
Long-Acting Extracellular Vesicle-Based Biologics in Osteoarthritis Immunotherapy




Abstract
1. Introduction
1.1. Osteoarthritis Pathophysiology
1.2. Monoclonal Antibodies and Cytokine Inhibitors in OA
1.3. Long-Acting Therapy and OA
2. EVs as an Emerging Therapeutic Strategy
2.1. Overview of EVs
2.2. Extended Duration and Effectiveness of Biologics When Combined with EV Technology
3. Formulation of EVs to Be Loaded with Specific Attributes
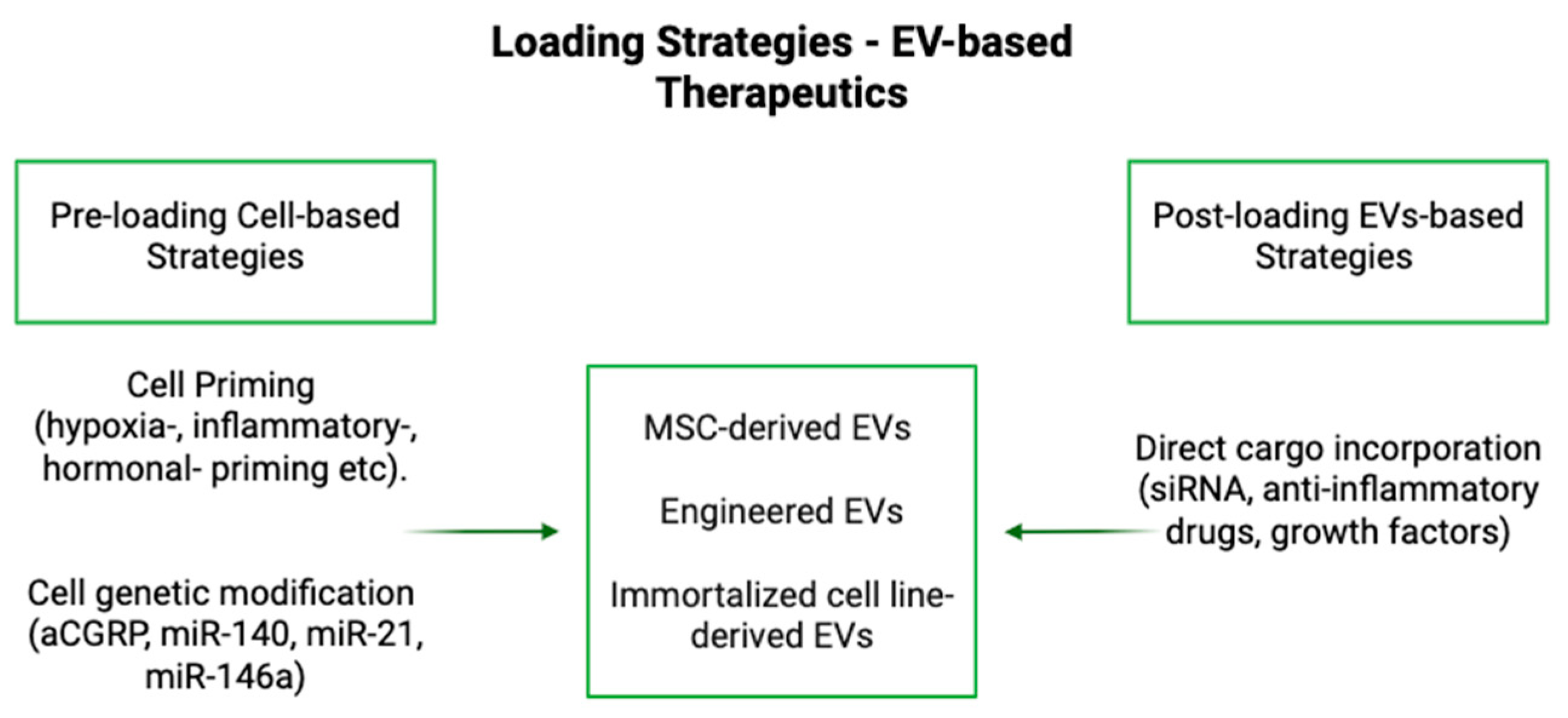
3.1. Loading
3.2. Properties and Modifications of EVs That Generate Specific Functions Unrelated to Cargo Effects
4. Considerations for the Use of EVs
4.1. Biocompatibility, Stability, and Delivery of EVs
4.2. Diffusion
4.3. Dosing and Specs
5. Logistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OA | Osteoarthritis |
| IL-1 | Interleukin-1 |
| TNF-α | Tumor Necrosis Factor Alpha |
| EVs | Extracellular Vesicles |
| MMP-13 | Matrix Metalloproteinase-13 |
| ADAMTS5 | A Disintegrin and Metalloproteinase with Thrombospondin Motifs 5 |
| KOA | Knee Osteoarthritis |
| IFP-MSC | Infrapatellar fat pad derived mesenchymal stem cells |
| IFP | Infrapatellar Fat Pad |
| IL-1β | Interleukin-1 Beta |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| SP | Substance P |
| MSC | Mesenchymal Stem/Stromal Cells |
| CD10 | Cluster of Differentiation 10 (Neprilysin) |
| IA | Intra-Articular |
| NGF | Nerve Growth Factor |
| WOMAC | Western Ontario and McMaster Universities Osteoarthritis Index |
| PGA | Patient Global Assessment |
| VEGF | Vascular Endothelial Growth Factor |
| RA | Rheumatoid Arthritis |
| KOOS | Knee Injury and Osteoarthritis Outcomes Score |
| PLGA | Poly(lactic-co-glycolic acid) |
| DSP | Dexamethasone Sodium Phosphate |
| iPSC | Induced Pluripotent Stem Cells |
| haMSC/hASC | Human Adipose-Derived Mesenchymal Stem Cells |
| haMPC | Human Adipose-Derived Mesenchymal Progenitor Cells |
| IL-10 | Interleukin-10 |
| AKI | Acute Kidney Injury |
| ARDS | Acute Respiratory Distress Syndrome |
| aCGRP | Antagonist Calcitonin Gene-Related Peptide |
| AAV | Adeno-Associated Virus |
| CGRP | Calcitonin Gene-Related Peptide |
| miRNA | MicroRNA |
| CAP | Chondrocyte Affinity Peptide |
| ASO | Antisense Oligonucleotide |
| sEVs | Small Extracellular Vesicles |
| FGF18 | Fibroblast Growth Factor 18 |
| HAMA | Methacrylic Anhydride-Modified Hyaluronic Acid |
| ECM | Extracellular Matrix |
| ESC | Embryonic stem cell-induced |
| BMSC | Bone Marrow-Derived Mesenchymal Stem Cells |
| BMP2 | Bone Morphogenetic Protein 2 |
| ApoVs | Apoptotic vesicles |
| PEG | Polyethylene Glycol |
| PBS | Phosphate-Buffered Saline |
| DMM | Destabilization of the medial meniscus |
| PTOA | Post-Traumatic Osteoarthritis |
| OARSI | Osteoarthritis Research Society International |
| PRP | Platelet-Rich Plasma |
| PRP-Exos | Platelet-Rich Plasma-Derived Exosomes |
| GMP | Good Manufacturing Practice |
References
- Iqbal, I.; Fleischmann, R. Treatment of osteoarthritis with anakinra. Curr. Rheumatol. Rep. 2007, 9, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Bihlet, A.; Byrjalsen, I.; Alexandersen, P.; Ladel, C.; Michaels, M.; Andersen, J.R.; Riis, B.J.; Kraus, V.; Bay-Jensen, A.C.; et al. OA phenotypes, rather than disease stage, drive structural progression—Identification of structural progressors from 2 phase III randomized clinical studies with symptomatic knee OA. Osteoarthr. Cartil. 2015, 23, 550–558. [Google Scholar] [CrossRef]
- Dell'Isola, A.; Steultjens, M. Classification of patients with knee osteoarthritis in clinical phenotypes: Data from the osteoarthritis initiative. PLoS ONE 2018, 13, e0191045. [Google Scholar] [CrossRef]
- Dell’Isola, A.; Allan, R.; Smith, S.L.; Marreiros, S.S.P.; Steultjens, M. Identification of clinical phenotypes in knee osteoarthritis: A systematic review of the literature. BMC Musculoskelet. Disord. 2016, 17, 425. [Google Scholar] [CrossRef] [PubMed]
- Driban, J.B.; Sitler, M.R.; Barbe, M.F.; Balasubramanian, E. Is osteoarthritis a heterogeneous disease that can be stratified into subsets? Clin. Rheumatol. 2009, 29, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T. Identifying different osteoarthritis phenotypes through epidemiology. Osteoarthr. Cartil. 2010, 18, 601–604. [Google Scholar] [CrossRef]
- Lieberthal, J.; Sambamurthy, N.; Scanzello, C.R. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1825–1834. [Google Scholar] [CrossRef]
- Li, Y.; Mai, Y.; Cao, P.; Wen, X.; Fan, T.; Wang, X.; Ruan, G.; Tang, S.; Ding, C.; Zhu, Z. Relative Efficacy and Safety of Anti-Inflammatory Biologic Agents for Osteoarthritis: A Conventional and Network Meta-Analysis. J. Clin. Med. 2022, 11, 3958. [Google Scholar] [CrossRef]
- Yang, B.; Li, X.; Fu, C.; Cai, W.; Meng, B.; Qu, Y.; Kou, X.; Zhang, Q. Extracellular vesicles in osteoarthritis of peripheral joint and temporomandibular joint. Front. Endocrinol. 2023, 14, 1158744. [Google Scholar] [CrossRef]
- Hou, M.; Zhang, Y.; Zhou, X.; Liu, T.; Yang, H.; Chen, X.; He, F.; Zhu, X. Kartogenin prevents cartilage degradation and alleviates osteoarthritis progression in mice via the miR-146a/NRF2 axis. Cell Death Dis. 2021, 12, 483. [Google Scholar] [CrossRef]
- Rizzo, M.G.; Best, T.M.; Huard, J.; Philippon, M.; Hornicek, F.; Duan, Z.; Griswold, A.J.; Kaplan, L.D.; Hare, J.M.; Kouroupis, D. Therapeutic Perspectives for Inflammation and Senescence in Osteoarthritis Using Mesenchymal Stem Cells, Mesenchymal Stem Cell-Derived Extracellular Vesicles and Senolytic Agents. Cells 2023, 12, 1421. [Google Scholar] [CrossRef] [PubMed]
- Kouroupis, D.; Kaplan, L.D.; Huard, J.; Best, T.M. CD10-Bound Human Mesenchymal Stem/Stromal Cell-Derived Small Extracellular Vesicles Possess Immunomodulatory Cargo and Maintain Cartilage Homeostasis under Inflammatory Conditions. Cells 2023, 12, 1824. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Jeong, S.; Kim, H.; Kang, D.; Lee, J.; Kang, S.-B.; Kim, J.-H. Disease-modifying therapeutic strategies in osteoarthritis: Current status and future directions. Exp. Mol. Med. 2021, 53, 1689–1696. [Google Scholar] [CrossRef]
- Wang, J. Efficacy and safety of adalimumab by intra-articular injection for moderate to severe knee osteoarthritis: An open-label randomized controlled trial. J. Int. Med. Res. 2018, 46, 326–334. [Google Scholar] [CrossRef]
- Estee, M.M.; Cicuttini, F.M.; Page, M.J.; Wluka, A.E.; Wang, Y. Efficacy of tumor necrosis factor inhibitors in hand osteoarthritis: A systematic review and meta-analysis of randomized controlled trials. Osteoarthr. Cartil. Open 2023, 5, 100404. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Merchan, E.C. The Current Role of Disease-modifying Osteoarthritis Drugs. Arch. Bone Jt. Surg. 2023, 11, 11–22. [Google Scholar]
- Schnitzer, T.J.; Lane, N.E.; Birbara, C.; Smith, M.D.; Simpson, S.L.; Brown, M.T. Long-term open-label study of tanezumab for moderate to severe osteoarthritic knee pain. Osteoarthr. Cartil. 2011, 19, 639–646. [Google Scholar] [CrossRef]
- Siddiq, M.A.B.; Oo, W.M.; Hunter, D.J. New therapeutic strategies in osteoarthritis. Jt. Bone Spine 2024, 91, 105739. [Google Scholar] [CrossRef]
- Dakin, P.; Dimartino, S.J.; Gao, H.; Maloney, J.; Kivitz, A.J.; Schnitzer, T.J.; Stahl, N.; Yancopoulos, G.D.; Geba, G.P. The Efficacy, Tolerability, and Joint Safety of Fasinumab in Osteoarthritis Pain: A Phase IIb/III Double-Blind, Placebo-Controlled, Randomized Clinical Trial. Arthritis Rheumatol. 2019, 71, 1824–1834. [Google Scholar] [CrossRef]
- Mayorga, A.J.; Wang, S.; Kelly, K.M.; Thipphawong, J. Efficacy and safety of fulranumab as monotherapy in patients with moderate to severe, chronic knee pain of primary osteoarthritis: A randomised, placebo- and active-controlled trial. Int. J. Clin. Pract. 2016, 70, 493–505. [Google Scholar] [CrossRef]
- Kelly, K.M.; Sanga, P.; Zaki, N.; Wang, S.; Haeussler, J.; Louie, J.; Thipphawong, J. Safety and efficacy of fulranumab in osteoarthritis of the hip and knee: Results from four early terminated phase III randomized studies. Curr. Med. Res. Opin. 2019, 35, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zeng, L.-F.; Liang, G.-H.; Pan, J.-K.; Luo, M.-H.; Han, Y.-H.; Liu, J.; Yang, W.-Y. Does anti-nerve growth factor monoclonal antibody treatment have the potential to replace nonsteroidal anti-inflammatory drugs and opioids in treating hip or knee osteoarthritis? A systematic review of randomized controlled trials. EFORT Open Rev. 2022, 7, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lin, J.; Wang, Z.; Ren, S.; Wu, X.; Yu, F.; Weng, J.; Zeng, H. Bevacizumab tested for treatment of knee osteoarthritis via inhibition of synovial vascular hyperplasia in rabbits. J. Orthop. Translat. 2019, 19, 38–46. [Google Scholar] [CrossRef]
- Vadalà, G.; Ambrosio, L.; Cattani, C.; Bernardini, R.; Giacalone, A.; Papalia, R.; Denaro, V. Bevacizumab Arrests Osteoarthritis Progression in a Rabbit Model: A Dose-Escalation Study. J. Clin. Med. 2021, 10, 2825. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Sato, M.; Kobayashi, M.; Yokoyama, M.; Tani, Y.; Mochida, J. Bevacizumab, an anti-vascular endothelial growth factor antibody, inhibits osteoarthritis. Arthritis Res. Ther. 2014, 16, 427. [Google Scholar] [CrossRef]
- Smakaj, A.; Gasbarra, E.; Cardelli, T.; Salvati, C.; Bonanni, R.; Cariati, I.; Iundusi, R.; Tarantino, U. Exploring Intra-Articular Administration of Monoclonal Antibodies as a Novel Approach to Osteoarthritis Treatment: A Systematic Review. Biomedicines 2024, 12, 2217. [Google Scholar] [CrossRef]
- Schieker, M.; Conaghan, P.G.; Mindeholm, L.; Praestgaard, J.; Solomon, D.H.; Scotti, C.; Gram, H.; Thuren, T.; Roubenoff, R.; Ridker, P.M. Effects of Interleukin-1β Inhibition on Incident Hip and Knee Replacement. Ann. Intern. Med. 2020, 173, 509–515. [Google Scholar] [CrossRef]
- Cohen, S.B.; Proudman, S.; Kivitz, A.J.; Burch, F.X.; Donohue, J.P.; Burstein, D.; Sun, Y.-N.; Banfield, C.; Vincent, M.S.; Ni, L. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 2011, 13, R125. [Google Scholar] [CrossRef]
- Bihlet, A.R.; Balchen, T.; Goteti, K.; Sonne, J.; Ladel, C.; Karsdal, M.A.; Ona, V.; Moreau, F.; Waterhouse, R.; Bay-Jensen, A. Safety, Tolerability, and Pharmacodynamics of the ADAMTS-5 Nanobody M6495: Two Phase 1, Single-Center, Double-Blind, Randomized, Placebo-Controlled Studies in Healthy Subjects and Patients with Osteoarthritis. ACR Open Rheumatol. 2024, 6, 205–213. [Google Scholar] [CrossRef]
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum 2009, 61, 344–352. [Google Scholar] [CrossRef]
- Yu, L.; Luo, R.; Qin, G.; Zhang, Q.; Liang, W. Efficacy and safety of anti-interleukin-1 therapeutics in the treatment of knee osteoarthritis: A systematic review and meta-analysis of randomized controlled trials. J. Orthop. Surg. Res. 2023, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ding, S.-L.; Zhao, X.; Sun, D.; Yang, Y.; Chen, M.; Zhu, C.; Jiang, B.; Gu, Q.; Liu, H.; et al. Self-Reinforced MOF-Based Nanogel Alleviates Osteoarthritis by Long-Acting Drug Release. Adv. Mater. 2024, 36, e2401094. [Google Scholar] [CrossRef]
- Abd-Allah, H.; Kamel, A.O.; Sammour, O.A. Injectable long acting chitosan/tripolyphosphate microspheres for the intra-articular delivery of lornoxicam: Optimization and in vivo evaluation. Carbohydr. Polym. 2016, 149, 263–273. [Google Scholar] [CrossRef]
- Bodick, N.; Lufkin, J.; Willwerth, C.; Blanks, R.C.; Inderjeeth, C.A.; Kumar, A.; Clayman, M.D. FX006 prolongs the residency of triamcinolone acetonide in the synovial tissues of patients with knee osteoarthritis. Osteoarthr. Cartil. 2013, 21, S144–S145. [Google Scholar] [CrossRef]
- Cushman, D.M.; Zurbuchen, E.; Elmer, A.; English, J.; Henrie, A.M.; Gee, C.; Monson, M.E.; Teramoto, M. Extended-release triamcinolone provides prolonged relief for patients who failed standard corticosteroid injection for knee osteoarthritis; a pragmatic retrospective study. Interv. Pain Med. 2022, 1, 100103. [Google Scholar] [CrossRef]
- Hunter, D.J.; Chang, C.-C.; Wei, J.C.-C.; Lin, H.-Y.; Brown, C.; Tai, T.-T.; Wu, C.-F.; Chuang, W.C.-M.; Shih, S.-F. TLC599 in patients with osteoarthritis of the knee: A phase IIa, randomized, placebo-controlled, dose-finding study. Arthritis Res. Ther. 2022, 24, 52. [Google Scholar] [CrossRef]
- Grigsby, E.; Rickam, M.; Thewlis, D.; Simon, L.; Chavez, R.; Huston, M.; Rieger, J.; Glover, D.; Collins, S. XT-150- A novel immunomodulatory gene therapy for osteoarthritis pain in phase 2b development. Osteoarthr. Cartil. 2021, 29, S12. [Google Scholar] [CrossRef]
- Liu, X.; Robbins, S.; Wang, X.; Virk, S.; Schuck, K.; Deveza, L.A.; Oo, W.M.; Carmichael, K.; Antony, B.; Eckstein, F.; et al. Efficacy and cost-effectiveness of Stem Cell injections for symptomatic relief and strUctural improvement in people with Tibiofemoral knee OsteoaRthritis: Protocol for a randomised placebo-controlled trial (the SCUlpTOR trial). BMJ Open 2021, 11, e056382. [Google Scholar] [CrossRef] [PubMed]
- Orozco, L.; Munar, A.; Soler, R.; Alberca, M.; Soler, F.; Huguet, M.; Sentis, J.; Garcia-Sancho, J. Treatment of knee osteoarthritis with autologous mesenchymal stem cells: Two-year follow-up results. Transplantation 2014, 97, e66–e68. [Google Scholar] [CrossRef]
- Song, Y.; Du, H.; Dai, C.; Zhnag, L.; Li, S.; Hunter, D.J.; Lu, L.; Bao, C. Human adipose-derived mesenchymal stem cells for osteoarthritis: A pilot study with long-term follow-up and repeated injections. Regen. Med. 2018, 13, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Dai, C.; Zhang, Z.; Du, H.; Li, S.; Ye, P.; Fu, Q.; Zhang, L.; Wu, X.; Dong, Y. Treatment of knee osteoarthritis with intra-articular injection of autologous adipose-derived mesenchymal progenitor cells: A prospective, randomized, double-blind, active-controlled, phase IIb clinical trial. Stem Cell Res. Ther. 2019, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Guan, Y.; Xie, A.; Yan, Z.; Gao, S.; Li, W.; Rao, L.; Chen, X.; Chen, T. Extracellular vesicles: A rising star for therapeutics and drug delivery. J. Nanobiotechnol. 2023, 21, 231. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Dang, X.T.T.; Kavishka, J.M.; Zhang, D.X.; Pirisinu, M.; Le, M.T.N. Extracellular Vesicles as an Efficient and Versatile System for Drug Delivery. Cells 2020, 9, 2191. [Google Scholar] [CrossRef]
- Sadeghi, S.; Tehrani, F.R.; Tahmasebi, S.; Shafiee, A.; Hashemi, S.M. Exosome engineering in cell therapy and drug delivery. Inflammopharmacology 2023, 31, 145–169. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; Van Der Meel, R.; Fens, M.H.A.M.; Heijnen, H.F.G.; Van Bergen En Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- Wang, Z.; Mo, H.; He, Z.; Chen, A.; Cheng, P. Extracellular vesicles as an emerging drug delivery system for cancer treatment: Current strategies and recent advances. Biomed. Pharmacother. 2022, 153, 113480. [Google Scholar] [CrossRef]
- Al Faruque, H.; Choi, E.-S.; Kim, J.-H.; Kim, E. Enhanced effect of autologous EVs delivering paclitaxel in pancreatic cancer. J. Control. Release 2022, 347, 330–346. [Google Scholar] [CrossRef]
- Yang, X.; Shi, G.; Guo, J.; Wang, C.; He, Y. Exosome-encapsulated antibiotic against intracellular infections of methicillin-resistant Staphylococcus aureus. Int. J. Nanomed. 2018, 13, 8095–8104. [Google Scholar] [CrossRef]
- Zarubova, J.; Hasani-Sadrabadi, M.M.; Dashtimoghadam, E.; Zhang, X.; Ansari, S.; Li, S.; Moshaverinia, A. Engineered Delivery of Dental Stem-Cell-Derived Extracellular Vesicles for Periodontal Tissue Regeneration. Adv. Health. Mater. 2022, 11, e2102593. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Fontana, S. Exosomes/Extracellular Vesicles and Targeted Tumor Immunotherapy. Int. J. Mol. Sci. 2024, 25, 5458. [Google Scholar] [CrossRef]
- Najafi, S.; Majidpoor, J.; Mortezaee, K. Extracellular vesicle–based drug delivery in cancer immunotherapy. Drug Deliv. Transl. Res. 2023, 13, 2790–2806. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.-T.; Wang, B.; Wu, M.; Li, Z.-L.; Feng, Y.; Cao, J.-Y.; Yin, D.; Liu, H.; Tang, R.-N.; Crowley, S.D.; et al. Extracellular vesicle–encapsulated IL-10 as novel nanotherapeutics against ischemic AKI. Sci. Adv. 2020, 6, eaaz0748. [Google Scholar] [CrossRef]
- Salazar-Puerta, A.I.; Rincon-Benavides, M.A.; Cuellar-Gaviria, T.Z.; Aldana, J.; Vasquez Martinez, G.; Ortega-Pineda, L.; Das, D.; Dodd, D.; Spencer, C.A.; Deng, B.; et al. Engineered Extracellular Vesicles Derived from Dermal Fibroblasts Attenuate Inflammation in a Murine Model of Acute Lung Injury. Adv. Mater. 2023, 35, e2210579. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liang, Y.; Li, X.; Ouyang, K.; Wang, M.; Cao, T.; Li, W.; Liu, J.; Xiong, J.; Li, B.; et al. Exosome-mediated delivery of kartogenin for chondrogenesis of synovial fluid-derived mesenchymal stem cells and cartilage regeneration. Biomaterials 2021, 269, 120539. [Google Scholar] [CrossRef]
- Lara-Barba, E.; Araya, M.J.; Hill, C.N.; Bustamante-Barrientos, F.A.; Ortloff, A.; García, C.; Galvez-Jiron, F.; Pradenas, C.; Luque-Campos, N.; Maita, G.; et al. Role of microRNA Shuttled in Small Extracellular Vesicles Derived from Mesenchymal Stem/Stromal Cells for Osteoarticular Disease Treatment. Front. Immunol. 2021, 12, 768771. [Google Scholar] [CrossRef]
- Liebmann, K.; Castillo, M.A.; Jergova, S.; Best, T.M.; Sagen, J.; Kouroupis, D. Modification of Mesenchymal Stem/Stromal Cell-Derived Small Extracellular Vesicles by Calcitonin Gene Related Peptide (CGRP) Antagonist: Potential Implications for Inflammation and Pain Reversal. Cells 2024, 13, 484. [Google Scholar] [CrossRef]
- Gardashli, M.; Baron, M.; Drohat, P.; Quintero, D.; Kaplan, L.D.; Szeto, A.; Mendez, A.J.; Best, T.M.; Kouroupis, D. The roles of regulatory-compliant media and inflammatory/oxytocin priming selection in enhancing human mesenchymal stem/stromal cell immunomodulatory properties. Sci. Rep. 2024, 14, 29438. [Google Scholar] [CrossRef]
- Liu, Z.; Zhuang, Y.; Fang, L.; Yuan, C.; Wang, X.; Lin, K. Breakthrough of extracellular vesicles in pathogenesis, diagnosis and treatment of osteoarthritis. Bioact. Mater. 2023, 22, 423–452. [Google Scholar] [CrossRef]
- Lu, X.; Fan, S.; Cao, M.; Liu, D.; Xuan, K.; Liu, A. Extracellular vesicles as drug delivery systems in therapeutics: Current strategies and future challenges. J. Pharm. Investig. 2024, 54, 785–802. [Google Scholar] [CrossRef]
- Stranford, D.M.; Simons, L.M.; Berman, K.E.; Cheng, L.; DiBiase, B.N.; Hung, M.E.; Lucks, J.B.; Hultquist, J.F.; Leonard, J.N. Genetically encoding multiple functionalities into extracellular vesicles for the targeted delivery of biologics to T cells. Nat. Biomed. Eng. 2024, 8, 397–414. [Google Scholar] [CrossRef]
- Kim, H.I.; Park, J.; Zhu, Y.; Wang, X.; Han, Y.; Zhang, D. Recent advances in extracellular vesicles for therapeutic cargo delivery. Exp. Mol. Med. 2024, 56, 836–849. [Google Scholar] [CrossRef]
- Miyaki, S.; Lotz, M.K. Extracellular vesicles in cartilage homeostasis and osteoarthritis. Curr. Opin. Rheumatol. 2018, 30, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xu, X.; Xu, L.; Iqbal, Z.; Ouyang, K.; Zhang, H.; Wen, C.; Duan, L.; Xia, J. Chondrocyte-specific genomic editing enabled by hybrid exosomes for osteoarthritis treatment. Theranostics 2022, 12, 4866–4878. [Google Scholar] [CrossRef]
- Liang, Y.; Xu, X.; Li, X.; Xiong, J.; Li, B.; Duan, L.; Wang, D.; Xia, J. Chondrocyte-Targeted MicroRNA Delivery by Engineered Exosomes toward a Cell-Free Osteoarthritis Therapy. ACS Appl. Mater. Interfaces 2020, 12, 36938–36947. [Google Scholar] [CrossRef]
- Yan, W.; Li, Y.; Xie, S.; Tao, W.A.; Hu, J.; Liu, H.; Zhang, G.; Liu, F.; Nie, Y.; Chen, X.; et al. Chondrocyte-Targeted Delivery System of Sortase A-Engineered Extracellular Vesicles Silencing MMP13 for Osteoarthritis Therapy. Adv. Health. Mater. 2024, 13, e2303510. [Google Scholar] [CrossRef]
- Liu, W.; Liu, A.; Li, X.; Sun, Z.; Sun, Z.; Liu, Y.; Wang, G.; Huang, D.; Xiong, H.; Yu, S.; et al. Dual-engineered cartilage-targeting extracellular vesicles derived from mesenchymal stem cells enhance osteoarthritis treatment via miR-223/NLRP3/pyroptosis axis: Toward a precision therapy. Bioact. Mater. 2023, 30, 169–183. [Google Scholar] [CrossRef]
- Chen, M.; Lu, Y.; Liu, Y.; Liu, Q.; Deng, S.; Liu, Y.; Cui, X.; Liang, J.; Zhang, X.; Fan, Y.; et al. Injectable Microgels with Hybrid Exosomes of Chondrocyte-Targeted FGF18 Gene-Editing and Self-Renewable Lubrication for Osteoarthritis Therapy. Adv. Mater. 2024, 36, e2312559. [Google Scholar] [CrossRef]
- Peruzzi, J.A.; Gunnels, T.F.; Edelstein, H.I.; Lu, P.; Baker, D.; Leonard, J.N.; Kamat, N.P. Enhancing extracellular vesicle cargo loading and functional delivery by engineering protein-lipid interactions. Nat. Commun. 2024, 15, 5618. [Google Scholar] [CrossRef]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, D.; Liu, Z.; Zhou, F.; Dai, J.; Wu, B.; Zhou, J.; Heng, B.C.; Zou, X.H.; Ouyang, H.; et al. Exosomes from embryonic mesenchymal stem cells alleviate osteoarthritis through balancing synthesis and degradation of cartilage extracellular matrix. Stem Cell Res. Ther. 2017, 8, 189. [Google Scholar] [CrossRef]
- He, L.; He, T.; Xing, J.; Zhou, Q.; Fan, L.; Liu, C.; Chen, Y.; Wu, D.; Tian, Z.; Liu, B.; et al. Bone marrow mesenchymal stem cell-derived exosomes protect cartilage damage and relieve knee osteoarthritis pain in a rat model of osteoarthritis. Stem Cell Res. Ther. 2020, 11, 276. [Google Scholar] [CrossRef]
- Li, F.; Wu, J.; Li, D.; Hao, L.; Li, Y.; Yi, D.; Yeung, K.W.K.; Chen, D.; Lu, W.W.; Pan, H.; et al. Engineering stem cells to produce exosomes with enhanced bone regeneration effects: An alternative strategy for gene therapy. J. Nanobiotechnol. 2022, 20, 135. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Ni, J.; Witherel, C.E.; Yang, M.; Burdick, J.A.; Wen, C.; Wong, S.H.D. Harnessing Tissue-derived Extracellular Vesicles for Osteoarthritis Theranostics. Theranostics 2022, 12, 207–231. [Google Scholar] [CrossRef]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and challenges of extracellular vesicle-based drug delivery system: Considering cell source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Jones, E.; Kouroupis, D. The Use of Mesenchymal Stem/Stromal Cell-Derived Extracellular Vesicles in the Treatment of Osteoarthritis: Insights from Preclinical Studies. Bioengineering 2024, 11, 961. [Google Scholar] [CrossRef]
- Chen, P.; Zheng, L.; Wang, Y.; Tao, M.; Xie, Z.; Xia, C.; Gu, C.; Chen, J.; Qiu, P.; Mei, S.; et al. Desktop-stereolithography 3D printing of a radially oriented extracellular matrix/mesenchymal stem cell exosome bioink for osteochondral defect regeneration. Theranostics 2019, 9, 2439–2459. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, Z.; Gao, R.; Yuan, J.; Zhang, J.; Li, H.; Xie, Z.; Wang, Y. Controlled release of MSC-derived small extracellular vesicles by an injectable Diels-Alder crosslinked hyaluronic acid/PEG hydrogel for osteoarthritis improvement. Acta Biomater. 2021, 128, 163–174. [Google Scholar] [CrossRef]
- Schwartz, G.; Rana, S.; Jackson, A.R.; Leñero, C.; Best, T.M.; Kouroupis, D.; Travascio, F. Human mesenchymal stem/stromal cell-derived extracellular vesicle transport in meniscus fibrocartilage. J. Orthop. Res. 2024, 43, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, J.; Yuan, H.; Xu, Z.; Li, Q.; Niu, X.; Hu, B.; Wang, Y.; Li, X. Exosomes Secreted by Human-Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Repair Critical-Sized Bone Defects through Enhanced Angiogenesis and Osteogenesis in Osteoporotic Rats. Int. J. Biol. Sci. 2016, 12, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.H.; Kim, H.K.; Jung, G.Y.; Jung, Y.J.; Lee, K.S.; Yun, Y.E.; Han, J.; Lee, J.; Kim, W.S.; Choi, J.S.; et al. Small extracellular vesicles from human adipose-derived stem cells attenuate cartilage degeneration. J. Extracell. Vesicles 2020, 9, 1735249. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, L.; Ma, C.; Wang, G.; Zhang, Y.; Sun, S. Exosomes derived from platelet-rich plasma present a novel potential in alleviating knee osteoarthritis by promoting proliferation and inhibiting apoptosis of chondrocyte via Wnt/β-catenin signaling pathway. J. Orthop. Surg. Res. 2019, 14, 470. [Google Scholar] [CrossRef]
- Nguyen, V.V.T.; Witwer, K.W.; Verhaar, M.C.; Strunk, D.; van Balkom, B.W.M. Functional assays to assess the therapeutic potential of extracellular vesicles. J. Extracell. Vesicles 2020, 10, e12033. [Google Scholar] [CrossRef]
- Tian, J.; Han, Z.; Song, D.; Peng, Y.; Xiong, M.; Chen, Z.; Duan, S.; Zhang, L. Engineered Exosome for Drug Delivery: Recent Development and Clinical Applications. Int. J. Nanomed. 2023, 18, 7923–7940. [Google Scholar] [CrossRef]
- Ramaswamy Reddy, S.H.; Reddy, R.; Babu, N.C.; Ashok, G.N. Stem-cell therapy and platelet-rich plasma in regenerative medicines: A review on pros and cons of the technologies. J. Oral Maxillofac. Pathol. 2018, 22, 367–374. [Google Scholar] [CrossRef]
- Labusek, N.; Mouloud, Y.; Köster, C.; Diesterbeck, E.; Tertel, T.; Wiek, C.; Hanenberg, H.; Horn, P.A.; Felderhoff-Müser, U.; Bendix, I.; et al. Extracellular vesicles from immortalized mesenchymal stromal cells protect against neonatal hypoxic-ischemic brain injury. Inflamm. Regen. 2023, 43, 24. [Google Scholar] [CrossRef]
- Wiest, E.F.; Zubair, A.C. Challenges of manufacturing mesenchymal stromal cell-derived extracellular vesicles in regenerative medicine. Cytotherapy 2020, 22, 606–612. [Google Scholar] [CrossRef]
- Valderrama, J.D.; Gutierrez, F.R. Chapter 5—Engineering of Bacterial Outer Membrane Vesicles: Potential Applications for the Development of Vaccines. In Lipid Nanocarriers for Drug Targeting; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 199–229. [Google Scholar]
- Busatto, S.; Vilanilam, G.; Ticer, T.; Lin, W.-L.; Dickson, D.W.; Shapiro, S.; Bergese, P.; Wolfram, J. Tangential Flow Filtration for Highly Efficient Concentration of Extracellular Vesicles from Large Volumes of Fluid. Cells 2018, 7, 273. [Google Scholar] [CrossRef]
- Welton, J.L.; Webber, J.P.; Botos, L.-A.; Jones, M.; Clayton, A. Ready-made chromatography columns for extracellular vesicle isolation from plasma. J. Extracell. Vesicles 2015, 4, 27269. [Google Scholar] [CrossRef] [PubMed]
- Gámez-Valero, A.; Monguió-Tortajada, M.; Carreras-Planella, L.; la Franquesa, M.; Beyer, K.; Borràs, F.E. Size-Exclusion Chromatography-based isolation minimally alters Extracellular Vesicles’ characteristics compared to precipitating agents. Sci. Rep. 2016, 6, 33641. [Google Scholar] [CrossRef]
- Gallart-Palau, X.; Serra, A.; Wong, A.S.W.; Sandin, S.; Lai, M.K.P.; Chen, C.P.; Kon, O.L.; Sze, S.K. Extracellular vesicles are rapidly purified from human plasma by PRotein Organic Solvent PRecipitation (PROSPR). Sci. Rep. 2015, 5, 14664. [Google Scholar] [CrossRef] [PubMed]
- Jeyaram, A.; Jay, S.M. Preservation and Storage Stability of Extracellular Vesicles for Therapeutic Applications. AAPS J. 2018, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Yamamoto, Y.; Sato, T.-A.; Ochiya, T. Extracellular vesicles as trans-genomic agents: Emerging roles in disease and evolution. Cancer Sci. 2017, 108, 824–830. [Google Scholar] [CrossRef]
- Fischer, S.; Cornils, K.; Speiseder, T.; Badbaran, A.; Reimer, R.; Indenbirken, D.; Grundhoff, A.; Brunswig-Spickenheier, B.; Alawi, M.; Lange, C. Indication of Horizontal DNA Gene Transfer by Extracellular Vesicles. PLoS ONE 2016, 11, e0163665. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Drug Name | Targeted Molecule | Mechanism of Action | Stage of Disease in Study | Effects/Findings | Clinical and/or Safety Considerations |
|---|---|---|---|---|---|
| Infliximab | TNF-α | Monoclonal antibody | Radiographic primary OA of any joint [8] or any hand OA [14] | Reduced pain in OA, improved structural outcomes (structural damage, erosive progression, and presence of active disease) in hand OA but no symptom relief [8,14] | Highlights role of TNF-α in OA-related inflammation and pain [8] |
| Adalimumab | TNF-α | Monoclonal antibody | Radiographic primary OA of any joint [8] or Moderate to severe KOA [15] | Reduced pain, particularly effective when administered intra-articularly [15] | Highlights role of TNF-α in OA-related inflammation and pain, more data needed on long-term safety profile [8,15] |
| Tanezumab | NGF | Monoclonal antibody | Patients with OA unresponsive to non-opioid medications [16] | Effective in relieving pain in hip and knee OA, with continued relief for up to 56 weeks [16,17,18] | NGF-targeting therapies have inconsistent safety profiles [18,19] |
| Fasinumab | NGF | Monoclonal antibody | Patients with moderate to severe knee or hip OA [20] | Reduced pain and improved function (as measured by WOMAC function score and PGA function score) in hip and knee OA in phase IIb/III trials [13,20] | More research needed due to safety concerns [19,20] |
| Fulranumab | NGF | Monoclonal antibody | Moderate to severe KOA [21] or moderate to severe knee or hip OA [22] | Comparable efficacy to oxycodone in phase II trials, further validated in phase III trials [21,22] | Safety concerns remain inconsistent across studies [19] |
| Bevacizumab | VEGF | Monoclonal antibody | Rabbit model of KOA [23,24,25] | Reduced joint inflammation, synovial proliferation, and cartilage degradation; local intra-articular administration was safer and more effective than systemic administration in animal models [23,24,25] | Most promising monoclonal antibody for slowing OA progression in animal studies [26] |
| Canakinumab | IL-1β | Monoclonal antibody | Patient with knee and hip OA [27] | Reduced need for hip or knee arthroplasty, suggesting disease-modifying potential [27] | IL-1 remains a key cytokine target in OA, but efficacy varies [27] |
| AMG 108 | IL-1 receptor | Monoclonal antibody | Patient with KOA [28] | Unsuccessful in alleviating pain in knee OA [28] | Minimal clinical benefit [28] |
| Anakinra | IL-1 receptor | IL-1 receptor antagonist | Patients with KAO [29] | Did not show significant improvement in OA symptoms compared to placebo [17,29] | Demonstrates differences in inflammatory pathways between RA and OA [17,29] |
| M6495 | ADAMTS5 | Monoclonal antibody | Patients with OA and pain greater than or equal to 40 on WOMAC scale [30] | Under investigation for OA therapy [30] | Emerging target for OA treatment [30] |
| IL-1 antibodies | IL-1 | Antibodies against IL-1 | Patients diagnosed with KOA [31] | Reduced OA symptoms, improved joint function (as measured with WOMAC and KOOS function scores) [31] | IL-1 inhibitors had safety concerns despite promising results [31] |
| IL-1 receptor antibodies | IL-1 receptor | Antibodies against IL-1 receptor | Patients diagnosed with KOA [31] | Did not show success in OA treatment [31] | Raises questions about effectiveness in different joints [31] |
| Loading Method | Endogenous or Exogenous | Active or Passive | Mechanism | Advantages/Disadvantages |
|---|---|---|---|---|
| Co-incubation | Endogenous or exogenous | Passive | Cells are incubated with drugs, allowing uptake before EV secretion, or EVs are incubated with drugs, allowing for simple diffusion of cargo into EV [60,63] | Simple, but efficiency depends on drug type and conditions [60,63] |
| Transfection | Endogenous or Exogenous | Active | Genetic modification of donor cells to express therapeutic cargo [60] | Allows precise cargo control, but has low loading efficiency [60] |
| Genetic Engineering | Endogenous or Exogenous | Active | Modifies donor cells or EV surface/cargo for precise targeting and therapeutic efficacy [60,61,62,63] | Enables specific therapeutic cargo production, but complex and may have safety concerns [60,61,62,63] |
| Hypoxic conditions | Endogenous | Active | Cells exposed to low oxygen, influencing EV content [63] | Can enhance therapeutic properties, but is complex and requires specific conditions [60] |
| Mechanical Stress | Endogenous | Active | Applying physical forces to modify EV composition [60] | Can enhance EV production, but may unpredictably alter cargo composition [60] |
| 3D co-culturing | Endogenous | Active | Culturing cells in a 3D environment to enhance EV loading [60] | More physiologically relevant, but complex to optimize [60] |
| Endocytosis/Receptor mediated uptake | Endogenous | Passive | Drugs enter cells and get packaged into EVs during biogenesis (can be regulated by cell signaling pathways) [60] | Natural process, but efficiency varies, can be optimized by utilizing receptors for specific targeting [60] |
| Ultrasonication | Exogenous | Active | Sound waves temporarily disrupt membranes for drug entry [60,61,62,63] | High efficiency, but may damage EVs [60,61,62,63] |
| Electroporation | Exogenous | Active | Electric fields create pores in membranes for cargo loading [60] | Effective for large molecules, but can cause aggregation [60] |
| Freeze-thaw cycles | Exogenous | Active | Cycles of freezing and thawing to facilitate cargo loading [60] | Simple, but can lead to cargo degradation [60] |
| Study | EV Source | Model | Dose & Route | Frequency/Duration | Key Outcomes |
|---|---|---|---|---|---|
| Wang et al., 2017 [72] | Embryonic stem cell-derived EVs | Rat OA model | 5 μL (1 × 106 EVs) intra-articular per joint | Every 3 days for 4 weeks | Improved cartilage integrity, ↓ MMPs/aggrecanases, ↑ collagen II, ↓ ADAMTS5 in IL-1β presence |
| Woo et al., 2020 [83] | hASC EVs | Rat OA (subacute & chronic), mouse PTOA (DMM surgery) | 1 × 108 particles in 30 μL per joint | Subacute OA: Weekly x3 (Day 7–28 post-induction) Chronic OA: Twice weekly x6 (Day 14–54 post-induction) DMM: Weekly post-surgery | ↓ Mankin and OARSI scores, preserved cartilage structure, ↓ NITEGE & MMP-13-positive chondrocytes |
| Liu et al., 2019 [84] | PRP-Exos | Rabbit OA model | 100 μg/mL intra-articular injections | Weekly x6 post-surgery | ↑ chondrocyte count, ↓ OARSI scores vs. OA and PRP-As groups |
| Clinical Trial (NCT06431152) | Allogeneic umbilical cord MSC-EVs | Knee OA (clinical trial) | 2 × 109, 6 × 109, 2 × 1010 particles per injection | Single IA injection, 1-year follow-up | Dose optimization trial; clinical outcomes being assessed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drohat, P.; Baron, M.; Kaplan, L.D.; Best, T.M.; Kouroupis, D. Long-Acting Extracellular Vesicle-Based Biologics in Osteoarthritis Immunotherapy. Bioengineering 2025, 12, 525. https://doi.org/10.3390/bioengineering12050525
Drohat P, Baron M, Kaplan LD, Best TM, Kouroupis D. Long-Acting Extracellular Vesicle-Based Biologics in Osteoarthritis Immunotherapy. Bioengineering. 2025; 12(5):525. https://doi.org/10.3390/bioengineering12050525
Chicago/Turabian StyleDrohat, Philip, Max Baron, Lee D. Kaplan, Thomas M. Best, and Dimitrios Kouroupis. 2025. "Long-Acting Extracellular Vesicle-Based Biologics in Osteoarthritis Immunotherapy" Bioengineering 12, no. 5: 525. https://doi.org/10.3390/bioengineering12050525
APA StyleDrohat, P., Baron, M., Kaplan, L. D., Best, T. M., & Kouroupis, D. (2025). Long-Acting Extracellular Vesicle-Based Biologics in Osteoarthritis Immunotherapy. Bioengineering, 12(5), 525. https://doi.org/10.3390/bioengineering12050525