
In Silico Identification of LSD1 Inhibition-Responsive Targets in Small Cell Lung Cancer



Abstract

1. Introduction
2. Material and Methods
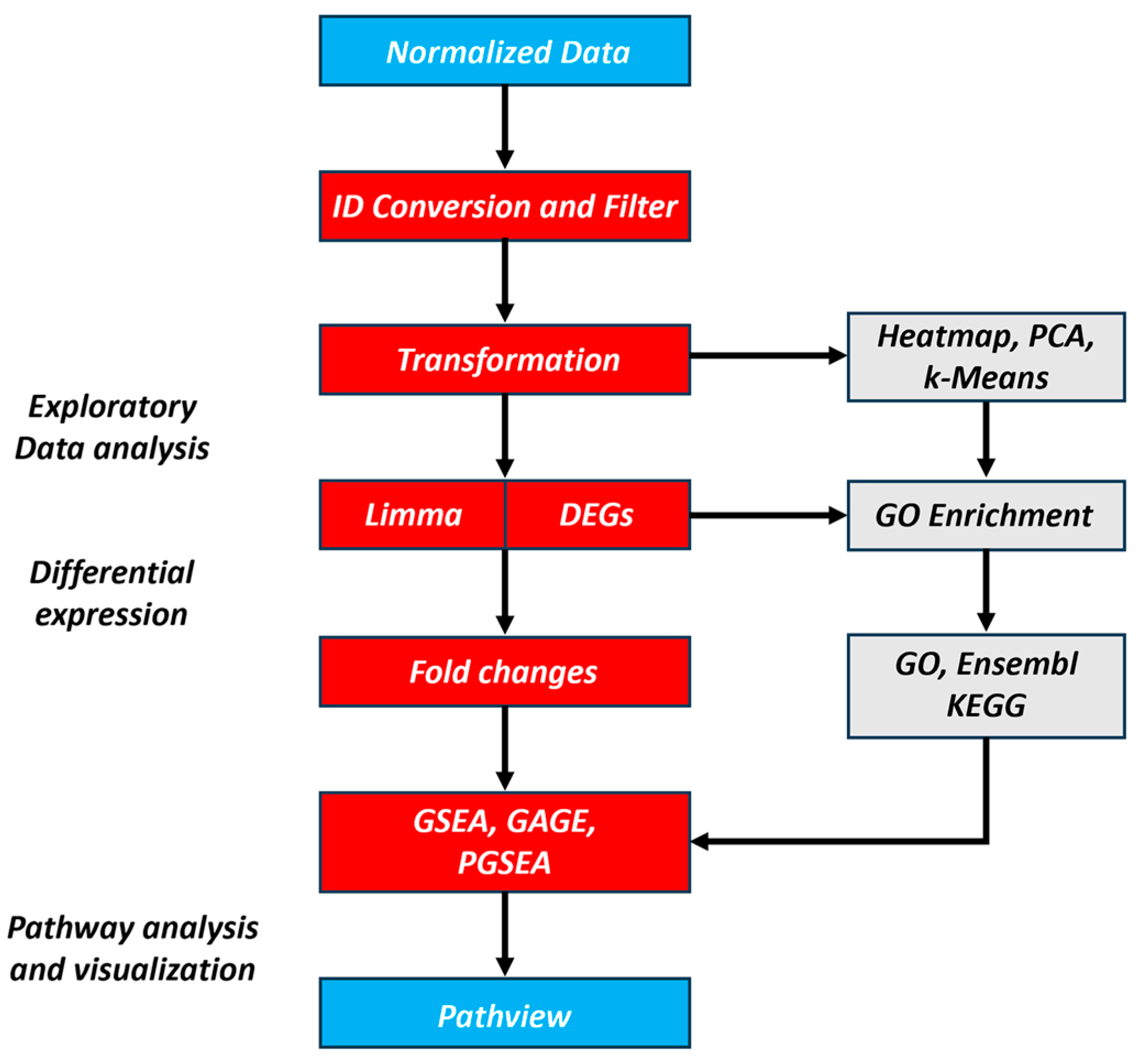
2.1. Bioinformatic Processing of Public RNA-Seq Data
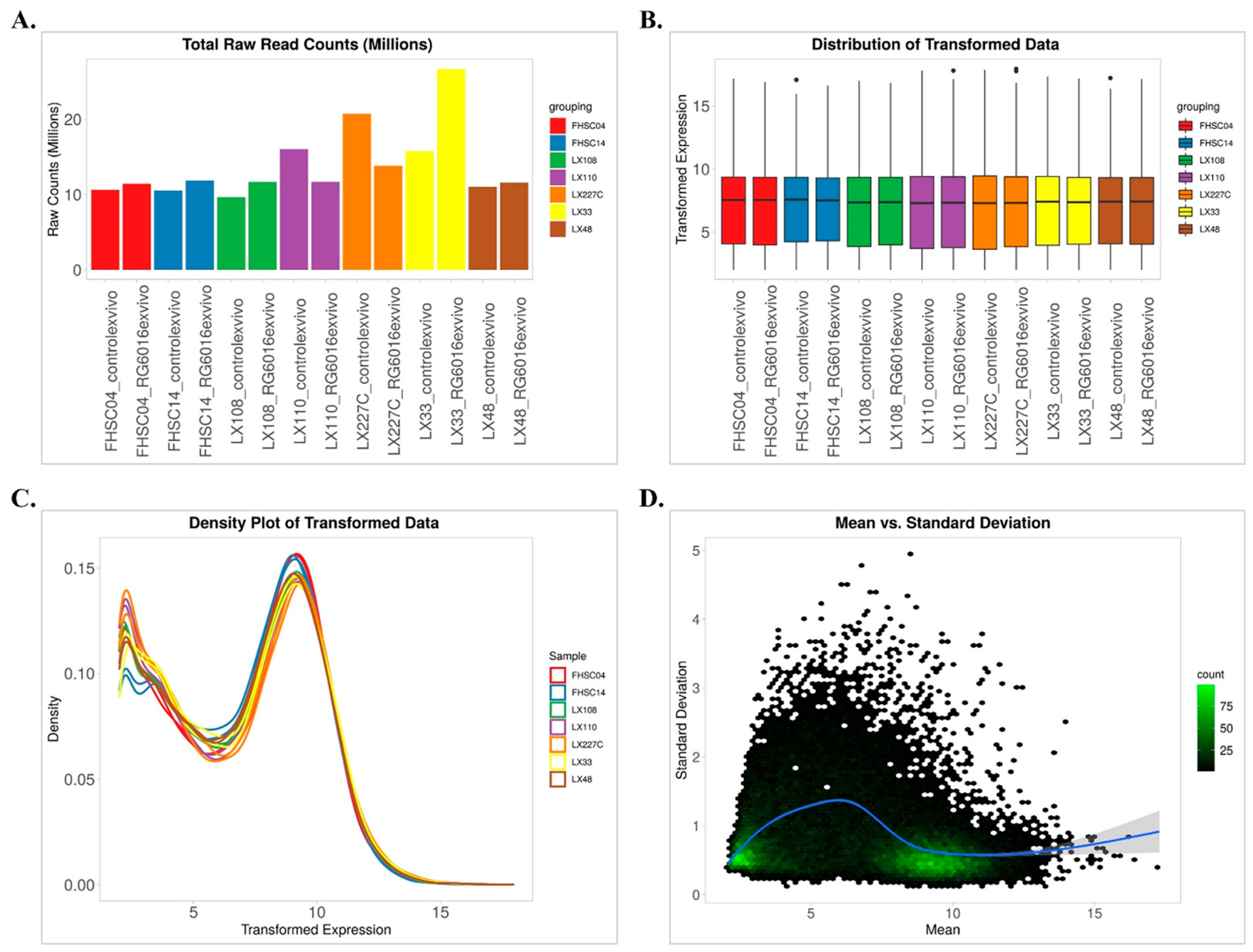
2.2. Data Preprocessing
2.3. Principal Component Analysis
2.4. Molecular Docking
3. Results
3.1. Correlation Matrix Among PDX Samples
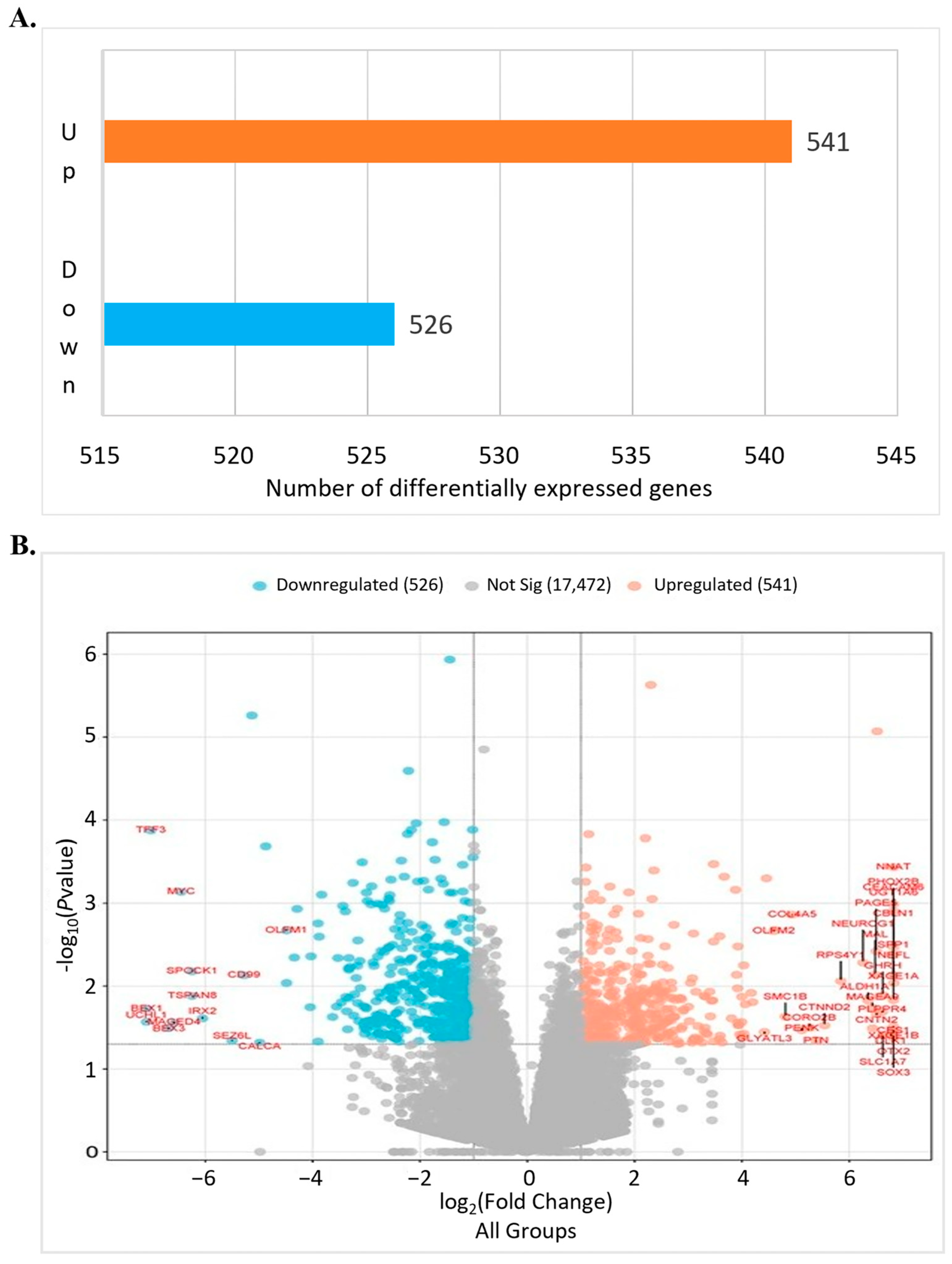
3.2. Differentially Expressed Genes2 (DEG2) Analysis
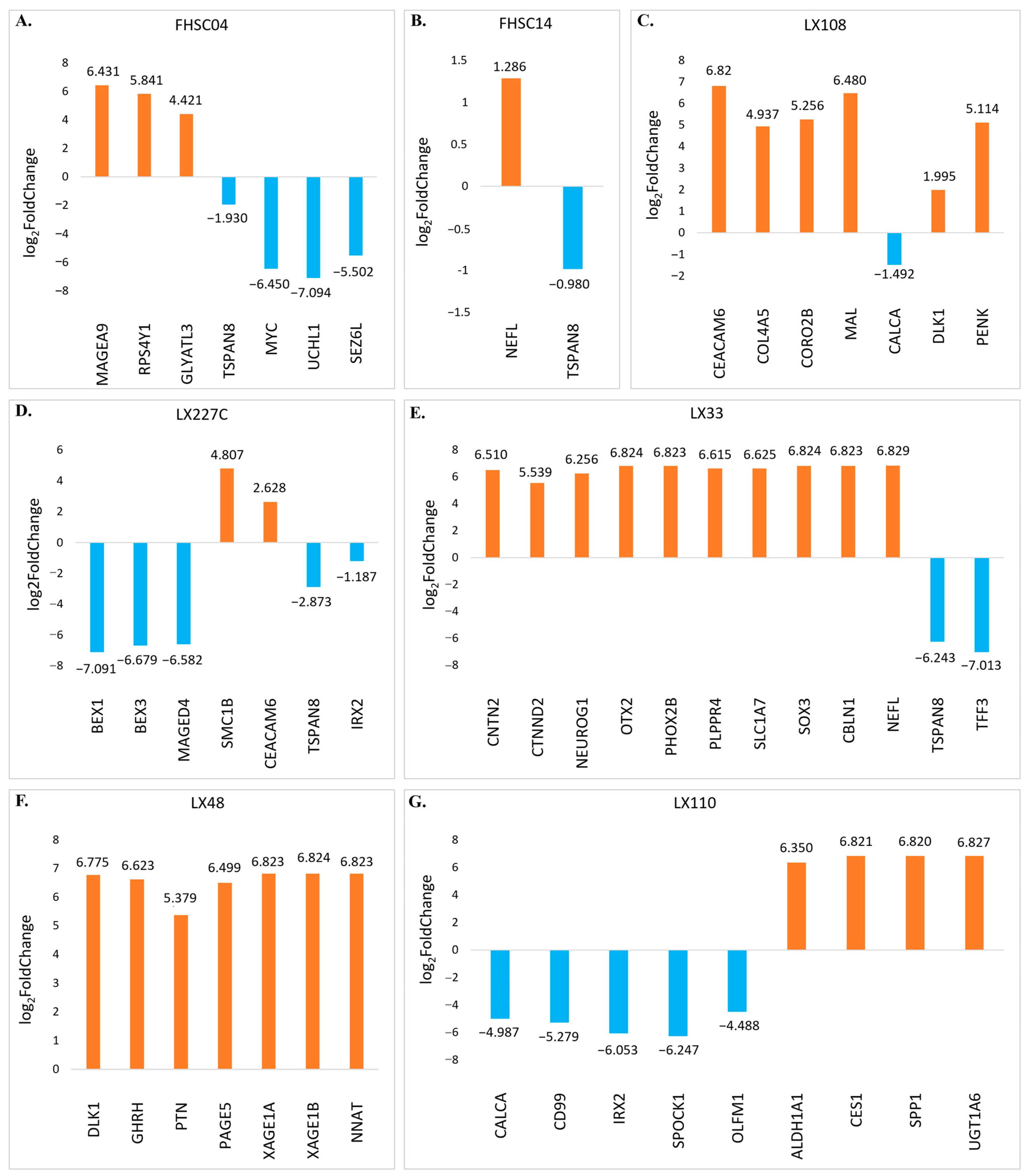
3.3. DEG2 Profiles Across All PDX Samples Following RG6016 Treatment
3.4. Pathway Enrichment Analysis of DEGs
3.4.1. Upregulated Pathways
3.4.2. Downregulated Pathways
3.5. Network Analysis
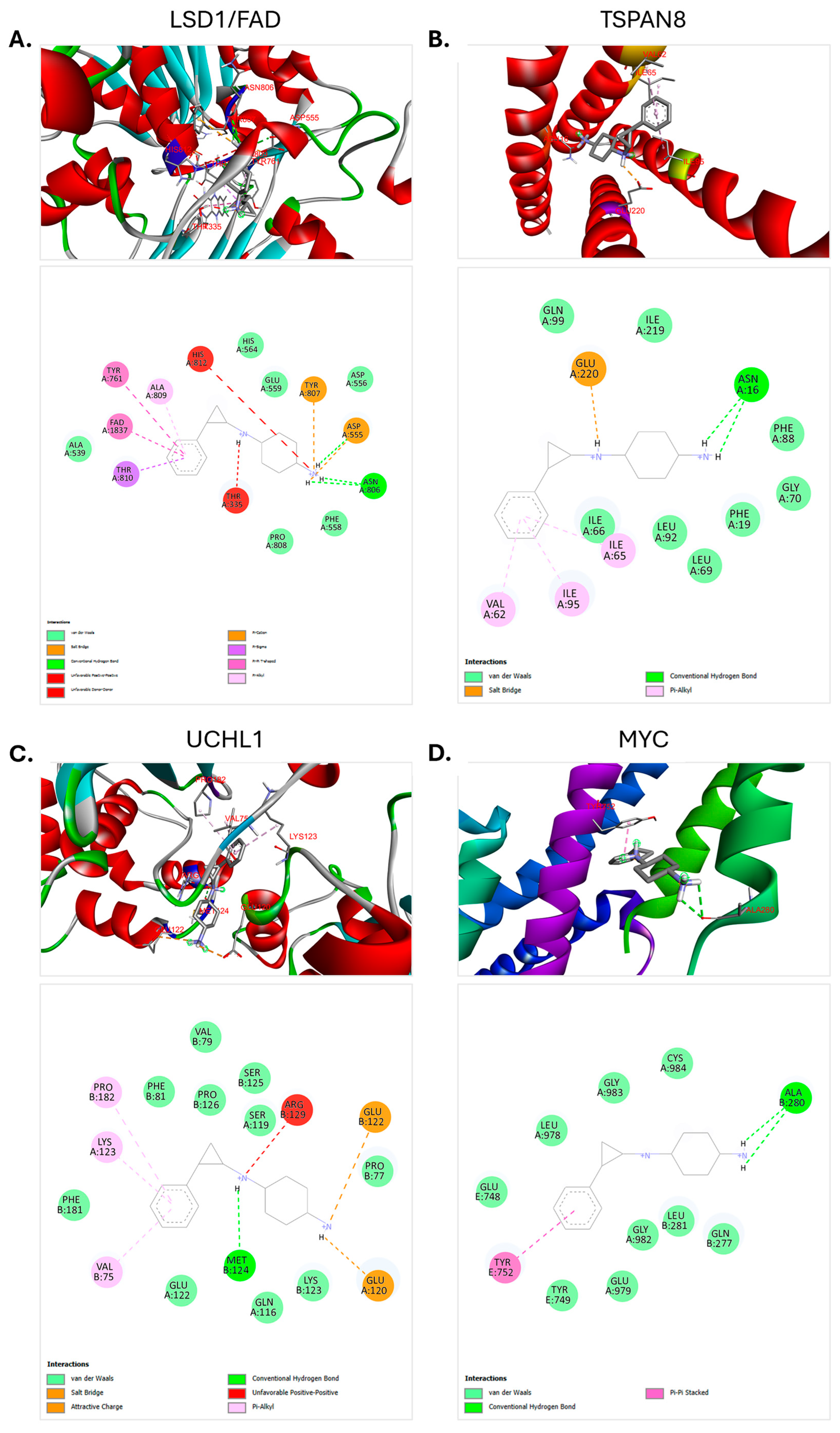
3.6. Molecular Docking Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Primers 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wei, Y.; Hung, R.J.; Liu, G.; Su, L.; Zhang, R.; Zong, X.; Zhang, Z.-F.; Morgenstern, H.; Brüske, I.; et al. Associated links among smoking, chronic obstructive pulmonary disease, and small cell lung cancer: A pooled analysis in the International Lung Cancer Consortium. EBioMedicine 2015, 2, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-B.; Dunn, C.T.; Park, K.-S. Recent progress in mapping the emerging landscape of the small-cell lung cancer genome. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Baine, M.K.; Hsieh, M.-S.; Lai, W.V.; Egger, J.V.; Jungbluth, A.A.; Daneshbod, Y.; Beras, A.; Spencer, R.; Lopardo, J.; Bodd, F.; et al. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: A comprehensive immunohistochemical and histopathologic characterization. J. Thorac. Oncol. 2020, 15, 1823–1835. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef]
- Yang, D.; Denny, S.K.; Greenside, P.G.; Chaikovsky, A.C.; Brady, J.J.; Ouadah, Y.; Granja, J.M.; Jahchan, N.S.; Lim, J.S.; Kwok, S.; et al. Intertumoral heterogeneity in SCLC is influenced by the cell type of origin. Cancer Discov. 2018, 8, 1316–1331. [Google Scholar] [CrossRef]
- Ko, J.; Winslow, M.M.; Sage, J. Mechanisms of small cell lung cancer metastasis. EMBO Mol. Med. 2021, 13, e13122. [Google Scholar] [CrossRef]
- Hodgkinson, C.L.; Morrow, C.J.; Li, Y.; Metcalf, R.L.; Rothwell, D.G.; Trapani, F.; Polanski, R.; Burt, D.J.; Simpson, K.L.; Morris, K.; et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat. Med. 2014, 20, 897–903. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- McGrath, J.P.; Williamson, K.E.; Balasubramanian, S.; Odate, S.; Arora, S.; Hatton, C.; Edwards, T.M.; O’Brien, T.; Magnuson, S.; Stokoe, D.; et al. Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 2016, 76, 1975–1988. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Sugino, N.; Kawahara, M.; Tatsumi, G.; Kanai, A.; Matsui, H.; Yamamoto, R.; Nagai, Y.; Fujii, S.; Shimazu, Y.; Hishizawa, M.; et al. A novel LSD1 inhibitor NCD38 ameliorates MDS-related leukemia with complex karyotype by attenuating leukemia programs via activating super-enhancers. Leukemia 2017, 31, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Rudneva, V.A.; Erkek, S.; Zapatka, M.; Chau, L.Q.; Tacheva-Grigorova, S.K.; Garancher, A.; Rusert, J.M.; Aksoy, O.; Lea, R.; et al. Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nat. Commun. 2019, 10, 332. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Ishikawa, Y.; Mizutani, A.; Iwasaki, S.; Matsumoto, S.; Kamada, Y.; Nomura, T.; Nakamura, K. LSD1 inhibitor T-3775440 inhibits SCLC cell proliferation by disrupting LSD1 interactions with SNAG domain proteins INSM1 and GFI1B. Cancer Res. 2017, 77, 4652–4662. [Google Scholar] [CrossRef]
- Li, M.; Dai, M.; Cheng, B.; Li, S.; Guo, E.; Fu, J.; Ma, T.; Yu, B. Strategies that regulate LSD1 for novel therapeutics. Acta Pharm. Sin. B 2024, 14, 1494–1507. [Google Scholar] [CrossRef]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 2019, 12, 129. [Google Scholar] [CrossRef]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.; Somervaille, T.C.; et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 2018, 33, 495–511.e412. [Google Scholar] [CrossRef]
- Salamero, O.; Montesinos, P.; Willekens, C.; Pérez-Simón, J.A.; Pigneux, A.; Récher, C.; Popat, R.; Carpio, C.; Molinero, C.; Mascaró, C.; et al. First-in-human phase I study of iadademstat (ORY-1001): A first-in-class lysine-specific histone demethylase 1A inhibitor, in relapsed or refractory acute myeloid leukemia. J. Clin. Oncol. 2020, 38, 4260–4273. [Google Scholar] [CrossRef]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci. Signal 2019, 12. [Google Scholar] [CrossRef]
- Kivrak, M.; Nalkiran, I.; Sevim Nalkiran, H. Exploring the Therapeutic Potential of the DOT1L Inhibitor EPZ004777 Using Bioinformatics and Molecular Docking Approaches in Acute Myeloid Leukemia. Curr. Issues Mol. Biol. 2025, 47, 173. [Google Scholar] [CrossRef]
- Jolliffe, I. Principal Component Analysis. In Encyclopedia of Statistics in Behavioral Science; Everitt, B.S., Howell, D.C., Eds.; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; et al. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Nalkiran, I.; Sevim Nalkiran, H. Repurposing ProTAME for Bladder Cancer: A Combined Therapeutic Approach Targeting Cell Migration and MMP Regulation. Biology 2025, 14, 263. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 2016, 893, 1–19. [Google Scholar]
- Kalemkerian, G.P.; Akerley, W.; Bogner, P.; Borghaei, H.; Chow, L.Q.; Downey, R.J.; Gandhi, L.; Ganti, A.K.P.; Govindan, R.; Grecula, J.C.; et al. Small cell lung cancer. J. Natl. Compr. Canc Netw. 2013, 11, 78–98. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef]
- George, J.L.J.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; Müller, C.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Mai, A. Targeting histone demethylases: A new avenue for the fight against cancer. Genes. Cancer 2011, 2, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Pharmacologic targeting of hypoxia-inducible factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Proline hydroxylation and gene expression. Annu. Rev. Biochem. 2005, 74, 115–128. [Google Scholar] [CrossRef]
- Wass, M.; Göllner, S.; Besenbeck, B.; Schlenk, R.F.; Mundmann, P.; Göthert, J.R.; Noppeney, R.; Schliemann, C.; Mikesch, J.-H.; Lenz, G.; et al. A proof of concept phase I/II pilot trial of LSD1 inhibition by tranylcypromine combined with ATRA in refractory/relapsed AML patients not eligible for intensive therapy. Leukemia 2021, 35, 701–711. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef]
- Claas, C.; Seiter, S.; Claas, A.; Savelyeva, L.; Schwab, M.; Zöller, M. Association between the rat homologue of CO-029, a metastasis-associated tetraspanin molecule and consumption coagulopathy. J. Cell Biol. 1998, 141, 267–280. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, Y.; Zheng, W.; Lu, S. CO-029 is overexpressed in gastric cancer and mediates the effects of EGF on gastric cancer cell proliferation and invasion. Int. J. Mol. Med. 2015, 35, 798–802. [Google Scholar] [CrossRef]
- Kanetaka, K.; Sakamoto, M.; Yamamoto, Y.; Yamasaki, S.; Lanza, F.; Kanematsu, T.; Hirohashi, S. Overexpression of tetraspanin CO-029 in hepatocellular carcinoma. J. Hepatol. 2001, 35, 637–642. [Google Scholar] [CrossRef]
- Berthier-Vergnes, O.; El Kharbili, M.; de la Fouchardière, A.; Pointecouteau, T.; Verrando, P.; Wierinckx, A.; Lachuer, J.; Le Naour, F.; Lamartine, J. Gene expression profiles of human melanoma cells with different invasive potential reveal TSPAN8 as a novel mediator of invasion. Br. J. Cancer 2011, 104, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.-J.; Wu, Y.-B.; Cai, S.; Pan, Y.-X.; Liu, W.; Bian, L.-G.; Sun, B.; Sun, Q.-F. Over-expression of tetraspanin 8 in malignant glioma regulates tumor cell progression. Biochem. Biophys. Res. Commun. 2015, 458, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Ngoc Anh Suong, D.; Thi Phuong Thao, D.; Masamitsu, Y.; Linh Thuoc, T. Ubiquitin carboxyl hydrolase L1 significance for human diseases. Protein Pept. Lett. 2014, 21, 624–630. [Google Scholar] [CrossRef]
- Bishop, P.; Rocca, D.; Henley, J.M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473, 2453–2462. [Google Scholar] [CrossRef]
- Hibi, K.; Westra, W.H.; Borges, M.; Goodman, S.; Sidransky, D.; Jen, J. PGP9. 5 as a candidate tumor marker for non-small-cell lung cancer. Am. J. Pathol. 1999, 155, 711–715. [Google Scholar] [CrossRef]
- Kim, H.; Kim, Y.; Lim, S.; Nam, Y.; Jeong, J.; Lee, K. Ubiquitin C-terminal hydrolase-L1 is a key regulator of tumor cell invasion and metastasis. Oncogene 2009, 28, 117–127. [Google Scholar] [CrossRef]
- Lien, H.-C.; Wang, C.-C.; Lin, C.-H.; Lu, Y.-S.; Huang, C.-S.; Hsiao, L.-P.; Yao, Y.-T. Differential expression of ubiquitin carboxy-terminal hydrolase L1 in breast carcinoma and its biological significance. Hum. Pathol. 2013, 44, 1838–1848. [Google Scholar] [CrossRef]
- Lien, H.-C.; Wang, C.-C.; Huang, C.-S.; Yang, Y.-W.; Kuo, W.-H.; Yao, Y.-T. Ubiquitin carboxy-terminal hydrolase L1 may be involved in the development of mammary phyllodes tumors. Virchows Arch. 2013, 462, 155–161. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, W.; Xu, J.; Wang, H.; Zhang, Z.; Chu, C.; Liu, X.; Zou, Q. UCH-L1 involved in regulating the degradation of EGFR and promoting malignant properties in drug-resistant breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 12500. [Google Scholar]
- Wulfänger, J.; Biehl, K.; Tetzner, A.; Wild, P.; Ikenberg, K.; Meyer, S.; Seliger, B. Heterogeneous expression and functional relevance of the ubiquitin carboxyl-terminal hydrolase L1 in melanoma. Int. J. Cancer 2013, 133, 2522–2532. [Google Scholar] [CrossRef]
- Kim, H.J.; Magesh, V.; Lee, J.-J.; Kim, S.; Knaus, U.G.; Lee, K.-J. Ubiquitin C-terminal hydrolase-L1 increases cancer cell invasion by modulating hydrogen peroxide generated via NADPH oxidase 4. Oncotarget 2015, 6, 16287. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zeng, B.; Ma, J.; Wan, C. Comparative proteomic analysis of osteosarcoma cell and human primary cultured osteoblastic cell. Cancer Investig. 2009, 27, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Qiao, G.; Min, D.; Zhang, Z.; Lin, F.; Yang, Q.; Feng, T.; Tang, L.; Sun, Y.; Zhao, H.; et al. Heterogeneous expression and biological function of ubiquitin carboxy-terminal hydrolase-L1 in osteosarcoma. Cancer Lett. 2015, 359, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Dang, C.V.; Resar, L.M.; Emison, E.; Kim, S.; Li, Q.; Prescott, J.E.; Wonsey, D.; Zeller, K. Function of the c-Myc oncogenic transcription factor. Exp. Cell Res. 1999, 253, 63–77. [Google Scholar] [CrossRef]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Den Hollander, J.; Rimpi, S.; Doherty, J.R.; Rudelius, M.; Buck, A.; Hoellein, A.; Kremer, M.; Graf, N.; Scheerer, M.; Hall, M.A.; et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010, 116, 1498–1505. [Google Scholar] [CrossRef]
- Kim, Y.; Nam, H.J.; Lee, J.; Park, D.Y.; Kim, C.; Yu, Y.S.; Kim, D.; Park, S.W.; Bhin, J.; Hwang, D.; et al. Methylation-dependent regulation of HIF-1α stability restricts retinal and tumour angiogenesis. Nat. Commun. 2016, 7, 10347. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Z.; Xu, C.; Leng, X.; Cao, H.; Ouyang, G.; Xiao, W. Repression of hypoxia-inducible factor α signaling by Set7-mediated methylation. Nucleic Acids Res. 2015, 43, 5081–5098. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, J.H.; Choi, H.J.; Won, H.; Joo, H.; Shin, D.-H.; Park, M.-K.; Han, B.; Kim, K.P.; Lee, T.J.; et al. LSD1 demethylates HIF1α to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 2017, 36, 5512–5521. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Park, Y.S.; Cho, J.H.; Moon, B.; An, H.J.; Lee, J.Y.; Xie, Z.; Wang, Y.; Pocalyko, D.; Lee, D.C.; et al. Regulation of hypoxia responses by flavin adenine dinucleotide-dependent modulation of HIF-1α protein stability. EMBO J. 2017, 36, 1011–1028. [Google Scholar] [CrossRef]
- Goto, Y.; Zeng, L.; Yeom, C.J.; Zhu, Y.; Morinibu, A.; Shinomiya, K.; Kobayashi, M.; Hirota, K.; Itasaka, S.; Yoshimura, M.; et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1α. Nat. Commun. 2015, 6, 6153. [Google Scholar] [CrossRef]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef]
- Liu, S.; Chai, T.; Garcia-Marques, F.; Yin, Q.; Hsu, E.C.; Shen, M.; Shaw Toland, A.M.; Bermudez, A.; Hartono, A.B.; Massey, C.F.; et al. UCHL1 is a potential molecular indicator and therapeutic target for neuroendocrine carcinomas. Cell Rep. Med. 2024, 5, 101381. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | PDX Sample | Treatment |
|---|---|---|
| FHSC04_Control | FHSC04 | CONTROL |
| FHSC04_RG6016 | FHSC04 | RG6016 |
| FHSC14_Control | FHSC14 | CONTROL |
| FHSC14_RG6016 | FHSC14 | RG6016 |
| LX108_Control | LX108 | CONTROL |
| LX108_RG6016 | LX108 | RG6016 |
| LX110_Control | LX110 | CONTROL |
| LX110_RG6016 | LX110 | RG6016 |
| LX227C_Control | LX227C | CONTROL |
| LX227C_RG6016 | LX227C | RG6016 |
| LX33_Control | LX33 | CONTROL |
| LX33_RG6016 | LX33 | RG6016 |
| LX48_Control | LX48 | CONTROL |
| LX48_RG6016 | LX48 | RG6016 |
| Protein | Center at (X, Y, Z) | Dimension (Å) |
|---|---|---|
| LSD1/FAD complex | X: −5.864, Y: 60.314, Z: 93.813 | 40 Å × 40 Å × 40 Å |
| TSPAN8 | X: −35.139, Y: −19.879, Z: 50.937 | 100 Å × 100 Å × 100 Å |
| UCHL1 | X: 84.171, Y: 25.697, Z: 19.341 | 40 Å × 40 Å × 40 Å |
| MYC | X: 53.448, Y: 47.445, Z: 58.079 | 40 Å × 40 Å × 40 Å |
| BEX1 | X: −35.261, Y: −20.238, Z: 50.826 | 100 Å × 100 Å × 100 Å |
| BEX3 | X: 74.024, Y: 0.969, Z: −16.177 | 100 Å × 100 Å × 100 Å |
| CALCA | X: −5.523, Y: −2.272, Z: 0.537 | 100 Å × 100 Å × 100 Å |
| CD99 | X: −1.135, Y: −2.164, Z: −6.97 | 100 Å × 100 Å × 100 Å |
| IRX2 | X: −4.002, Y: 0.413, Z: −4.138 | 40 Å × 40 Å × 40 Å |
| MAGED4 | X: −2.805, Y: 5.328, Z: −3.031 | 40 Å × 40 Å × 40 Å |
| OLFM1 | X: 4.006, Y: 1.697, Z: −2.998 | 40 Å × 40 Å × 40 Å |
| SEZL6 | X: −6.505, Y: 3.144, Z: 16.417 | 100 Å × 100 Å × 100 Å |
| TFF3 | X: −1.078, Y: 2.169, Z: −4.762 | 40 Å × 40 Å × 40 Å |
| SPOCK1 | X: 0.809, Y: 3.861, Z: −0.615 | 40 Å × 40 Å × 40 Å |
| Up/Downregulated | Ensembl ID | Symbol | Entrez-Gene ID | log2FC Values | Description |
|---|---|---|---|---|---|
| Up | ENSG00000277586 | NEFL | 4747 | 6.829 | Neurofilament light chain |
| Up | ENSG00000167165 | UGT1A6 | 54578 | 6.827 | UDP glucuronosyltransferase family 1 member A6 |
| Up | ENSG00000086548 | CEACAM6 | 4680 | 6.825 | CEA cell adhesion molecule 6 |
| Up | ENSG00000134595 | SOX3 | 6658 | 6.824 | SRY-box transcription factor 3 |
| Up | ENSG00000204382 | XAGE1B | 653067 | 6.824 | X antigen family member 1B |
| Up | ENSG00000165588 | OTX2 | 5015 | 6.824 | Orthodenticle homeobox 2 |
| Up | ENSG00000102924 | CBLN1 | 869 | 6.823 | Cerebellin 1 precursor |
| Up | ENSG00000204379 | XAGE1A | 653220 | 6.823 | X antigen family member 1A |
| Up | ENSG00000109132 | PHOX2B | 8929 | 6.823 | Paired like homeobox 2B |
| Up | ENSG00000053438 | NNAT | 4826 | 6.823 | Neuronatin |
| Up | ENSG00000198848 | CES1 | 1066 | 6.821 | Carboxylesterase 1 |
| Up | ENSG00000118785 | SPP1 | 6696 | 6.820 | Secreted phosphoprotein 1 |
| Up | ENSG00000185559 | DLK1 | 8788 | 6.775 | Delta like non-canonical Notch ligand 1 |
| Up | ENSG00000162383 | SLC1A7 | 6512 | 6.625 | Solute carrier family 1 member 7 |
| Up | ENSG00000118702 | GHRH | 2691 | 6.623 | Growth hormone releasing hormone |
| Up | ENSG00000117600 | PLPPR4 | 9890 | 6.615 | Phospholipid phosphatase related 4 |
| Up | ENSG00000184144 | CNTN2 | 6900 | 6.510 | Contactin 2 |
| Up | ENSG00000158639 | PAGE5 | 90737 | 6.499 | PAGE family member 5 |
| Up | ENSG00000172005 | MAL | 4118 | 6.480 | Mal, T cell differentiation protein |
| Up | ENSG00000123584 | MAGEA9 | 4108 | 6.431 | MAGE family member A9 |
| Up | ENSG00000165092 | ALDH1A1 | 216 | 6.350 | Aldehyde dehydrogenase 1 family member A1 |
| Up | ENSG00000181965 | NEUROG1 | 4762 | 6.256 | Neurogenin 1 |
| Up | ENSG00000129824 | RPS4Y1 | 6192 | 5.841 | Ribosomal protein S4 Y-linked 1 |
| Up | ENSG00000169862 | CTNND2 | 1501 | 5.539 | Catenin delta 2 |
| Up | ENSG00000105894 | PTN | 5764 | 5.379 | Pleiotrophin |
| Up | ENSG00000103647 | CORO2B | 10391 | 5.256 | Coronin 2B |
| Up | ENSG00000181195 | PENK | 5179 | 5.114 | proenkephalin |
| Up | ENSG00000188153 | COL4A5 | 1287 | 4.937 | Collagen type IV alpha 5 chain |
| Up | ENSG00000077935 | SMC1B | 27127 | 4.807 | Structural maintenance of chromosomes 1B |
| Up | ENSG00000203972 | GLYATL3 | 389396 | 4.421 | Glycine-N-acyltransferase like 3 |
| Down | ENSG00000130558 | OLFM1 | 10439 | −4.488 | Olfactomedin 1 |
| Down | ENSG00000110680 | CALCA | 796 | −4.987 | Calcitonin related polypeptide alpha |
| Down | ENSG00000002586 | CD99 | 4267 | −5.279 | CD99 molecule (Xg blood group) |
| Down | ENSG00000100095 | SEZ6L | 23544 | −5.502 | Seizure related 6 homolog like |
| Down | ENSG00000170561 | IRX2 | 153572 | −6.053 | Iroquois homeobox 2 |
| Down | ENSG00000127324 | TSPAN8 | 7103 | −6.243 | Tetraspanin 8 |
| Down | ENSG00000152377 | SPOCK1 | 6695 | −6.247 | SPARC (osteonectin), cwcv and kazal like domains proteoglycan 1 |
| Down | ENSG00000136997 | MYC | 4609 | −6.450 | MYC proto-oncogene, bHLH transcription factor |
| Down | ENSG00000154545 | MAGED4 | 728239 | −6.582 | MAGE family member D4 |
| Down | ENSG00000166681 | BEX3 | 27018 | −6.679 | Brain expressed X-linked 3 |
| Down | ENSG00000160180 | TFF3 | 7033 | −7.013 | Trefoil factor 3 |
| Down | ENSG00000133169 | BEX1 | 55859 | −7.091 | Brain expressed X-linked 1 |
| Down | ENSG00000154277 | UCHL1 | 7345 | −7.094 | Ubiquitin C-terminal hydrolase L1 |
| Direction | DEG2 Analysis: All PDX Sample Pathways | FoldEnriched | nGenes | −log10(FDR) |
|---|---|---|---|---|
| Up | Nicotine addiction | 4.09 | 16 | 4.23 |
| Protein digestion and absorption | 3.26 | 35 | 7.86 | |
| ECM-receptor interaction | 3.14 | 30 | 6.29 | |
| Down | Histidine metabolism | 3.52 | 12 | 2.87 |
| Steroid hormone biosynthesis | 2.36 | 19 | 2.31 | |
| Metabolism of xenobiotics by cytochrome P450 | 2.29 | 25 | 2.87 |
| Protein | Docking Score (Kcal/mol) | Amino Acid Interaction | Hydrogen BondDistance (Å) | Number of Conventional Hydrogen Bonds |
|---|---|---|---|---|
| LSD1/FAD complex | −7.2 | ASP555 | 2.21 | 3 |
| ASN806 | 2.57 | |||
| ASN806 | 2.60 | |||
| TSPAN8 | −7.4 | ASN16 | 2.44 | 2 |
| ASN16 | 2.48 | |||
| UCHL1 | −7.2 | MET124 | 2.54 | 1 |
| MYC | −7.0 | ALA280 | 2.63 | 2 |
| ALA280 | 2.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nalkiran, I.; Sevim Nalkiran, H.; Ozcelik, N.; Kivrak, M. In Silico Identification of LSD1 Inhibition-Responsive Targets in Small Cell Lung Cancer. Bioengineering 2025, 12, 504. https://doi.org/10.3390/bioengineering12050504
Nalkiran I, Sevim Nalkiran H, Ozcelik N, Kivrak M. In Silico Identification of LSD1 Inhibition-Responsive Targets in Small Cell Lung Cancer. Bioengineering. 2025; 12(5):504. https://doi.org/10.3390/bioengineering12050504
Chicago/Turabian StyleNalkiran, Ihsan, Hatice Sevim Nalkiran, Neslihan Ozcelik, and Mehmet Kivrak. 2025. "In Silico Identification of LSD1 Inhibition-Responsive Targets in Small Cell Lung Cancer" Bioengineering 12, no. 5: 504. https://doi.org/10.3390/bioengineering12050504
APA StyleNalkiran, I., Sevim Nalkiran, H., Ozcelik, N., & Kivrak, M. (2025). In Silico Identification of LSD1 Inhibition-Responsive Targets in Small Cell Lung Cancer. Bioengineering, 12(5), 504. https://doi.org/10.3390/bioengineering12050504