-neovascular age-related macular degeneration. Nat. Commun. 2019, 10, 3347. [Google Scholar] [CrossRef]
- Copland, D.A.; Theodoropoulou, S.; Liu, J.; Dick, A.D. A Perspective of AMD Through the Eyes of Immunology. Investig. Ophthalmol. Vis. Sci. 2018, 59, Amd83–Amd92. [Google Scholar] [CrossRef]
- Liu, J.; Copland, D.A.; Horie, S.; Wu, W.K.; Chen, M.; Xu, Y.; Paul Morgan, B.; Mack, M.; Xu, H.; Nicholson, L.B.; et al. Myeloid cells expressing VEGF and arginase-1 following uptake of damaged retinal pigment epithelium suggests potential mechanism that drives the onset of choroidal angiogenesis in mice. PLoS ONE 2013, 8, e72935. [Google Scholar] [CrossRef]
- Theodoropoulou, S.; Copland, D.A.; Liu, J.; Wu, J.; Gardner, P.J.; Ozaki, E.; Doyle, S.L.; Campbell, M.; Dick, A.D. Interleukin-33 regulates tissue remodelling and inhibits angiogenesis in the eye. J. Pathol. 2017, 241, 45–56. [Google Scholar] [CrossRef]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef]
- de Jong, S.; Tang, J.; Clark, S.J. Age-related macular degeneration: A disease of extracellular complement amplification. Immunol. Rev. 2023, 313, 279–297. [Google Scholar] [CrossRef]
- Choudhary, M.; Malek, G. Potential therapeutic targets for age-related macular degeneration: The nuclear option. Prog. Retin. Eye Res. 2023, 94, 101130. [Google Scholar] [CrossRef]
- Manai, F.; Smedowski, A.; Kaarniranta, K.; Comincini, S.; Amadio, M. Extracellular vesicles in degenerative retinal diseases: A new therapeutic paradigm. J. Control Release 2024, 365, 448–468. [Google Scholar] [CrossRef]
- Kumbhar, P.; Kolekar, K.; Vishwas, S.; Shetti, P.; Kumbar, V.; Andreoli Pinto, T.J.; Paiva-Santos, A.C.; Veiga, F.; Gupta, G.; Singh, S.K.; et al. Treatment avenues for age-related macular degeneration: Breakthroughs and bottlenecks. Ageing Res. Rev. 2024, 98, 102322. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.A.; Apte, R.S. Complement regulation in the eye: Implications for age-related macular degeneration. J. Clin. Investig. 2024, 134, e178296. [Google Scholar] [CrossRef]
- Yang, J.H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305–326.e327. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, S.; Xie, B.; Zhong, Y. Aging, Cellular Senescence, and Glaucoma. Aging Dis. 2024, 15, 546–564. [Google Scholar] [CrossRef]
- Chaudhry, S.; Dunn, H.; Carnt, N.; White, A. Nutritional supplementation in the prevention and treatment of glaucoma. Surv. Ophthalmol. 2022, 67, 1081–1098. [Google Scholar] [CrossRef]
- Santoro, M.M. The Antioxidant Role of Non-mitochondrial CoQ10: Mystery Solved! Cell Metab. 2020, 31, 13–15. [Google Scholar] [CrossRef]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef]
- Guillaumet-Adkins, A.; Yañez, Y.; Peris-Diaz, M.D.; Calabria, I.; Palanca-Ballester, C.; Sandoval, J. Epigenetics and Oxidative Stress in Aging. Oxid. Med. Cell Longev. 2017, 2017, 9175806. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, H.I.; Chang, W.S.; Choi, J.S. Triphenylphosphonium-conjugated glycol chitosan microspheres for mitochondria-targeted drug delivery. Int. J. Biol. Macromol. 2021, 167, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Iomdina, E.N.; Khoroshilova-Maslova, I.P.; Robustova, O.V.; Averina, O.A.; Kovaleva, N.A.; Aliev, G.; Reddy, V.P.; Zamyatnin, A.A., Jr.; Skulachev, M.V.; Senin, I.I.; et al. Mitochondria-targeted antioxidant SkQ1 reverses glaucomatous lesions in rabbits. Front. Biosci. 2015, 20, 892–901. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Ahmed, Z.; Morgan-Warren, P.J.; Berry, M.; Scott, R.A.H.; Logan, A. Effects of siRNA-Mediated Knockdown of GSK3β on Retinal Ganglion Cell Survival and Neurite/Axon Growth. Cells 2019, 8, 956. [Google Scholar] [CrossRef]
- Simpson, D.J.; Olova, N.N.; Chandra, T. Cellular reprogramming and epigenetic rejuvenation. Clin. Epigenetics 2021, 13, 170. [Google Scholar] [CrossRef]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Gao, F.J.; Zhang, S.H.; Xu, P.; Yang, B.Q.; Zhang, R.; Cheng, Y.; Zhou, X.J.; Huang, W.J.; Wang, M.; Chen, J.Y.; et al. Quercetin Declines Apoptosis, Ameliorates Mitochondrial Function and Improves Retinal Ganglion Cell Survival and Function in In Vivo Model of Glaucoma in Rat and Retinal Ganglion Cell Culture In Vitro. Front. Mol. Neurosci. 2017, 10, 285. [Google Scholar] [CrossRef]
- Zhou, X.; Li, G.; Yang, B.; Wu, J. Quercetin Enhances Inhibitory Synaptic Inputs and Reduces Excitatory Synaptic Inputs to OFF- and ON-Type Retinal Ganglion Cells in a Chronic Glaucoma Rat Model. Front. Neurosci. 2019, 13, 672. [Google Scholar] [CrossRef]
- Rocha, L.R.; Nguyen Huu, V.A.; Palomino La Torre, C.; Xu, Q.; Jabari, M.; Krawczyk, M.; Weinreb, R.N.; Skowronska-Krawczyk, D. Early removal of senescent cells protects retinal ganglion cells loss in experimental ocular hypertension. Aging Cell 2020, 19, e13089. [Google Scholar] [CrossRef]
- El-Nimri, N.W.; Moore, S.M.; Zangwill, L.M.; Proudfoot, J.A.; Weinreb, R.N.; Skowronska-Krawczyk, D.; Baxter, S.L. Evaluating the neuroprotective impact of senolytic drugs on human vision. Sci. Rep. 2020, 10, 21752. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lou, W.; Zhang, Y.; Chen, Z.; Huang, Y.; Jin, H. HO-1-Mediated Autophagic Restoration Protects Lens Epithelial Cells Against Oxidative Stress and Cellular Senescence. Investig. Ophthalmol. Vis. Sci. 2023, 64, 6. [Google Scholar] [CrossRef]
- Wei, X.; Luo, D.; Yan, Y.; Yu, H.; Sun, L.; Wang, C.; Song, F.; Ge, H.; Qian, H.; Li, X.; et al. Kojic acid inhibits senescence of human corneal endothelial cells via NF-κB and p21 signaling pathways. Exp. Eye Res. 2019, 180, 174–183. [Google Scholar] [CrossRef]
- Bae, Y.; Hwang, J.S.; Shin, Y.J. miR-30c-1 encourages human corneal endothelial cells to regenerate through ameliorating senescence. Aging 2021, 13, 9348–9372. [Google Scholar] [CrossRef]
- Gidfar, S.; Milani, F.Y.; Milani, B.Y.; Shen, X.; Eslani, M.; Putra, I.; Huvard, M.J.; Sagha, H.; Djalilian, A.R. Rapamycin Prolongs the Survival of Corneal Epithelial Cells in Culture. Sci. Rep. 2017, 7, 40308. [Google Scholar] [CrossRef]
- Yamane, M.; Sato, S.; Shimizu, E.; Shibata, S.; Hayano, M.; Yaguchi, T.; Kamijuku, H.; Ogawa, M.; Suzuki, T.; Mukai, S.; et al. Senescence-associated secretory phenotype promotes chronic ocular graft-vs-host disease in mice and humans. FASEB J. 2020, 34, 10778–10800. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e116. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
| Inducer | Senescence Pathway in Ocular Context |
|---|---|
| Oxidative stress | Chronic exposure to oxidative stress in RPE cells induces DNA damage and mitochondrial dysfunction, leading to p53–p21-mediated senescence. |
| Chronic inflammation | Prolonged inflammatory signaling in the retina or uveal tract activates NF-κB and cytokine secretion (e.g., IL-6, IL-8), reinforcing the SASP and promoting senescence. |
| Accumulation of metabolic waste | In diseases like AMD, impaired autophagy and accumulation of lipofuscin or drusen contribute to lysosomal stress, mitochondrial dysfunction, and subsequent senescence. |
| Light-induced damage | High-energy light exposure (e.g., blue light) causes photo-oxidative damage to RPE cells, triggering DNA damage response and activation of the p16–RB pathway. |
| Age-related epigenetic changes | Epigenetic drift with age (e.g., histone modification and DNA methylation changes) disrupts gene expression in retinal cells, promoting p16 or p21 expression and senescence. |
| Genetic alterations |
|
| Hypoxia or ischemia | Retinal ischemia induces oxidative stress and HIF-1α signaling, which may lead to endothelial cell senescence and neovascular dysfunction. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Liu, X.; Liu, Y.; Su, W.; Zhuo, Y. New Insights into the Role of Cellular Senescence and Its Therapeutic Implications in Ocular Diseases. Bioengineering 2025, 12, 563. https://doi.org/10.3390/bioengineering12060563
Wu J, Liu X, Liu Y, Su W, Zhuo Y. New Insights into the Role of Cellular Senescence and Its Therapeutic Implications in Ocular Diseases. Bioengineering. 2025; 12(6):563. https://doi.org/10.3390/bioengineering12060563
Chicago/Turabian StyleWu, Junying, Xiuxing Liu, Yidan Liu, Wenru Su, and Yehong Zhuo. 2025. "New Insights into the Role of Cellular Senescence and Its Therapeutic Implications in Ocular Diseases" Bioengineering 12, no. 6: 563. https://doi.org/10.3390/bioengineering12060563
APA StyleWu, J., Liu, X., Liu, Y., Su, W., & Zhuo, Y. (2025). New Insights into the Role of Cellular Senescence and Its Therapeutic Implications in Ocular Diseases. Bioengineering, 12(6), 563. https://doi.org/10.3390/bioengineering12060563