

Peptide–Drug Conjugates as Next-Generation Therapeutics: Exploring the Potential and Clinical Progress

 ,
,  ,
,  ,
,  , , , , ,
, , , , , 
Abstract
1. Introduction
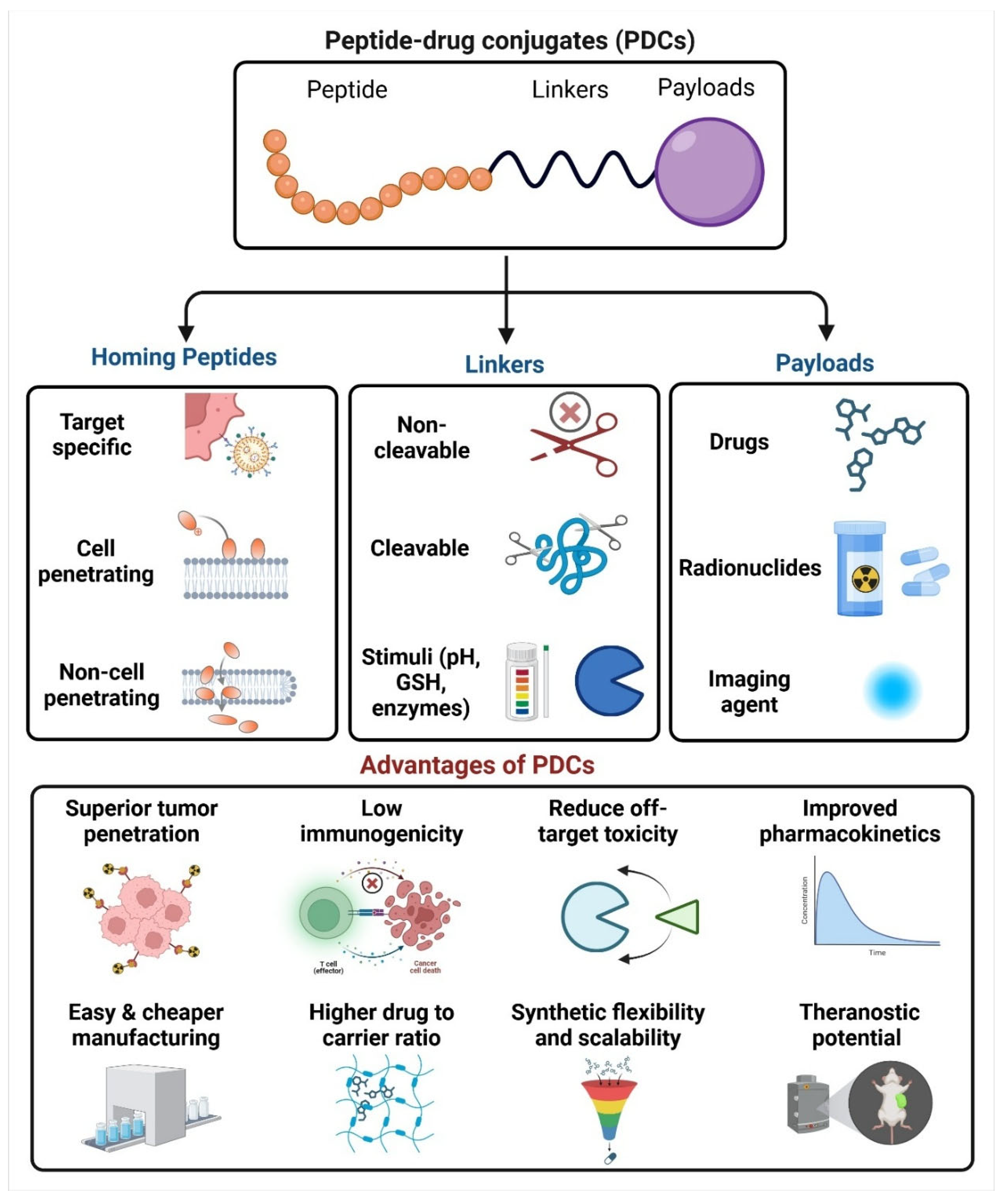
2. Fundamentals of Peptide-Based Drug Conjugates
2.1. Homing Peptides
2.2. Linkers in PDC
2.2.1. Non-Cleavable Linkers
2.2.2. Cleavable Linkers
2.2.3. pH-Sensitive Linkers
2.2.4. Redox-Sensitive Linkers
2.2.5. Enzyme-Sensitive Linkers
2.3. Payloads of PDC
3. Targeting, Binding, Internalization, and Drug Release at Target Sites
4. Applications of Peptide–Drug Conjugates (PDCs)
4.1. Cancer Therapy
4.1.1. Cell Targeting Peptide–Drug Conjugates
4.1.2. Integrins
4.1.3. Somatostatin Receptors
4.1.4. Epidermal Growth Factor Receptor (EGFR)
4.1.5. Bombesin Receptor Family
4.1.6. PDCs Acting on Immune Regulation (Immune Checkpoint Blockade)
4.1.7. Other Advantages of PDC in Delivering Chemotherapeutic Agents
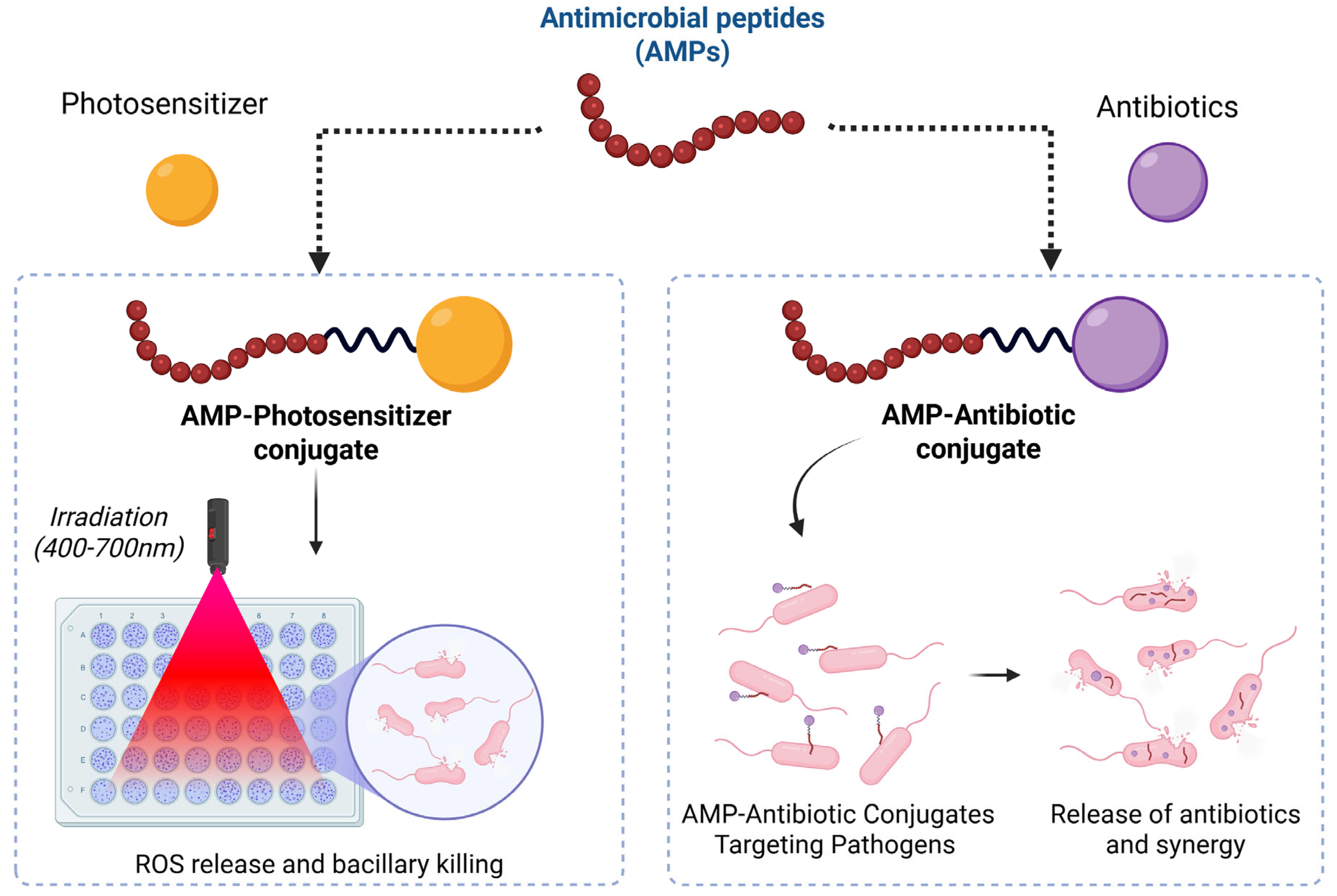
4.2. Antimicrobial Peptide–Drug Conjugate
4.3. Antiviral Peptide–Drug Conjugates

4.4. Neurological Disorders
4.5. Inflammatory Diseases
4.6. Theranostic Peptide Drug Conjugate (TPDC)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeting Peptide | Payload | Therapeutic Target | Indication | Reference |
|---|---|---|---|---|
| Heptapeptide (P7) | Docetaxel | Hsp98 | Non-Small Cell Lung Cancer (NSCLC) | [75] |
| RGD | Paclitaxel | αv integrin receptors | Metastatic breast cancer | [76] |
| Cyclic peptide 3207-86 | Camptothecin (CPT) | SSTR2 | Anticancer effect | [79] |
| Octreotide | Doxorubicin | SSTR2 | Breast cancer | [83] |
| [D-Phe6, β-Ala11, Sta13, Nle14] BBN (6–14) | Daunorubicin | Gastrin-Releasing Peptide Receptor (GRP-R) | Prostate and breast cancer | [90] |
| PD-L1-targeting peptide (PPA1) | Doxorubicin | PD-L1 | Colon Cancer | [96] |
| Oligomeric peptides (KGFRWR) | Doxorubicin | MMP-2 and MMP-9. | Hepatocellular carcinoma (HCC) | [106] |
| Pep-4 | Levofloxacin | Membrane Disruption | Antibacterial | [107] |
| P14LRR (Fl-PRPRPL-4) | Kanamycin | - | Antibacterial | [112] |
| T20 peptide (enfuvirtide) | Sapogenin | gp41-specific | Antiviral | [123] |
| Sulfonium-tethered peptide | GRL0617 | Papain-like cysteine protease (PLpro) | Antiviral | [124] |
| Cell-penetrating peptides (CPPs) | Porphyrins | - | HIV-associated neurocognitive disorders (HAND) | [128] |
| Arg-Gly-Asx (RGD) and Asn-Gly-Arg (NGR) | Naproxen | Aminopeptidase N | Cancer therapy | [132] |
| UBI (29–41) | Gallium-68 (68Ga) | - | Theranostic | [136] |
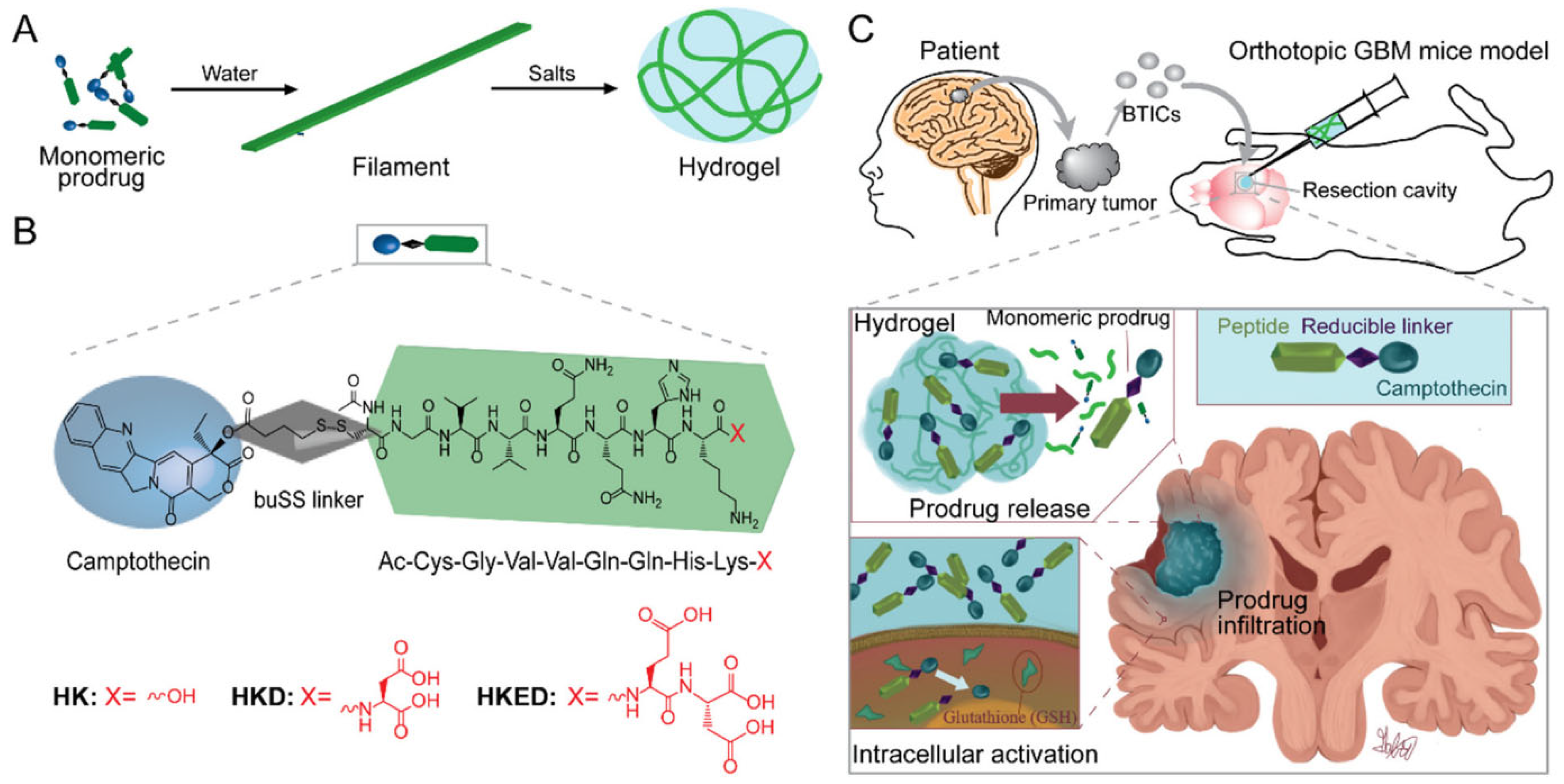
5. PDC-Based Formulations
6. Challenges in Development of PDCs
7. Clinical Status of Peptide–Drug Conjugates
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, M.; Huang, W.; Yang, N.; Liu, Y. Learn from antibody–drug conjugates: Consideration in the future construction of peptide-drug conjugates for cancer therapy. Exp. Hematol. Oncol. 2022, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.M.; Iegre, J.; O’ Donovan, D.H.; Ölwegård Halvarsson, M.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide–drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, M.; Sun, L.; Liu, J.; Yin, L.; Xia, M.; Zhang, L.; Liu, X.; Cheng, Y. Recent Advances in Targeted Cancer Therapy: Are PDCs the Next Generation of ADCs? J. Med. Chem. 2024, 67, 11469–11487. [Google Scholar] [CrossRef] [PubMed]
- Rossino, G.; Marchese, E.; Galli, G.; Verde, F.; Finizio, M.; Serra, M.; Linciano, P.; Collina, S. Peptides as Therapeutic Agents: Challenges and Opportunities in the Green Transition Era. Molecules 2023, 28, 7165. [Google Scholar] [CrossRef]
- Simić, S.; Zukić, E.; Schmermund, L.; Faber, K.; Winkler, C.K.; Kroutil, W. Shortening Synthetic Routes to Small Molecule Active Pharmaceutical Ingredients Employing Biocatalytic Methods. Chem. Rev. 2022, 122, 1052–1126. [Google Scholar] [CrossRef]
- Ray, E.; Jadhav, K.; Kadian, M.; Sharma, G.; Sharma, K.; Jhilta, A.; Singh, R.; Kumar, A.; Verma, R.K. Targeted delivery of the metastasis-specific tumour homing TMTP1 peptide to non-small-cell lung cancer (NSCLC) using inhalable hybrid nano-assemblies. J. Mater. Chem. B 2024, 12, 9740–9759. [Google Scholar] [CrossRef]
- Rizvi, S.F.A.; Zhang, L.; Zhang, H.; Fang, Q. Peptide-Drug Conjugates: Design, Chemistry, and Drug Delivery System as a Novel Cancer Theranostic. ACS Pharmacol. Transl. Sci. 2024, 7, 309–334. [Google Scholar] [CrossRef]
- Vrettos, E.I.; Mező, G.; Tzakos, A.G. On the design principles of peptide–drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein J. Org. Chem. 2018, 14, 930–954. [Google Scholar] [CrossRef]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide–Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Dean, T.T.; Jelú-Reyes, J.; Allen, A.C.; Moore, T.W. Peptide–Drug Conjugates: An Emerging Direction for the Next Generation of Peptide Therapeutics. J. Med. Chem. 2024, 67, 1641–1661. [Google Scholar] [CrossRef]
- Wang, M.; Liu, J.; Xia, M.; Yin, L.; Zhang, L.; Liu, X.; Cheng, Y. Peptide-drug conjugates: A new paradigm for targeted cancer therapy. Eur. J. Med. Chem. 2024, 265, 116119. [Google Scholar] [CrossRef] [PubMed]
- Lamb, H.O.; Benfield, A.H.; Henriques, S.T. Peptides as innovative strategies to combat drug resistance in cancer therapy. Drug Discov. Today 2024, 29, 104206. [Google Scholar] [CrossRef]
- He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules 2019, 24, 1855. [Google Scholar] [CrossRef]
- Alobaid, A.A.; Skoda, M.W.A.; Harris, L.K.; Campbell, R.A. Translational use of homing peptides: Tumor and placental targeting. J. Colloid Interface Sci. 2024, 662, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Milewska, S.; Sadowska, A.; Stefaniuk, N.; Misztalewska-Turkowicz, I.; Wilczewska, A.Z.; Car, H.; Niemirowicz-Laskowska, K. Tumor-Homing Peptides as Crucial Component of Magnetic-Based Delivery Systems: Recent Developments and Pharmacoeconomical Perspective. Int. J. Mol. Sci. 2024, 25, 6219. [Google Scholar] [CrossRef]
- Ghorai, S.M.; Deep, A.; Magoo, D.; Gupta, C.; Gupta, N. Cell-Penetrating and Targeted Peptides Delivery Systems as Potential Pharmaceutical Carriers for Enhanced Delivery across the Blood–Brain Barrier (BBB). Pharmaceutics 2023, 15, 1999. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Tumor-Penetrating Peptides. Front. Oncol. 2013, 3, 59276. [Google Scholar] [CrossRef]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 3–12. [Google Scholar] [CrossRef]
- David, A. Peptide ligand-modified nanomedicines for targeting cells at the tumor microenvironment. Adv. Drug Deliv. Rev. 2017, 119, 120–142. [Google Scholar] [CrossRef]
- Jiang, Z.; Guan, J.; Qian, J.; Zhan, C. Peptide ligand-mediated targeted drug delivery of nanomedicines. Biomater. Sci. 2019, 7, 461–471. [Google Scholar] [CrossRef]
- Vadevoo, S.M.P.; Gurung, S.; Lee, H.-S.; Gunassekaran, G.R.; Lee, S.-M.; Yoon, J.-W.; Lee, Y.-K.; Lee, B. Peptides as multifunctional players in cancer therapy. Exp. Mol. Med. 2023, 55, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Szabó, I.; Yousef, M.; Soltész, D.; Bató, C.; Mező, G.; Bánóczi, Z. Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System. Pharmaceutics 2022, 14, 907. [Google Scholar] [CrossRef]
- Lamers, C. Overcoming the Shortcomings of Peptide-Based Therapeutics. Futur. Drug Discov. 2022, 4. [Google Scholar] [CrossRef]
- Bottens, R.A.; Yamada, T. Cell-Penetrating Peptides (CPPs) as Therapeutic and Diagnostic Agents for Cancer. Cancers 2022, 14, 5546. [Google Scholar] [CrossRef] [PubMed]
- Gori, A.; Lodigiani, G.; Colombarolli, S.G.; Bergamaschi, G.; Vitali, A. Cell Penetrating Peptides: Classification, Mechanisms, Methods of Study, and Applications. ChemMedChem 2023, 18, e202300236. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Ludwig, B.S.; Kessler, H.; Kossatz, S.; Reuning, U. RGD-Binding Integrins Revisited: How Recently Discovered Functions and Novel Synthetic Ligands (Re-)Shape an Ever-Evolving Field. Cancers 2021, 13, 1711. [Google Scholar] [CrossRef]
- Javid, H.; Oryani, M.A.; Rezagholinejad, N.; Esparham, A.; Tajaldini, M.; Karimi-Shahri, M. RGD peptide in cancer targeting: Benefits, challenges, solutions, and possible integrin–RGD interactions. Cancer Med. 2024, 13, e6800. [Google Scholar] [CrossRef]
- Yamada, Y.; Onda, T.; Wada, Y.; Hamada, K.; Kikkawa, Y.; Nomizu, M. Structure–Activity Relationships of RGD-Containing Peptides in Integrin αvβ5-Mediated Cell Adhesion. ACS Omega 2023, 8, 4687–4693. [Google Scholar] [CrossRef]
- Wang, F.; Li, Y.; Shen, Y.; Wang, A.; Wang, S.; Xie, T. The Functions and Applications of RGD in Tumor Therapy and Tissue Engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [Google Scholar] [CrossRef] [PubMed]
- Anderluzzi, G.; Ghitti, M.; Gasparri, A.M.; Taiè, G.; Sacchi, A.; Gori, A.; Andolfo, A.; Pozzi, F.; Musco, G.; Curnis, F.; et al. A novel aminopeptidase N/CD13 inhibitor selectively targets an endothelial form of CD13 after coupling to proteins. Cell. Mol. Life Sci. 2024, 81, 68. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Amin, M.A.; Fox, D.A. CD13/Aminopeptidase N Is a Potential Therapeutic Target for Inflammatory Disorders. J. Immunol. 2020, 204, 3–11. [Google Scholar] [CrossRef]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer Treatment by Targeted Drug Delivery to Tumor Vasculature in a Mouse Model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef]
- Garde, S.V.; Forté, A.J.; Ge, M.; Lepekhin, E.A.; Panchal, C.J.; Rabbani, S.A.; Wu, J.J. Binding and internalization of NGR-peptide-targeted liposomal doxorubicin (TVT-DOX) in CD13-expressing cells and its antitumor effects. Anticancer. Drugs 2007, 18, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Lingasamy, P.; Teesalu, T. Homing Peptides for Cancer Therapy. In Bio-Nanomedicine for Cancer Therapy; Advances in Experimental Medicine and Biology (AEMB); Fontana, F., Santos, H.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2021; Volume 1295, pp. 29–48. [Google Scholar]
- Abhang, A.; Katari, O.; Ghadi, R.; Chaudhari, D.; Jain, S. Exploring the synergistic behavior of paclitaxel and vorinostat upon co-loading in albumin nanoparticles for breast cancer management. Drug Deliv. Transl. Res. 2024, 14, 510–523. [Google Scholar] [CrossRef]
- Ayo, A.; Laakkonen, P. Peptide-Based Strategies for Targeted Tumor Treatment and Imaging. Pharmaceutics 2021, 13, 481. [Google Scholar] [CrossRef]
- Roy, S.; Ghosh, P.; Sekhar Roy, N.; Mazumder, A.; Roy, K.; Kumar Manna, A.; Mallick, S.; Ahmed, I. Peptide based Molecules as Protein-Protein Interaction Inhibitors: Tools for Chemical Genetics and Therapy. Curr. Chem. Biol. 2012, 6, 145–163. [Google Scholar] [CrossRef]
- Jiang, K.; Song, X.; Yang, L.; Li, L.; Wan, Z.; Sun, X.; Gong, T.; Lin, Q.; Zhang, Z. Enhanced antitumor and anti-metastasis efficacy against aggressive breast cancer with a fibronectin-targeting liposomal doxorubicin. J. Control. Release 2018, 271, 21–30. [Google Scholar] [CrossRef]
- Kole, E.; Jadhav, K.; Singh, R.; Mandpe, S.; Abhang, A.; Verma, R.K.; Naik, J. Recent Developments in Tyrosine Kinase Inhibitor-based Nanotherapeutics for EGFR-resistant Non-small Cell Lung Cancer. Curr. Drug Deliv. 2024, 21, 249–260. [Google Scholar] [CrossRef]
- Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials. Cancers 2019, 11, 1826. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Yang, W.; Cheng, L.; Meng, F.; Zhang, J.; Zhong, Z. EGFR-targeted multifunctional polymersomal doxorubicin induces selective and potent suppression of orthotopic human liver cancer in vivo. Acta Biomater. 2017, 64, 323–333. [Google Scholar] [CrossRef]
- Balogh, B.; Ivánczi, M.; Nizami, B.; Beke-Somfai, T.; Mándity, I.M. ConjuPepDB: A database of peptide–drug conjugates. Nucleic Acids Res. 2021, 49, D1102–D1112. [Google Scholar] [CrossRef]
- Sun, X.; Li, H.; Chen, Z.; Zhang, Y.; Wei, Z.; Xu, H.; Liao, Y.; Jiang, W.; Ge, Y.; Zheng, L.; et al. PDCdb: The biological activity and pharmaceutical information of peptide–drug conjugate (PDC). Nucleic Acids Res. 2025, 53, D1476–D1485. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Yu, L.; Miao, Y.; Liu, X.; Yu, Z.; Wei, M. Peptide–drug conjugates (PDCs): A novel trend of research and development on targeted therapy, hype or hope? Acta Pharm. Sin. B 2023, 13, 498–516. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Jafari, B.; Pourseif, M.M.; Barar, J.; Rafi, M.A.; Omidi, Y. Peptide-mediated drug delivery across the blood-brain barrier for targeting brain tumors. Expert Opin. Drug Deliv. 2019, 16, 583–605. [Google Scholar] [CrossRef]
- Mező, G.; Gomena, J.; Ranđelović, I.; Dókus, E.; Kiss, K.; Pethő, L.; Schuster, S.; Vári, B.; Vári-Mező, D.; Lajkó, E.; et al. Oxime-Linked Peptide–Daunomycin Conjugates as Good Tools for Selection of Suitable Homing Devices in Targeted Tumor Therapy: An Overview. Int. J. Mol. Sci. 2024, 25, 1864. [Google Scholar] [CrossRef]
- Gong, L.; Zhao, H.; Liu, Y.; Wu, H.; Liu, C.; Chang, S.; Chen, L.; Jin, M.; Wang, Q.; Gao, Z.; et al. Research advances in peptide—drug conjugates. Acta Pharm. Sin. B 2023, 13, 3659–3677. [Google Scholar] [CrossRef]
- Liang, Y.; Li, S.; Wang, X.; Zhang, Y.; Sun, Y.; Wang, Y.; Wang, X.; He, B.; Dai, W.; Zhang, H.; et al. A comparative study of the antitumor efficacy of peptide-doxorubicin conjugates with different linkers. J. Control. Release 2018, 275, 129–141. [Google Scholar] [CrossRef]
- Heh, E.; Allen, J.; Ramirez, F.; Lovasz, D.; Fernandez, L.; Hogg, T.; Riva, H.; Holland, N.; Chacon, J. Peptide Drug Conjugates and Their Role in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 829. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, V.M.; Bellmann-Sickert, K.; Beck-Sickinger, A.G. Peptides And Peptide Conjugates: Therapeutics On The Upward Path. Future Med. Chem. 2012, 4, 1567–1586. [Google Scholar] [CrossRef] [PubMed]
- Gillies, E.R.; Goodwin, A.P.; Fréchet, J.M.J. Acetals as pH-Sensitive Linkages for Drug Delivery. Bioconjug. Chem. 2004, 15, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xu, S.; Xiang, T.; Liu, H.; Chen, L.; Jiang, B.; Yao, J.; Zhu, H.; Hu, R.; Chen, Z. Multifunctional building elements for the construction of peptide drug conjugates. Eng. Regen. 2022, 3, 92–109. [Google Scholar] [CrossRef]
- Wu, C.; Cheng, Z.; Lu, D.; Liu, K.; Cheng, Y.; Wang, P.; Zhou, Y.; Li, M.; Shao, X.; Li, H.; et al. Novel N -Methylated Cyclodepsipeptide Prodrugs for Targeted Cancer Therapy. J. Med. Chem. 2021, 64, 991–1000. [Google Scholar] [CrossRef]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide–drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 112–126. [Google Scholar] [CrossRef]
- Böhme, D.; Krieghoff, J.; Beck-Sickinger, A.G. Double Methotrexate-Modified Neuropeptide Y Analogues Express Increased Toxicity and Overcome Drug Resistance in Breast Cancer Cells. J. Med. Chem. 2016, 59, 3409–3417. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef]
- Janardhanam, L.S.L.; Indukuri, V.V.; Verma, P.; Dusane, A.C.; Venuganti, V.V.K. Functionalized layer-by-layer assembled film with directional 5-fluorouracil release to target colon cancer. Mater. Sci. Eng. C 2020, 115, 111118. [Google Scholar] [CrossRef]
- Ma, L.; Wang, C.; He, Z.; Cheng, B.; Zheng, L.; Huang, K. Peptide-Drug Conjugate: A Novel Drug Design Approach. Curr. Med. Chem. 2017, 24, 3373–3396. [Google Scholar] [CrossRef]
- Chavda, V.P.; Solanki, H.K.; Davidson, M.; Apostolopoulos, V.; Bojarska, J. Peptide-Drug Conjugates: A New Hope for Cancer Management. Molecules 2022, 27, 7232. [Google Scholar] [CrossRef]
- Ma, J.; Hu, X.; Li, L.; Rao, Z.; Zhang, C. Efficacy and safety of 177Lu-DOTATATE targeted therapy in advanced/metastatic pulmonary neuroendocrine tumors: A systematic review and meta-analysis. Front. Oncol. 2022, 12, 993182. [Google Scholar] [CrossRef]
- You, X.; Guo, W.; Wang, L.; Hou, Y.; Zhang, H.; Pan, Y.; Han, R.; Huang, M.; Liao, L.; Chen, Y. Subcellular distribution of RAD23B controls XPC degradation and DNA damage repair in response to chemotherapy drugs. Cell. Signal. 2017, 36, 108–116. [Google Scholar] [CrossRef]
- López-garcía, G.; Dublan-garcía, O.; Arizmendi-cotero, D.; Oliván, L.M.G. Antioxidant and Antimicrobial Peptides Derived from Food Proteins. Molecules 2022, 27, 1343. [Google Scholar] [CrossRef]
- Deslouches, B.; Peter Di, Y. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef]
- Rizzo, M.G.; Palermo, N.; D’amora, U.; Oddo, S.; Guglielmino, S.P.P.; Conoci, S.; Szychlinska, M.A.; Calabrese, G. Multipotential Role of Growth Factor Mimetic Peptides for Osteochondral Tissue Engineering. Int. J. Mol. Sci. 2022, 23, 7388. [Google Scholar] [CrossRef]
- Dai, M.Y.; Shi, Y.Y.; Wang, A.J.; Liu, X.L.; Liu, M.; Cai, H.B. High-potency PD-1/PD-L1 degradation induced by Peptide-PROTAC in human cancer cells. Cell Death Dis. 2022, 13, 924. [Google Scholar] [CrossRef]
- Hao, X.; Yan, Q.; Zhao, J.; Wang, W.; Huang, Y.; Chen, Y. TAT modification of alpha-helical anticancer peptides to improve specificity and efficacy. PLoS ONE 2015, 10, e0138911. [Google Scholar] [CrossRef]
- Najm, A.A.K.; Azfaralariff, A.; Dyari, H.R.E.; Othman, B.A.; Shahid, M.; Khalili, N.; Law, D.; Syed Alwi, S.S.; Fazry, S. Anti-breast cancer synthetic peptides derived from the Anabas testudineus skin mucus fractions. Sci. Rep. 2021, 11, 23182. [Google Scholar] [CrossRef]
- Yamada, K.H.; Kang, H.; Malik, A.B. Antiangiogenic Therapeutic Potential of Peptides Derived from the Molecular Motor KIF13B that Transports VEGFR2 to Plasmalemma in Endothelial Cells. Am. J. Pathol. 2017, 187, 214–224. [Google Scholar] [CrossRef]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef]
- De Berardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Jiang, Y.; Huang, W.; Sun, X.; Yang, X.; Wu, Y.; Shi, J.; Zheng, J.; Fan, S.; Liu, J.; Wang, J.; et al. DTX-P7, a peptide-drug conjugate, is highly effective for non-small cell lung cancer. J. Hematol. Oncol. 2022, 15, 73. [Google Scholar] [CrossRef]
- Chen, X.; Plasencia, C.; Hou, Y.; Neamati, N. Synthesis and Biological Evaluation of Dimeric RGD Peptide-Paclitaxel Conjugate as a Model for Integrin-Targeted Drug Delivery. J. Med. Chem. 2005, 48, 1098–1106. [Google Scholar] [CrossRef]
- Burkhart, D.J.; Kalet, B.T.; Coleman, M.P.; Post, G.C.; Koch, T.H. Doxorubicin-formaldehyde conjugates targeting alphavbeta3 integrin. Mol. Cancer Ther. 2004, 3, 1593–1604. [Google Scholar] [CrossRef]
- Davis, R.A.; Ganguly, T.; Harris, R.; Hausner, S.H.; Kovacs, L.; Sutcliffe, J.L. Synthesis and Evaluation of a Monomethyl Auristatin E─Integrin α v β 6 Binding Peptide–Drug Conjugate for Tumor Targeted Drug Delivery. J. Med. Chem. 2023, 66, 9842–9852. [Google Scholar] [CrossRef]
- Redko, B.; Ragozin, E.; Andreii, B.; Helena, T.; Amnon, A.; Talia, S.Z.; Mor, O.; Genady, K.; Gary, G. Synthesis, drug release, and biological evaluation of new anticancer drug–bioconjugates containing somatostatin backbone cyclic analog as a targeting moiety. Pept. Sci. 2015, 104, 743–752. [Google Scholar] [CrossRef]
- Whalen, K.A.; White, B.H.; Quinn, J.M.; Kriksciukaite, K.; Alargova, R.; Yeung, T.P.A.; Bazinet, P.; Brockman, A.; Dupont, M.M.; Oller, H.; et al. Targeting the Somatostatin Receptor 2 with the Miniaturized Drug Conjugate, PEN-221: A Potent and Novel Therapeutic for the Treatment of Small Cell Lung Cancer. Mol. Cancer Ther. 2019, 18, 1926–1936. [Google Scholar] [CrossRef]
- Bauer, W.; Briner, U.; Doepfner, W.; Halier, R.; Huguenin, R.; Marbach, P.; Petcher, T.J.; Pless, J. SMS 201-995: A very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci. 1982, 31, 1133–1140. [Google Scholar] [CrossRef]
- Forssell-Aronsson, E.; Bernhardt, P.; Nilsson, O.; Tisell, L.E.; Wängberg, B.; Ahlman, H. Biodistribution data from 100 patients i.v. injected with 111In-DTPA-D-Phe1-octreotide. Acta Oncol. 2004, 43, 436–442. [Google Scholar] [CrossRef]
- Lelle, M.; Kaloyanova, S.; Freidel, C.; Theodoropoulou, M.; Musheev, M.; Niehrs, C.; Stalla, G.; Peneva, K. Octreotide-mediated tumor-targeted drug delivery via a cleavable doxorubicin-peptide conjugate. Mol. Pharm. 2015, 12, 4290–4300. [Google Scholar] [CrossRef]
- Sun, L.C.; Mackey, L.V.; Luo, J.; Fuselier, J.A.; Coy, D.H. Targeted chemotherapy using a cytotoxic somatostatin conjugate to inhibit tumor growth and metastasis in nude mice. Clin. Med. Oncol. 2008, 2, 491–499. [Google Scholar] [CrossRef]
- Panosa, C.; Tebar, F.; Ferrer-Batallé, M.; Fonge, H.; Seno, M.; Reilly, R.M.; Massaguer, A.; de Llorens, R. Development of an Epidermal Growth Factor Derivative with EGFR Blocking Activity. PLoS ONE 2013, 8, e69325. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, R.; Wu, X.; Sun, Y.; Yao, M.; Li, J.; Xu, Y.; Gu, J. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005, 19, 1978–1985. [Google Scholar] [CrossRef]
- Ahsa, A.; Ramanand, S.G.; Bergin, I.L.; Zha, L.; Whitehead, C.E.; Rehemtulla, A.; Ray, D.; Pratt, W.B.; Lawrence, T.S.; Nyati, M.K. Efficacy of an EGFR-specific peptide against EGFR-dependent cancer cell lines and tumor xenografts. Neoplasia 2014, 16, 105–114. [Google Scholar] [CrossRef]
- Ramos-Álvarez, I.; Moreno, P.; Mantey, S.A.; Nakamura, T.; Nuche-Berenguer, B.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Insights into bombesin receptors and ligands: Highlighting recent advances. Peptides 2015, 72, 128–144. [Google Scholar] [CrossRef]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef]
- Gomena, J.; Vári, B.; Oláh-Szabó, R.; Biri-Kovács, B.; Bősze, S.; Borbély, A.; Soós, Á.; Ranđelović, I.; Tóvári, J.; Mező, G. Targeting the Gastrin-Releasing Peptide Receptor (GRP-R) in Cancer Therapy: Development of Bombesin-Based Peptide–Drug Conjugates. Int. J. Mol. Sci. 2023, 24, 3400. [Google Scholar] [CrossRef]
- Moody, T.W.; Pradhan, T.; Mantey, S.A.; Jensen, R.T.; Dyba, M.; Moody, D.; Tarasova, N.I.; Michejda, C.J. Bombesin marine toxin conjugates inhibit the growth of lung cancer cells. Life Sci. 2008, 82, 855–861. [Google Scholar] [CrossRef]
- Ahmed, H.; Mahmud, A.R.; Siddiquee, M.F.R.; Shahriar, A.; Biswas, P.; Shimul, M.E.K.; Ahmed, S.Z.; Ema, T.I.; Rahman, N.; Khan, M.A.; et al. Role of T cells in cancer immunotherapy: Opportunities and challenges. Cancer Pathog. Ther. 2023, 1, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jang, H.; Choi, J.; Choi, Y.; Yang, Y.; Shim, M.K.; Kim, S.H. Immune checkpoint-targeted drug conjugates: A promising tool for remodeling tumor immune microenvironment. J. Control. Release 2023, 359, 85–96. [Google Scholar] [CrossRef]
- Kubli, S.P.; Berger, T.; Araujo, D.V.; Siu, L.L.; Mak, T.W. Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov. 2021, 20, 899–919. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Shu, X.S.; Li, M.; Zhang, Y.; Yao, Y.; Huang, X.; Li, J.; Wei, P.; He, Z.; Lu, J.; et al. A Novel Strategy Conjugating PD-L1 Polypeptide With Doxorubicin Alleviates Chemotherapeutic Resistance and Enhances Immune Response in Colon Cancer. Front. Oncol. 2021, 11, 737323. [Google Scholar] [CrossRef]
- Pang, L.; Pei, Y.; Uzunalli, G.; Hyun, H.; Lyle, L.T.; Yeo, Y. Surface Modification of Polymeric Nanoparticles with M2pep Peptide for Drug Delivery to Tumor-Associated Macrophages. Pharm. Res. 2019, 36, 65. [Google Scholar] [CrossRef]
- Moon, Y.; Shim, M.K.; Choi, J.; Yang, S.; Kim, J.; Yun, W.S.; Cho, H.; Park, J.Y.; Kim, Y.; Seong, J.K.; et al. Anti-PD-L1 peptide-conjugated prodrug nanoparticles for targeted cancer immunotherapy combining PD-L1 blockade with immunogenic cell death. Theranostics 2022, 12, 1919–2014. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). M9 Biopharmaceutics Classification SystemBased Biowaivers: Guidance for Industry; ICH: Geneva, Switzerland, 2021; Volume 20.
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, H. Molecular Engineering of Peptide–Drug Conjugates for Therapeutics. Pharmaceutics 2022, 14, 212. [Google Scholar] [CrossRef]
- Soepenberg, O.; Sparreboom, A.; Verweij, J. Clinical Studies of Camptothecin and Derivatives. Alkaloids Chem. Biol. 2003, 60, 1–50. [Google Scholar] [CrossRef]
- Cheetham, A.G.; Ou, Y.C.; Zhang, P.; Cui, H. Linker-determined drug release mechanism of free camptothecin from self-assembling drug amphiphiles. Chem. Commun. 2014, 50, 6039–6042. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Su, H.; Lin, R.; Chakroun, R.W.; Monroe, M.K.; Wang, Z.; Porter, M.; Cui, H. Supramolecular Tubustecan Hydrogel as Chemotherapeutic Carrier to Improve Tumor Penetration and Local Treatment Efficacy. ACS Nano 2020, 14, 10083–10094. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Kuang, Y.; Guo, Z.F.; Guo, Z.; Krauss, I.J.; Xu, B. Enzyme-instructed molecular self-assembly confers nanofibers and a supramolecular hydrogel of taxol derivative. J. Am. Chem. Soc. 2009, 131, 13576–13577. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Xiao, Y.; Xu, L.; He, J.; Qian, C.; Li, W.; Wu, L.; Chen, R.; Wang, J.; Hu, R.; et al. Drug-Bearing Supramolecular MMP Inhibitor Nanofibers for Inhibition of Metastasis and Growth of Liver Cancer. Adv. Sci. 2018, 5, 1700867. [Google Scholar] [CrossRef]
- Rodriguez, C.A.; Papanastasiou, E.A.; Juba, M.; Bishop, B. Covalent modification of a ten-residue cationic antimicrobial peptide with levofloxacin. Front. Chem. 2014, 2, 71. [Google Scholar] [CrossRef]
- Desgranges, S.; Ruddle, C.C.; Burke, L.P.; McFadden, T.M.; O’Brien, J.E.; Fitzgerald-Hughes, D.; Humphreys, H.; Smyth, T.P.; Devocelle, M. β-Lactam-host defence peptide conjugates as antibiotic prodrug candidates targeting resistant bacteria. RSC Adv. 2012, 2, 2480. [Google Scholar] [CrossRef]
- Li, W.; O’Brien-Simpson, N.M.; Holden, J.A.; Otvos, L.; Reynolds, E.C.; Separovic, F.; Hossain, M.A.; Wade, J.D. Covalent conjugation of cationic antimicrobial peptides with a β-lactam antibiotic core. Pept. Sci. 2018, 110, e24059. [Google Scholar] [CrossRef]
- Ghaffar, K.; Hussein, W.; Khalil, Z.; Capon, R.; Skwarczynski, M.; Toth, I. Levofloxacin and Indolicidin for Combination Antimicrobial Therapy. Curr. Drug Deliv. 2015, 12, 108–114. [Google Scholar] [CrossRef]
- Umstätter, F.; Werner, J.; Zerlin, L.; Mühlberg, E.; Kleist, C.; Klika, K.D.; Hertlein, T.; Beijer, B.; Domhan, C.; Zimmermann, S.; et al. Impact of Linker Modification and PEGylation of Vancomycin Conjugates on Structure-Activity Relationships and Pharmacokinetics. Pharmaceuticals 2022, 15, 159. [Google Scholar] [CrossRef]
- Brezden, A.; Mohamed, M.F.; Nepal, M.; Harwood, J.S.; Kuriakose, J.; Seleem, M.N.; Chmielewski, J. Dual Targeting of Intracellular Pathogenic Bacteria with a Cleavable Conjugate of Kanamycin and an Antibacterial Cell-Penetrating Peptide. J. Am. Chem. Soc. 2016, 138, 10945–10949. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Brezden, A.; Mohammad, H.; Chmielewski, J.; Seleem, M.N. Targeting biofilms and persisters of ESKAPE pathogens with P14KanS, a kanamycin peptide conjugate. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Ptaszyńska, N.; Gucwa, K.; Olkiewicz, K.; Łȩgowska, A.; Okońska, J.; Ruczyński, J.; Gitlin-Domagalska, A.; Dȩbowski, D.; Milewski, S.; Rolka, K. Antibiotic-Based Conjugates Containing Antimicrobial HLopt2 Peptide: Design, Synthesis, Antimicrobial and Cytotoxic Activities. ACS Chem. Biol. 2019, 14, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, R.; Kawano, K.; Yamaoka, Y.; Taniguchi, A.; Yano, Y.; Takasu, K.; Matsuzaki, K. Development of Antimicrobial Peptide–Antibiotic Conjugates to Improve the Outer Membrane Permeability of Antibiotics Against Gram-Negative Bacteria. ACS Infect. Dis. 2022, 8, 2339–2347. [Google Scholar] [CrossRef]
- Azuma, E.; Choda, N.; Odaki, M.; Yano, Y.; Matsuzaki, K. Improvement of Therapeutic Index by the Combination of Enhanced Peptide Cationicity and Proline Introduction. ACS Infect. Dis. 2020, 6, 2271–2278. [Google Scholar] [CrossRef]
- Etayash, H.; Alford, M.; Akhoundsadegh, N.; Drayton, M.; Straus, S.K.; Hancock, R.E.W. Multifunctional Antibiotic–Host Defense Peptide Conjugate Kills Bacteria, Eradicates Biofilms, and Modulates the Innate Immune Response. J. Med. Chem. 2021, 64, 16854–16863. [Google Scholar] [CrossRef]
- Yang, K.; Gitter, B.; Rüger, R.; Wieland, G.D.; Chen, M.; Liu, X.; Albrecht, V.; Fahr, A. Antimicrobial peptide-modified liposomes for bacteria targeted delivery of temoporfin in photodynamic antimicrobial chemotherapy. Photochem. Photobiol. Sci. 2011, 10, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Judzewitsch, P.R.; Corrigan, N.; Wong, E.H.H.; Boyer, C. Photo-Enhanced Antimicrobial Activity of Polymers Containing an Embedded Photosensitiser. Angew. Chemie Int. Ed. 2021, 60, 24248–24256. [Google Scholar] [CrossRef]
- Dosselli, R.; Millioni, R.; Puricelli, L.; Tessari, P.; Arrigoni, G.; Franchin, C.; Segalla, A.; Teardo, E.; Reddi, E. Molecular targets of antimicrobial photodynamic therapy identified by a proteomic approach. J. Proteomics 2012, 77, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Todorovski, T.; Kalafatovic, D.; Andreu, D. Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives. Pharmaceutics 2023, 15, 357. [Google Scholar] [CrossRef]
- Liang, G.; Wang, H.; Chong, H.; Cheng, S.; Jiang, X.; He, Y.; Wang, C.; Liu, K. An effective conjugation strategy for designing short peptide-based HIV-1 fusion inhibitors. Org. Biomol. Chem. 2016, 14, 7875–7882. [Google Scholar] [CrossRef]
- Wang, C.; Lu, L.; Na, H.; Li, X.; Wang, Q.; Jiang, X.; Xu, X.; Yu, F.; Zhang, T.; Li, J.; et al. Conjugation of a Nonspecific Antiviral Sapogenin with a Specific HIV Fusion Inhibitor: A Promising Strategy for Discovering New Antiviral Therapeutics. J. Med. Chem. 2014, 57, 7342–7354. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, Y.; Lei, Y.; Wang, R.; Zhan, M.; Liu, J.; An, Y.; Zhou, Y.; Zhan, J.; Yin, F.; et al. Design and Evaluation of a Novel Peptide–Drug Conjugate Covalently Targeting SARS-CoV-2 Papain-like Protease. J. Med. Chem. 2022, 65, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Liotard, J.-F.; Mehiri, M.; Di Giorgio, A.; Boggetto, N.; Reboud-Ravaux, M.; Aubertin, A.-M.; Condom, R.; Patino, N. AZT and AZT-monophosphate Prodrugs Incorporating HIV-protease Substrate Fragment: Synthesis and Evaluation as Specific Drug Delivery Systems. Antivir. Chem. Chemother. 2006, 17, 193–213. [Google Scholar] [CrossRef]
- Zhou, X.; Smith, Q.R.; Liu, X. Brain penetrating peptides and peptide–drug conjugates to overcome the blood–brain barrier and target CNS diseases. WIREs Nanomed. Nanobiotechnology 2021, 13, e1695. [Google Scholar] [CrossRef] [PubMed]
- Chapeau, D.; Beekman, S.; Piet, A.; Li, L.; de Ridder, C.; Stuurman, D.; Seimbille, Y. eSOMA-DM1, a Maytansinoid-Based Theranostic Small-Molecule Drug Conjugate for Neuroendocrine Tumors. Bioconjug. Chem. 2024, 35, 1823–1834. [Google Scholar] [CrossRef]
- Mendonça, D.A.; Bakker, M.; Cruz-Oliveira, C.; Neves, V.; Jiménez, M.A.; Defaus, S.; Cavaco, M.; Veiga, A.S.; Cadima-Couto, I.; Castanho, M.A.R.B.; et al. Penetrating the Blood-Brain Barrier with New Peptide–Porphyrin Conjugates Having anti-HIV Activity. Bioconjug. Chem. 2021, 32, 1067–1077. [Google Scholar] [CrossRef]
- Liang, Y.; Yang, Y.; Huang, R.; Ning, J.; Bao, X.; Yan, Z.; Chen, H.; Ding, L.; Shu, C. Conjugation of sulpiride with a cell penetrating peptide to augment the antidepressant efficacy and reduce serum prolactin levels. Biomed. Pharmacother. 2024, 174, 116610. [Google Scholar] [CrossRef]
- Zheng, M.-Z.; Yang, Z.-Q.; Cai, S.-L.; Zheng, L.-T.; Xue, Y.; Chen, L.; Lin, J. Blood-brain barrier and blood-brain tumor barrier penetrating peptide-drug conjugate as targeted therapy for the treatment of lung cancer brain metastasis. Lung Cancer 2024, 196, 107957. [Google Scholar] [CrossRef]
- Bi, J.; Wang, W.; Du, J.; Chen, K.; Cheng, K. Structure-activity relationship study and biological evaluation of SAC-Garlic acid conjugates as novel anti-inflammatory agents. Eur. J. Med. Chem. 2019, 179, 233–245. [Google Scholar] [CrossRef]
- Shokri, B.; Zarghi, A.; Shahhoseini, S.; Mohammadi, R.; Kobarfard, F. Design, synthesis and biological evaluation of peptide-NSAID conjugates for targeted cancer therapy. Arch. Pharm. 2019, 352, e1800379. [Google Scholar] [CrossRef]
- Rakesh, K.P.; Suhas, R.; Gowda, D.C. Anti-inflammatory and Antioxidant Peptide-Conjugates: Modulation of Activity by Charged and Hydrophobic Residues. Int. J. Pept. Res. Ther. 2019, 25, 227–234. [Google Scholar] [CrossRef]
- Pratihar, S.; Bhagavath, K.K.; Govindaraju, T. Small molecules and conjugates as theranostic agents. RSC Chem. Biol. 2023, 4, 826–849. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; No, Y.H.; Sluyter, R.; Konstantinov, K.; Kim, Y.H.; Kim, J.H. Peptide-nanoparticle conjugates as a theranostic platform. Coord. Chem. Rev. 2024, 500, 215530. [Google Scholar] [CrossRef]
- Bhatt Mitra, J.; Chatterjee, S.; Kumar, A.; Khatoon, E.; Chandak, A.; Rakshit, S.; Bandyopadhyay, A.; Mukherjee, A. Expanding a peptide-covalent probe hybrid for PET imaging of S. aureus driven focal infections. EJNMMI Radiopharm. Chem. 2024, 9, 25. [Google Scholar] [CrossRef]
- Pham, T.T.; Hungnes, I.N.; Rivas, C.; Cleaver, J.; Firth, G.; Blower, P.J.; Sosabowski, J.; Cook, G.J.R.; Livieratos, L.; Young, J.D.; et al. Receptor-Targeted Peptide Conjugates Based on Diphosphines Enable Preparation of 99m Tc and 188 Re Theranostic Agents for Prostate Cancer. J. Nucl. Med. 2024, 65, 1087–1094. [Google Scholar] [CrossRef]
- Qin, Y.; Tang, X.; Chen, J.; Huang, J.; Wang, D.; Zhang, X.; Zhang, Y.; Wu, F.; Wang, J. An LHRH peptide-conjugated ruthenium(II) complex as tumor-targeted theranostic anticancer agent. Inorg. Chem. Commun. 2022, 136, 109166. [Google Scholar] [CrossRef]
- Khan, A.; Tripathi, A.; Gandhi, M.; Bellare, J.; Srivastava, R. Development of injectable upconversion nanoparticle-conjugated doxorubicin theranostics electrospun nanostructure for targeted photochemotherapy in breast cancer. J. Biomed. Mater. Res. Part A 2024, 112, 1612–1626. [Google Scholar] [CrossRef]
- Shang, Q.; Su, Y.; Leslie, F.; Sun, M.; Wang, F. Advances in peptide-drug conjugate-based supramolecular hydrogel systems for local drug delivery. Med. Drug Discov. 2022, 14, 100125. [Google Scholar] [CrossRef]
- Schiapparelli, P.; Zhang, P.; Lara-Velazquez, M.; Guerrero-Cazares, H.; Lin, R.; Su, H.; Chakroun, R.W.; Tusa, M.; Quiñones-Hinojosa, A.; Cui, H. Self-assembling and self-formulating prodrug hydrogelator extends survival in a glioblastoma resection and recurrence model. J. Control. Release 2020, 319, 311–321. [Google Scholar] [CrossRef]
- Webber, M.J.; Matson, J.B.; Tamboli, V.K.; Stupp, S.I. Controlled release of dexamethasone from peptide nanofiber gels to modulate inflammatory response. Biomaterials 2012, 33, 6823–6832. [Google Scholar] [CrossRef]
- Chakroun, R.W.; Wang, F.; Lin, R.; Wang, Y.; Su, H.; Pompa, D.; Cui, H. Fine-Tuning the Linear Release Rate of Paclitaxel-Bearing Supramolecular Filament Hydrogels through Molecular Engineering. ACS Nano 2019, 13, 7780–7790. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Song, H.; Feng, Z.; Wang, C.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. Synthetic, Supramolecular, and Self-Adjuvanting CD8+ T-Cell Epitope Vaccine Increases the Therapeutic Antitumor Immunity. Adv. Ther. 2019, 2, 1900010. [Google Scholar] [CrossRef]
- Li, J.; Fang, Y.; Zhang, Y.; Wang, H.; Yang, Z.; Ding, D. Supramolecular Self-Assembly-Facilitated Aggregation of Tumor-Specific Transmembrane Receptors for Signaling Activation and Converting Immunologically Cold to Hot Tumors. Adv. Mater. 2021, 33, 2008518. [Google Scholar] [CrossRef] [PubMed]
- Sagar, B.; Gupta, S.; Verma, S.K.; Reddy, Y.V.M.; Shukla, S. Navigating cancer therapy: Harnessing the power of peptide-drug conjugates as precision delivery vehicles. Eur. J. Med. Chem. 2025, 283, 117131. [Google Scholar] [CrossRef]
- Fetse, J.; Zhao, Z.; Liu, H.; Mamani, U.F.; Mustafa, B.; Adhikary, P.; Ibrahim, M.; Liu, Y.; Patel, P.; Nakhjiri, M.; et al. Discovery of Cyclic Peptide Inhibitors Targeting PD-L1 for Cancer Immunotherapy. J. Med. Chem. 2022, 65, 12002–12013. [Google Scholar] [CrossRef]

| Clinical Trials Gov. ID | Peptide–Drug Conjugate | Application | Phase | Drug Component | Sponsors |
|---|---|---|---|---|---|
| NCT02048059 | ANG1005 | Targets breast cancer cells with relapsing brain metastases | II | Paclitaxel | Angiochem Inc |
| NCT03613181 | For newly diagnosed leptomeningeal carcinomatosis with prior brain metastases | III | |||
| NCT04706962 | TH1902 | Therapy for solid tumors or cancers expressing the SORT1 receptor | I | Docetaxel | Theratechnologies |
| NCT05465590 | MB1707 | Targets the SDF-1/CXCR4 pathway to inhibit tumor growth and metastasis | I | Paclitaxel | Mainline Biosciences, Inc. |
| NCT05725070 | 212Pb-NG001 | Theranostic salvage therapy for metastatic castration-resistant prostate cancer using PSMA-targeted 212Pb-NG001 | 0/I | 212Pb-NG001 | ARTBIO Inc. |
| NCT01480583 | GRN1005 | Potential monotherapy or combination with trastuzumab for breast cancer brain metastases | II | Paclitaxel | Angiochem Inc |
| NCT01497665 | Patients with non-small cell lung cancer and brain metastases. | II | |||
| NCT05518071 | FLUOPANC | Fluorescent marker for bile duct and pancreatic tumor surgery | I | Fluorophore ZW800-1 | Leiden University Medical Center |
| NCT01698281 | AEZS-108 (Zoptarelin DOX) | Chemotherapy for triple-negative breast cancer | II | DOX | AEterna Zentaris |
| NCT01767155 | Second-line treatment for endometrial cancer | III | |||
| NCT03486730 | BT1718 | Treatment of advanced solid tumors | I/IIa | DM1 | Cancer Research UK |
| NCT00710125 | GPX-150 | Curing solid tumors | I | Modified analog of DOX | Gem Pharmaceuticals |
| NCT02267083 | Therapy of soft tissue sarcoma | II | |||
| NCT06326190 | 177Lu-DOTATATE | Recurrent Meningioma | II | 177Lu | European Organization for Research and Treatment of Cancer—EORTC |
| NCT04529044 | Treating recurrent or stage 4 breast cancer | II | OHSU Knight Cancer Institute | ||
| NCT02489604 | Curing advanced gastroenteropancreatic neuroendocrine tumors | II | Istituto Scientifico Romagnolo per lo Studio e la cura dei Tumori | ||
| NCT04385992 | Post-surgical treatment for resectable pancreatic neuroendocrine tumors | II | IRCCS San Raffaele | ||
| NCT06460467 | Dosimetric calculation for treating neuroendocrine tumors or meningiomas | I | Central Hospital, Nancy, France | ||
| NCT02736500 | Treating aggressive gastroenteropancreatic neuroendocrine tumors | I-II | Istituto Scientifico Romagnolo per lo Studio e la cura dei Tumori | ||
| NCT04544098 | Gastroenteropancreatic, bronchial, or unknown primary neuroendocrine tumors metastasized to the liver | I | Memorial Sloan Kettering Cancer Center | ||
| NCT04180371 | BT5528 | Treating advanced solid tumors exhibiting EphA2 expression | I/II | Monomethyl auristatin E | BicycleTx Limited |
| NCT04552847 | [18F]AlF-NOTA-octreotide | PET imaging of neuroendocrine tumors | II/III | 18F | Universitaire Ziekenhuizen KU Leuven |
| NCT00918281 | [18F]Fluciclatide | Solid tumor PET imaging | II | 18F | GE Healthcare |
| NCT01633255 | Imaging kidney cancer | I/II | National Cancer Institute | ||
| NCT01633255 | [18F]RGD-K5 | PET imaging | II | 18F | Siemens Molecular Imaging |
| NCT02381236 | G-202 (mipsagargin) | Non-invasive multiparametric prostate magnetic resonance imaging (mpMRI) | II | Thapsigargin | GenSpera, Inc. |
| NCT03445884 | 68Ga-NODAGA-E[cyclo(RGDyK)] | PET imaging | II | 68Ga | Rigshospitalet, Denmark |
| NCT02749019 | 68Ga-NOTA-BBN-RGD | PET imaging | I | 68Ga | Peking Union Medical College Hospital |
| NCT02936323 | PEN-221 | For somatostatin receptor 2 expressing higher stages of cancers, including neuroendocrine and small cell lung cancers | I/IIa | DM-1 | Tarveda Therapeutics |
| NCT03273712 | 90Y-DOTATOC | Radionuclide therapy for patients with somatostatin receptor-positive tumors | II | 90Y | University of Iowa |
| NCT04740398 | CBP-1008 | Later stages of solid tumors | I | MMAE | Coherent Biopharma (Suzhou) Co., Ltd. |
| NCT04928612 | CBP-1018 | Later stages of solid tumors | I | MMAE | Coherent Biopharma (Suzhou) Co., Ltd. |
| NCT03784677 | SOR-C13 | Later stages of malignant solid neoplasm | I | MMAE | M.D. Anderson Cancer Center |
| NCT05079698 | 177Lu-PSMA-617 | Prostate cancer | I | DOTA | Memorial Sloan Kettering Cancer Center |
| NCT02742168 | 99mTc-3PRGD2 | Breast cancer | I | 99mTc | First Affiliated Hospital of Fujian Medical University |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jadhav, K.; Abhang, A.; Kole, E.B.; Gadade, D.; Dusane, A.; Iyer, A.; Sharma, A.; Rout, S.K.; Gholap, A.D.; Naik, J.; et al. Peptide–Drug Conjugates as Next-Generation Therapeutics: Exploring the Potential and Clinical Progress. Bioengineering 2025, 12, 481. https://doi.org/10.3390/bioengineering12050481
Jadhav K, Abhang A, Kole EB, Gadade D, Dusane A, Iyer A, Sharma A, Rout SK, Gholap AD, Naik J, et al. Peptide–Drug Conjugates as Next-Generation Therapeutics: Exploring the Potential and Clinical Progress. Bioengineering. 2025; 12(5):481. https://doi.org/10.3390/bioengineering12050481
Chicago/Turabian StyleJadhav, Krishna, Ashwin Abhang, Eknath B. Kole, Dipak Gadade, Apurva Dusane, Aditya Iyer, Ankur Sharma, Saroj Kumar Rout, Amol D. Gholap, Jitendra Naik, and et al. 2025. "Peptide–Drug Conjugates as Next-Generation Therapeutics: Exploring the Potential and Clinical Progress" Bioengineering 12, no. 5: 481. https://doi.org/10.3390/bioengineering12050481
APA StyleJadhav, K., Abhang, A., Kole, E. B., Gadade, D., Dusane, A., Iyer, A., Sharma, A., Rout, S. K., Gholap, A. D., Naik, J., Verma, R. K., & Rojekar, S. (2025). Peptide–Drug Conjugates as Next-Generation Therapeutics: Exploring the Potential and Clinical Progress. Bioengineering, 12(5), 481. https://doi.org/10.3390/bioengineering12050481