

Three-Dimensional Trilineage Differentiation Conditions for Human Induced Pluripotent Stem Cells

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and 3D Cell Culture Conditions
2.2. 3D Embedded hiPSC Culture
2.3. hiPSC Spheroid Harvesting from 3D Culture
2.4. hiPSC Spheroid Counting and Dissociation
2.5. Trilineage Differentiation of hiPSC in 3D Embedded Conditions
2.5.1. Plating hiPSC Without EB Induction
2.5.2. Plating hiPSC Spheroids with EB Induction
2.5.3. Harvesting of Differentiated hiPSC Spheroids
2.6. Cell Spheroid Morphology and Image Analysis
2.7. Single Cell Count and Viability Measurement
2.8. Expression of Gene Biomarkers via RT-qPCR
2.9. Immunochemistry Staining
2.10. Flow Cytometry
2.11. Statistical Analysis
3. Results
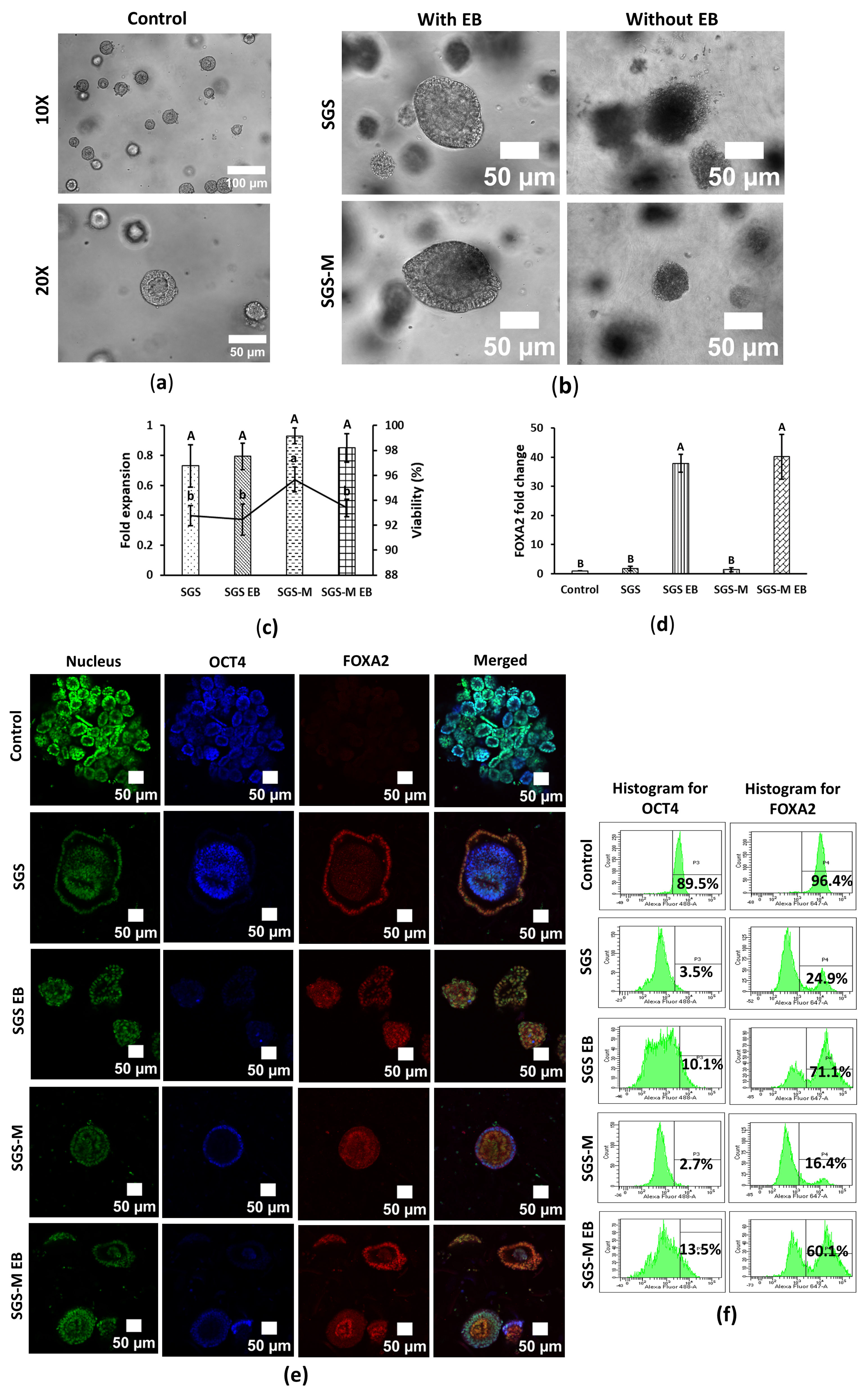
3.1. Synthegel Spheroid (SGS) Matrix with EB Medium Induction Determined a Viable Scaffold for Endoderm Differentiation of hiPSC Spheroids
3.2. Synthegel Spheroid (SGS) Matrix Without EB Induction Presented an Effective 3D Scaffold for Mesoderm Differentiation of hiPSC Spheroids
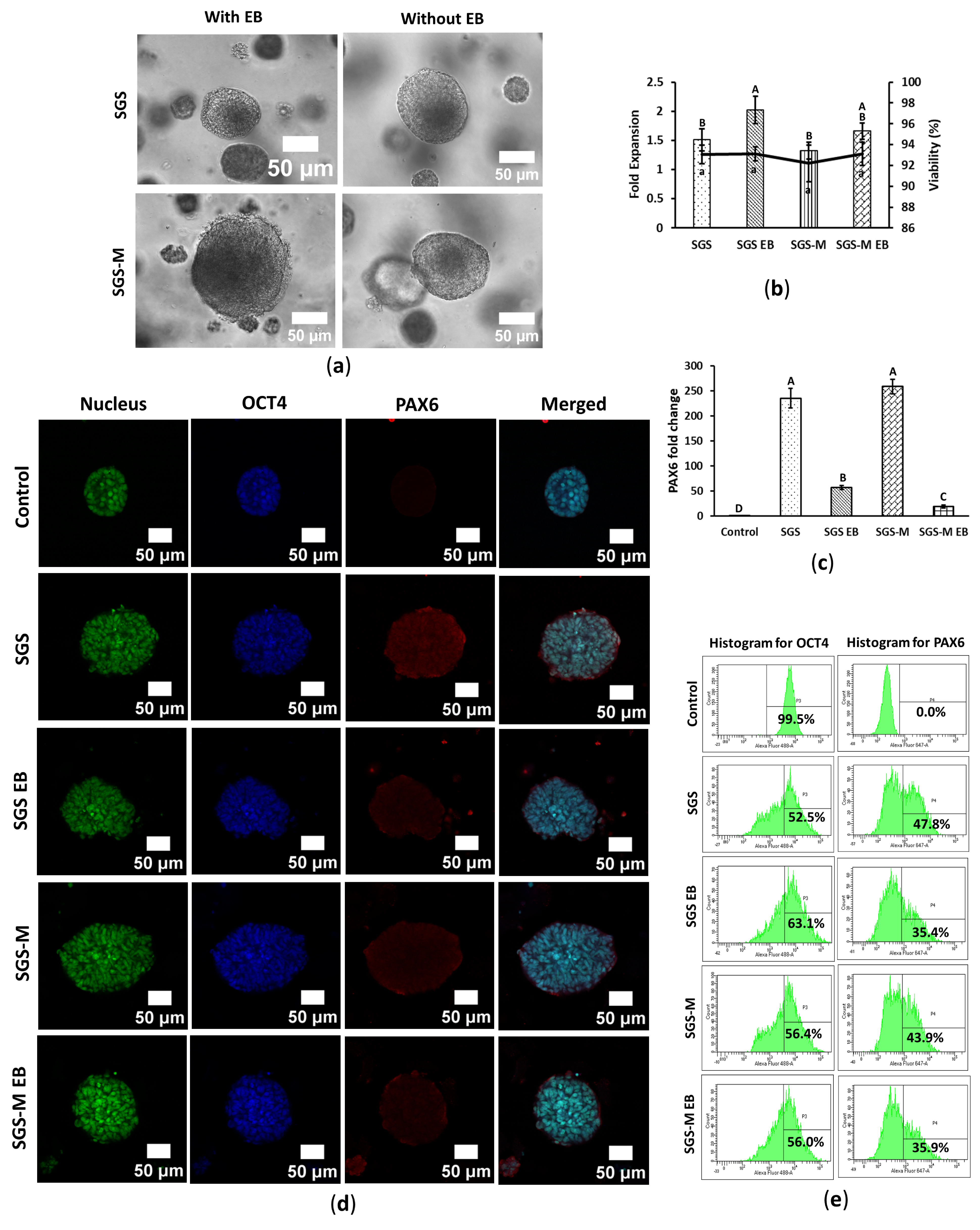
3.3. Synthegel Spheroid (SGS) Matrix Without EB Induction Determined an Effective Scaffold for Ectoderm Differentiation of hiPSC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mao, J.; Saiding, Q.; Qian, S.; Liu, Z.; Zhao, B.; Zhao, Q.; Lu, B.; Mao, X.; Zhang, L.; Zhang, Y.; et al. Reprogramming Stem Cells in Regenerative Medicine. Smart Med. 2022, 1, e20220005. [Google Scholar] [CrossRef] [PubMed]
- Mehra, V.; Chhetri, J.B.; Ali, S.; Roddie, C. The Emerging Role of Induced Pluripotent Stem Cells as Adoptive Cellular Immunotherapeutics. Biology 2023, 12, 1419. [Google Scholar] [CrossRef]
- Yiangou, L.; Ross, A.D.; Goh, K.J.; Vallier, L. Human pluripotent stem cell-derived endoderm for modeling development and clinical applications. Cell Stem Cell. 2018, 22, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Zahumenska, R.; Nosal, V.; Smolar, M.; Okajcekova, T.; Skovierova, H.; Strnadel, J.; Halasova, E. Induced Pluripotency: A Powerful Tool for In Vitro Modeling. Int. J. Mol. Sci. 2020, 21, 8910. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef]
- Li, Q.; Qi, G.; Liu, X.; Bai, J.; Zhao, J.; Tang, G.; Zhang, Y.S.; Chen-Tsai, R.; Zhang, M.; Wang, D.; et al. Universal Peptide Hydrogel for Scalable Physiological Formation and Bioprinting of 3D Spheroids from Human Induced Pluripotent Stem Cells. Adv. Funct. Mater. 2021, 31, e202104046. [Google Scholar] [CrossRef]
- Chaicharoenaudomrung, N.; Kunhorm, P.; Noisa, P. Three-Dimensional Cell Culture Systems as an in Vitro Platform for Cancer and Stem Cell Modeling. World J. Stem Cells 2019, 11, 1065–1083. [Google Scholar] [CrossRef]
- Lee, S.Y.; Koo, I.S.; Hwang, H.J.; Lee, D.W. In Vitro Three-Dimensional (3D) Cell Culture Tools for Spheroid and Organoid Models. SLAS Discov. 2023, 28, 119–137. [Google Scholar] [CrossRef]
- Ma, J.; Huang, C. Composition and Mechanism of Three-Dimensional Hydrogel System in Regulating Stem Cell Fate. Tissue Eng. Part B Rev. 2020, 26, 498–518. [Google Scholar] [CrossRef]
- Tchieu, J.; Zimmer, B.; Fattahi, F.; Amin, S.; Zeltner, N.; Chen, S.; Studer, L. A modular platform for differentiation of human PSCs into all major ectodermal lineages. Cell Stem Cell 2017, 21, 399–410. [Google Scholar] [CrossRef]
- Loh, K.M.; Ang, L.T.; Zhang, J.; Kumar, V.; Ang, J.; Auyeong, J.Q.; Lee, K.L.; Choo, S.H.; Lim, C.Y.; Nichane, M.; et al. Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 2014, 14, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.; Doan, M.T.; Roy, I.J.; Torres, A., Jr.; Teitell, M.A. Protocol for germ lineage differentiation of primed human pluripotent stem cells using chemically defined, nutrient-balanced media. STAR Protoc. 2022, 3, 101568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef] [PubMed]
- Alsobaie, S.; Alsobaie, T.; Alshammary, A.; Mantalaris, S. Differentiation of human induced pluripotent stem cells into functional lung alveolar epithelial cells in 3D dynamic culture. Front. Bioeng. Biotechnol. 2023, 11, 1173149. [Google Scholar] [CrossRef]
- Tabata, Y.; Joanna, I.; Higuchi, A. Stem Cell Culture and Differentiation in 3-D Scaffolds; Elsevier: Amsterdam, The Netherlands, 2023; Volume 199, ISBN 9780443134111. [Google Scholar]
- Maji, S.; Lee, H. Engineering Hydrogels for the Development of Three-Dimensional In Vitro Models. Int. J. Mol. Sci. 2022, 23, 2662. [Google Scholar] [CrossRef]
- Nickolls, A.R.; Lee, M.M.; Zukosky, K.; Mallon, B.S.; Bönnemann, C.G. Human Embryoid Bodies as a 3D Tissue Model of the Extracellular Matrix and α-Dystroglycanopathies. DMM Dis. Models Mech. 2020, 13, dmm042986. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carrera, I.; Frise, E.; Verena, K.; Mark, L.; Tobias, P.; Stephan, P.; Curtis, R.; Stephan, S.; Benjamin, S.; et al. Fiji—An Open Platform for Biological Image Analysis. Nat. Methods 2009, 9, 676–682. [Google Scholar] [CrossRef]
- Holmgren, G.; Ghosheh, N.; Zeng, X.; Bogestål, Y.; Sartipy, P.; Synnergren, J. Identification of stable reference genes in differentiating human pluripotent stem cells. Physiol. Genom. 2015, 47, 232–239. [Google Scholar] [CrossRef]
- Liu, W.; Saint, D.A. A New Quantitative Method of Real Time Reverse Transcription Polymerase Chain Reaction Assay Based on Simulation of Polymerase Chain Reaction Kinetics. Anal. Biochem. 2002, 302, 52–59. [Google Scholar] [CrossRef]
- Chen, C.X.Q.; Abdian, N.; Maussion, G.; Thomas, R.A.; Demirova, I.; Cai, E.; Tabatabaei, M.; Beitel, L.K.; Karamchandani, J.; Fon, E.A.; et al. A multistep workflow to evaluate newly generated iPSCs and their ability to generate different cell types. Methods Protoc. 2021, 4, 50. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Guo, N.N.; Liu, L.P.; Zheng, Y.W.; Li, Y.M. Inducing Human Induced Pluripotent Stem Cell Differentiation through Embryoid Bodies: A Practical and Stable Approach. World J. Stem Cells 2020, 12, 25–34. [Google Scholar] [CrossRef]
- Sohn, E.J.; Moon, H.J.; Lim, J.K.; Kim, D.S.; Kim, J.H. Regulation of the Protein Stability and Transcriptional Activity of OCT4 in Stem Cells. Adv. Biol. Regul. 2021, 79, 100777. [Google Scholar] [CrossRef]
- Mehravar, M.; Ghaemimanesh, F.; Poursani, E.M. An Overview on the Complexity of OCT4: At the Level of DNA, RNA and Protein. Stem Cell Rev. Rep. 2021, 17, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Wu, G.; Chen, R.; Drexler, H.C.A.; MacCarthy, C.M.; Kim, K.P.; Adachi, K.; Gerovska, D.; Mavrommatis, L.; Bedzhov, I.; et al. A Balanced Oct4 Interactome Is Crucial for Maintaining Pluripotency. Sci. Adv. 2022, 8, eabe4375. [Google Scholar] [CrossRef] [PubMed]
- Patel, I.; Parchem, R.J. Regulation of Oct4 in Stem Cells and Neural Crest Cells. Birth Defects Res. 2022, 114, 983–1002. [Google Scholar] [CrossRef]
- Palpant, N.J.; Pabon, L.; Friedman, C.E.; Roberts, M.; Hadland, B.; Zaunbrecher, R.J.; Bernstein, I.; Zheng, Y.; Murry, C.E. Generating High-Purity Cardiac and Endothelial Derivatives from Patterned Mesoderm Using Human Pluripotent Stem Cells. Nat. Protoc. 2017, 12, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Vahdat, S.; Pahlavan, S.; Mahmoudi, E.; Barekat, M.; Ansari, H.; Bakhshandeh, B.; Aghdami, N.; Baharvand, H. Expansion of Human Pluripotent Stem Cell-Derived Early Cardiovascular Progenitor Cells by a Cocktail of Signaling Factors. Sci. Rep. 2019, 9, 16006. [Google Scholar] [CrossRef]
- Poh, Y.C.; Chen, J.; Hong, Y.; Yi, H.; Zhang, S.; Chen, J.; Wu, D.C.; Wang, L.; Jia, Q.; Singh, R.; et al. Generation of Organized Germ Layers from a Single Mouse Embryonic Stem Cell. Nat. Commun. 2014, 5, 4000. [Google Scholar] [CrossRef]
- Youssef, K.K.; Nieto, M.A. Epithelial–Mesenchymal Transition in Tissue Repair and Degeneration. Nat. Rev. Mol. Cell Biol. 2024, 25, 720–739. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Plygawko, A.T.; Kan, S.; Campbell, K. Epithelial-Mesenchymal Plasticity: Emerging Parallels between Tissue Morphogenesis and Cancer Metastasis. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20200087. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.S.; Qi, G.; Li, Q.; Liu, X.; Bai, J.; Chen, M.; Atala, A.; Sun, X.S. Three-Dimensional Trilineage Differentiation Conditions for Human Induced Pluripotent Stem Cells. Bioengineering 2025, 12, 503. https://doi.org/10.3390/bioengineering12050503
Rahman MS, Qi G, Li Q, Liu X, Bai J, Chen M, Atala A, Sun XS. Three-Dimensional Trilineage Differentiation Conditions for Human Induced Pluripotent Stem Cells. Bioengineering. 2025; 12(5):503. https://doi.org/10.3390/bioengineering12050503
Chicago/Turabian StyleRahman, Md Sharifur, Guangyan Qi, Quan Li, Xuming Liu, Jianfa Bai, Mingshun Chen, Anthony Atala, and Xiuzhi Susan Sun. 2025. "Three-Dimensional Trilineage Differentiation Conditions for Human Induced Pluripotent Stem Cells" Bioengineering 12, no. 5: 503. https://doi.org/10.3390/bioengineering12050503
APA StyleRahman, M. S., Qi, G., Li, Q., Liu, X., Bai, J., Chen, M., Atala, A., & Sun, X. S. (2025). Three-Dimensional Trilineage Differentiation Conditions for Human Induced Pluripotent Stem Cells. Bioengineering, 12(5), 503. https://doi.org/10.3390/bioengineering12050503