
Biomechanical Properties and Cellular Responses in Pulmonary Fibrosis


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Mechanical Properties of Fibrotic Lung
2.1. Elastic Properties of Fibrotic Lung
2.2. Viscoelastic Properties of Fibrotic Lungs
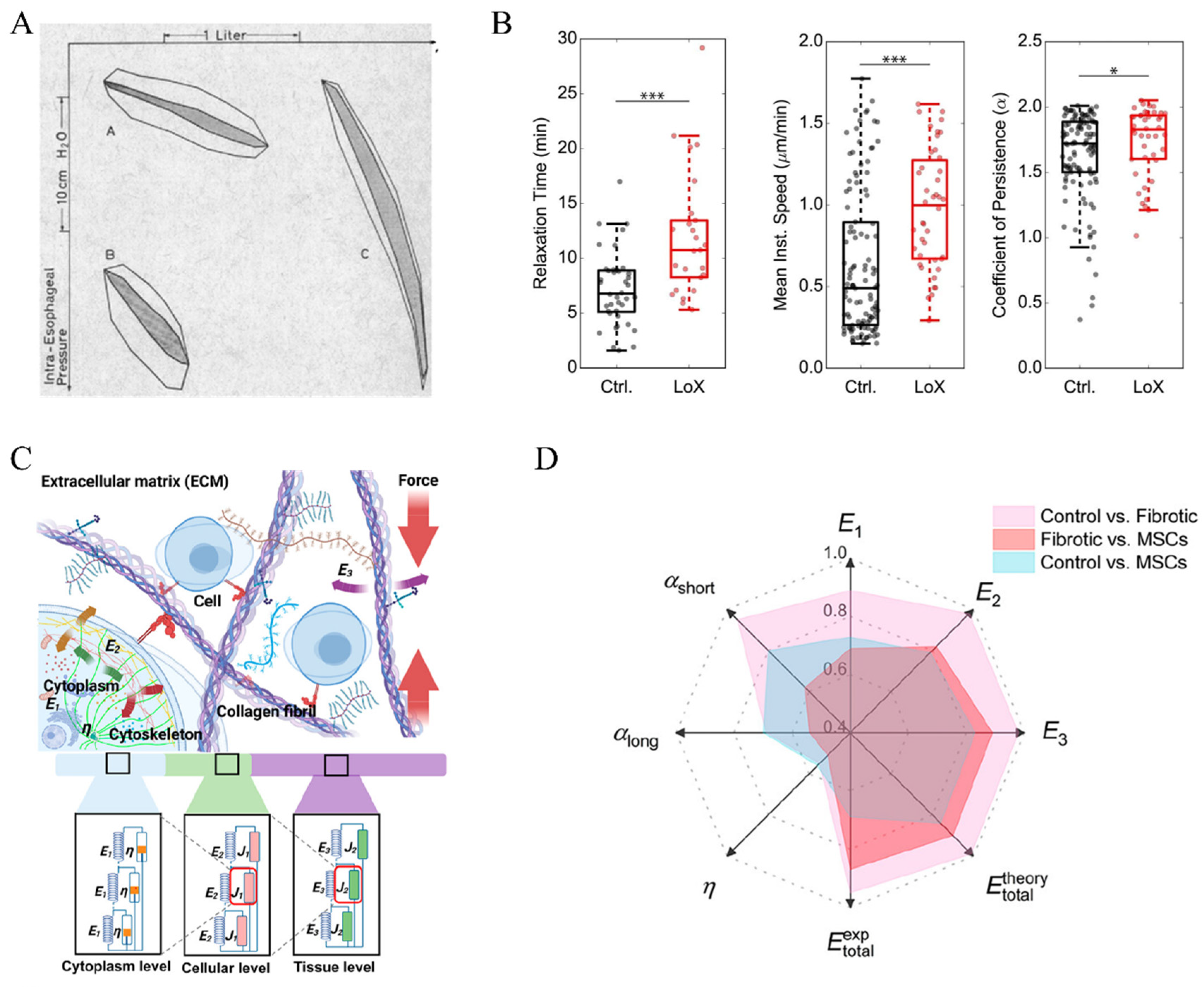
2.3. Surface Tension in Fibrotic Lungs
3. Cells Respond to Alterations in Mechanical Properties to Exacerbate Pulmonary Fibrosis
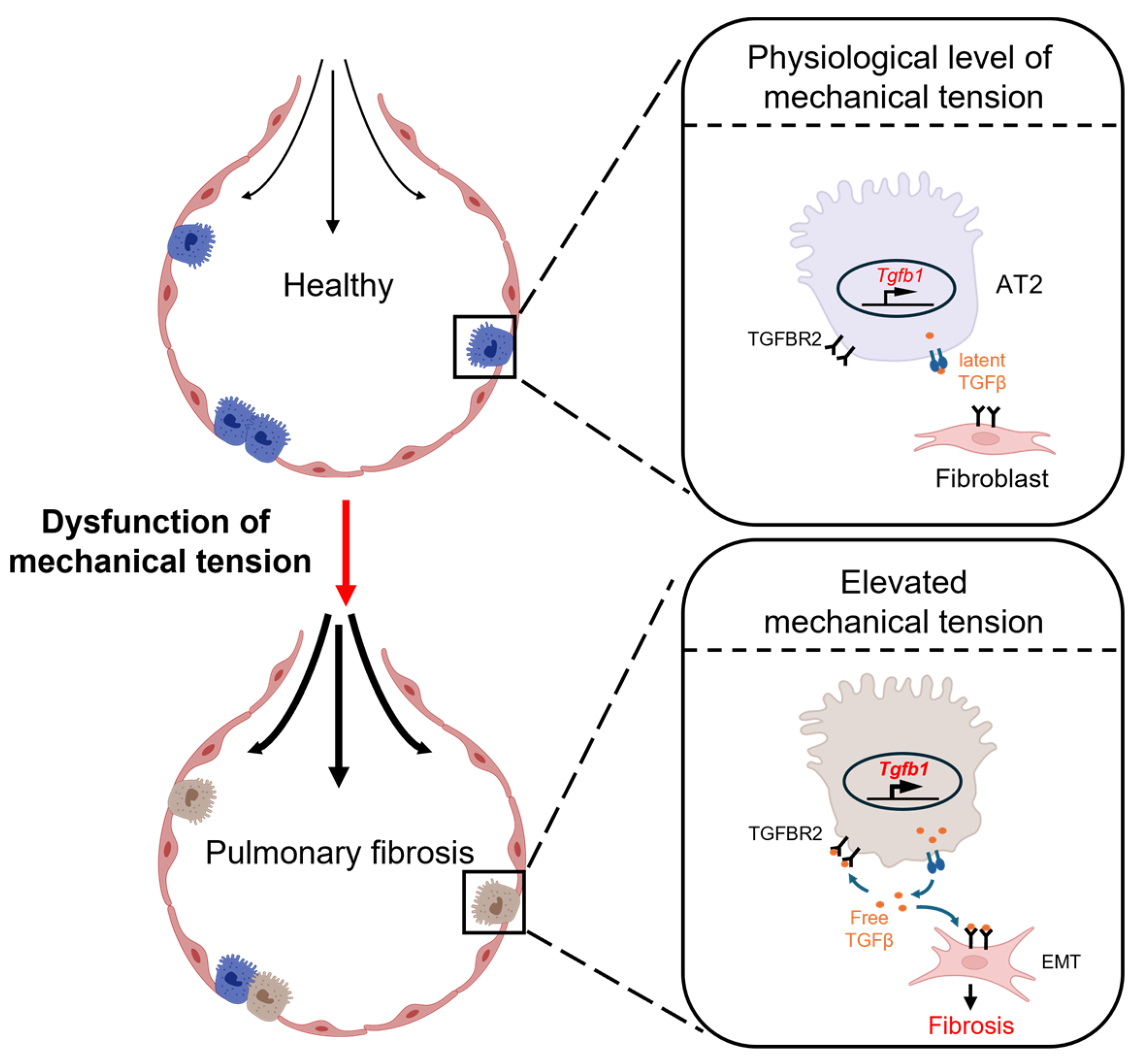
3.1. Alveolar Epithelial Cells
3.2. Fibroblast
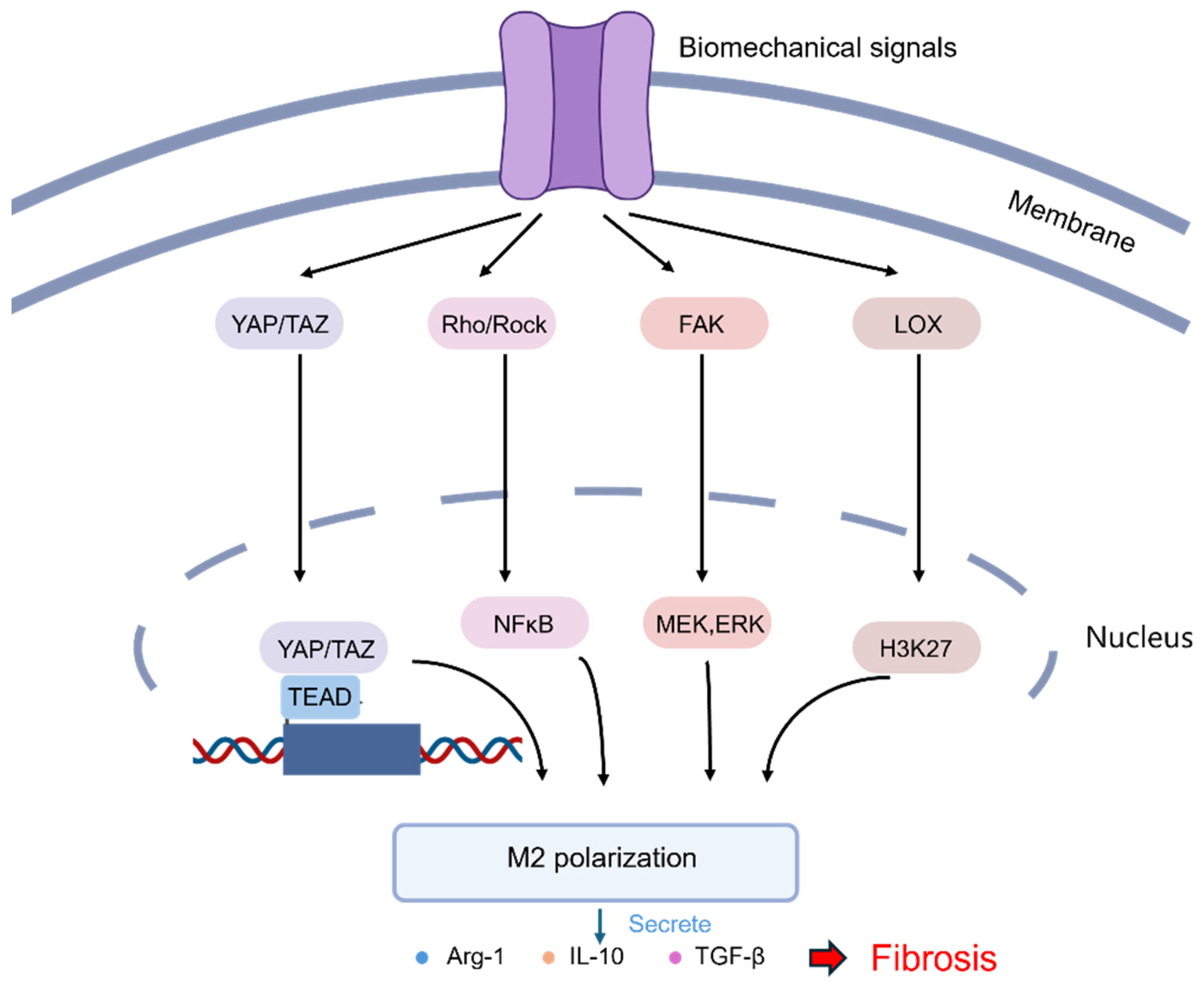
3.3. Macrophage
4. Future Perspective

5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ECM | Extracellular matrix |
| 3D | Three-dimensional |
| AFM | Atomic force microscopy |
| a-SMA | A-smooth muscle actin |
| TGF-B | Transforming growth factor β |
| PNX | Pneumonectomy |
| LOX | Lysyl oxidase |
| DMA | Dynamic mechanical analysis |
| GelMA | Methacrylate Gelatin |
| MSC | Mesenchymal stem cell |
| ROC | Receiver operating characteristic |
| BALF | Bronchoalveolar lavage fluid |
| SP-A | Surfactant protein A |
| PHMG | Polyhexamethylene guanidine |
| CFD | Computational fluid dynamics |
| AT1 | Alveolar type 1 cells |
| AT2 | Alveolar type 2 cells |
| ARDS | Acute respiratory distress syndrome |
| EMT | Epithelial–mesenchymal transition |
| TMEM63 | Transmembrane 63 |
| DAMP | Damage-associated molecular patterns |
| HMGB1 | High-mobility group box 1 |
| YAP | Yes-associated protein |
| TAZ | Transcriptional co-activator with PDZ-binding motif |
| NF-KB | Nuclear factor kappa-B |
| MAPK | Mitogen-activated protein kinase |
| PI3K | Phosphatidylin-ositol-3-kinase |
| ACTA2 | Actin alpha 2 |
| Col3A1 | Collagen alpha-1 (III) chain |
| Col4A1 | Collagen alpha-1 (IV) chain |
| ASK1 | Apoptosis signal-regulating kinase-1 |
| NUAK | NUAK family kinase 1 |
| ECAR | Extracellular acidification rate |
| MtRNA | Mitochondrial RNA |
| RhoA | Ras Homolog Family Member A |
| TNF-A | Tumor necrosis factor-alpha |
| IL-1B | Interleukin-1 beta |
| ROS | Reactive oxygen species |
| PDGF | Platelet-derived growth factor |
| CCL2 | Chemokine ligands 2 |
| CCL8 | Chemokine ligands 8 |
| MMPs | Matrix metalloproteinases |
| BMDM | Bone marrow-derived macrophages |
| MMP-1 | Matrix metallopeptidase 1 |
| MTG | Microbial transglutaminase |
| DLP | Digital light processing |
| ScRNA-seq | Single-cell RNA sequencing |
References
- Liu, G.Y.; Budinger, G.R.S.; Dematte, J.E. Advances in the management of idiopathic pulmonary fibrosis and progressive pulmonary fibrosis. BMJ 2022, 377, e066354. [Google Scholar] [CrossRef] [PubMed]
- Wijsenbeek, M.; Suzuki, A.; Maher, T.M. Interstitial lung diseases. Lancet 2022, 400, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Podolanczuk, A.J.; Thomson, C.C.; Remy-Jardin, M.; Richeldi, L.; Martinez, F.J.; Kolb, M.; Raghu, G. Idiopathic pulmonary fibrosis: State of the art for 2023. Eur. Respir. J. 2023, 61, 2200957. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; He, A.; Li, H.; Chen, H.; Xie, H.; Luo, L.; Huang, Y.; Chen, J.; Guan, J.; He, Q.; et al. TSSK4 upregulation in alveolar epithelial type-II cells facilitates pulmonary fibrosis through HSP90-AKT signaling restriction and AT-II apoptosis. Cell Death Dis. 2021, 12, 938. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Young, L.R. Insights into the Pathogenesis of Pulmonary Fibrosis from Genetic Diseases. Am. J. Respir. Cell Mol. Biol. 2022, 67, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Odell, I.D.; Steach, H.; Gauld, S.B.; Reinke-Breen, L.; Karman, J.; Carr, T.L.; Wetter, J.B.; Phillips, L.; Hinchcliff, M.; Flavell, R.A. Epiregulin is a dendritic cell–derived EGFR ligand that maintains skin and lung fibrosis. Sci. Immunol. 2022, 7, eabq6691. [Google Scholar] [CrossRef] [PubMed]
- Perrot, C.Y.; Karampitsakos, T.; Herazo-Maya, J.D. Monocytes and macrophages: Emerging mechanisms and novel therapeutic targets in pulmonary fibrosis. Am. J. Physiol. Physiol. 2023, 325, C1046–C1057. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.L.; Stokes, C.A.; Marciniak, S.J.; Parker, L.C.; Condliffe, A.M. Role of unfolded proteins in lung disease. Thorax 2021, 76, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Koudstaal, T.; Funke-Chambour, M.; Kreuter, M.; Molyneaux, P.L.; Wijsenbeek, M.S. Pulmonary fibrosis: From pathogenesis to clinical decision-making. Trends Mol. Med. 2023, 29, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef] [PubMed]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2018, 27, 180033. [Google Scholar] [CrossRef] [PubMed]
- Hesse, C.; Beneke, V.; Konzok, S.; Diefenbach, C.; Sand, J.M.B.; Rønnow, S.R.; Karsdal, M.A.; Jonigk, D.; Sewald, K.; Braun, A.; et al. Nintedanib modulates type III collagen turnover in viable precision-cut lung slices from bleomycin-treated rats and patients with pulmonary fibrosis. Respir. Res. 2022, 23, 201. [Google Scholar] [CrossRef] [PubMed]
- Janmey, P.A.; Fletcher, D.A.; Reinhart-King, C.A. Stiffness Sensing by Cells. Physiol. Rev. 2020, 100, 695–724. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Niu, Y.; Liang, K.; Du, Y. Mechanical communication in fibrosis progression. Trends Cell Biol. 2021, 32, 70–90. [Google Scholar] [CrossRef] [PubMed]
- de Hilster, R.H.J.; Sharma, P.K.; Jonker, M.R.; White, E.S.; Gercama, E.A.; Roobeek, M.; Timens, W.; Harmsen, M.C.; Hylkema, M.N.; Burgess, J.K. Human lung extracellular matrix hydrogels resemble the stiffness and viscoelasticity of native lung tissue. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L698–L704. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Lyu, C.; Liao, H.; Du, Y. Collagen crosslinking: Effect on structure, mechanics and fibrosis progression. Biomed. Mater. 2021, 16, 062005. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.; Du, C.; Jiang, Y.; Zhang, W.; Wang, S.; Zhu, X.; Gao, J.; Zhang, X.; Ren, D.; et al. Polyhexamethylene guanidine aerosol triggers pulmonary fibrosis concomitant with elevated surface tension via inhibiting pulmonary surfactant. J. Hazard. Mater. 2021, 420, 126642. [Google Scholar] [CrossRef]
- Cao, S.; Feng, H.; Yi, H.; Pan, M.; Lin, L.; Zhang, Y.S.; Feng, Z.; Liang, W.; Cai, B.; Li, Q.; et al. Single-cell RNA sequencing reveals the developmental program underlying proximal–distal patterning of the human lung at the embryonic stage. Cell Res. 2023, 33, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Haak, A.J.; Tan, Q.; Tschumperlin, D.J. Matrix biomechanics and dynamics in pulmonary fibrosis. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 73, 64–76. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Rosen, J.; Dagan, A.; Swanson, P.; Sinanan, M.; Hannaford, B. Assessment of tissue damage due to mechanical stresses. In Proceedings of the 2006 1st IEEE Ras-Embs International Conference on Biomedical Robotics and Biomechatronics, Pisa, Italy, 20–22 February 2006; Volume 1–3, p. 860. [Google Scholar]
- Bowman, K. Mechanical Properties: Elastic Behavior. In Encyclopedia of Condensed Matter Physics, 2nd ed.; Chakraborty, T., Ed.; Academic Press: Oxford, UK, 2005; pp. 838–843. [Google Scholar]
- Nakamura, T. Roles of short fibulins, a family of matricellular proteins, in lung matrix assembly and disease. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 73, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.H.; Madison, M.C.; Corry, D.; Kheradmand, F. Matrix remodeling in chronic lung diseases. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 73, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Shimbori, C.; Upagupta, C.; Bellaye, P.-S.; A Ayaub, E.; Sato, S.; Yanagihara, T.; Zhou, Q.; Ognjanovic, A.; Ask, K.; Gauldie, J.; et al. Mechanical stress-induced mast cell degranulation activates TGF-β1 signalling pathway in pulmonary fibrosis. Thorax 2019, 74, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.R.; Shimbori, C.; Bellaye, P.S.; Inman, M.; Obex, S.; Fatima, S.; Jenkins, G.; Gauldie, J.; Gauldie, K.; Kolb, M. Stretch-Induced Activation of Transforming Growth Factor-β1 in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 194, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.J.; Hadley, R.; Cornett, A.M.; Dreffs, A.A.; Matthes, S.A.; Tsui, J.L.; Weiss, K.; Horowitz, J.C.; Fiore, V.F.; Barker, T.H.; et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 2013, 186, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Fiore, V.F.; A Sulchek, T.; Barker, T.H. Physical and chemical microenvironmental cues orthogonally control the degree and duration of fibrosis-associated epithelial-to-mesenchymal transitions. J. Pathol. 2012, 229, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J. Cell Biol. 2010, 190, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2019, 180, 107–121.e17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, X.; Zhang, X.; Chen, S.; Shen, Y.; Song, L. Modeling the mechanical properties of liver fibrosis in rats. J. Biomech. 2016, 49, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Birzle, A.M.; Wall, W.A. A viscoelastic nonlinear compressible material model of lung parenchyma—Experiments and numerical identification. J. Mech. Behav. Biomed. Mater. 2019, 94, 164–175. [Google Scholar] [CrossRef]
- Naini, A.S.; Patel, R.V.; Samani, A. Measurement of lung hyperelastic properties using inverse finite element approach. IEEE Trans. Biomed. Eng. 2011, 58, 2852–2859. [Google Scholar] [CrossRef] [PubMed]
- Aghasafari, P.; Ibrahim, I.B.M.; Pidaparti, R. Strain-induced inflammation in pulmonary alveolar tissue due to mechanical ventilation. Biomech. Model. Mechanobiol. 2017, 16, 1103–1118. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, M.; Nordgren, T.M.; O’connell, G.D. Mechanics of pulmonary airways: Linking structure to function through constitutive modeling, biochemistry, and histology. Acta Biomater. 2019, 97, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wang, Z.; Liu, Y.; Eickhoff, J.C.; Eliceiri, K.W.; Chesler, N.C. Validation of an arterial constitutive model accounting for collagen content and crosslinking. Acta Biomater. 2015, 31, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Cacopardo, L.; Guazzelli, N.; Ahluwalia, A. Characterizing and Engineering Biomimetic Materials for Viscoelastic Mechanotransduction Studies. Tissue Eng. Part B Rev. 2022, 28, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, L.E.; Robertson, G.W. The visco-elastic properties of the lungs. Q. J. Exp. Physiol. Cogn. Med. Sci. Transl. Integr. 1939, 29, 27–47. [Google Scholar] [CrossRef]
- Bachofen, H.; Scherrer, M. Lung tissue resistance in diffuse interstitial pulmonary fibrosis. J. Clin. Investig. 1967, 46, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Venkatesan, N.; Tanaka, R.; Ludwig, M.S. Changes in extracellular matrix and tissue viscoelasticity in bleomycin–induced lung fibrosis. Temporal Asp. Am. J. Respir. Crit. Care Med. 2000, 162 Pt 1, 1569–1576. [Google Scholar] [CrossRef]
- Fredberg, J.J.; Stamenovic, D. On the imperfect elasticity of lung tissue. J. Appl. Physiol. 1985, 67, 2408–2419. [Google Scholar] [CrossRef] [PubMed]
- Pinart, M.; Serrano-Mollar, A.; Negri, E.; Cabrera, R.; Rocco, P.; Romero, P. Inflammatory related changes in lung tissue mechanics after bleomycin-induced lung injury. Respir. Physiol. Neurobiol. 2008, 160, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Bera, K.; Kiepas, A.; Godet, I.; Li, Y.; Mehta, P.; Ifemembi, B.; Paul, C.D.; Sen, A.; Serra, S.A.; Stoletov, K.; et al. Extracellular fluid viscosity enhances cell migration and cancer dissemination. Nature 2022, 611, 365–373. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Maitra, A.; Jacques, C.; Bergert, M.; Pérez-González, C.; Simon, A.; Lederer, L.; Diz-Muñoz, A.; Trepat, X.; Voituriez, R.; et al. Self-generated gradients steer collective migration on viscoelastic collagen networks. Nat. Mater. 2022, 21, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, S.; Evans, D.; Sparks, J.L. Poroviscoelastic Modeling of liver biomechanical response in unconfined compression. Ann. Biomed. Eng. 2010, 38, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Abuhattum, S.; Mokbel, D.; Müller, P.; Soteriou, D.; Guck, J.; Aland, S. An explicit model to extract viscoelastic properties of cells from AFM force-indentation curves. iScience 2022, 25, 104016. [Google Scholar] [CrossRef] [PubMed]
- Efremov, Y.M.; Okajima, T.; Raman, A. Measuring viscoelasticity of soft biological samples using atomic force microscopy. Soft Matter 2019, 16, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Tosini, M.; Tänzer, T.; Villata, S.; Baruffaldi, D.; Monica, V.; Peracino, B.; Primo, L.; Frascella, F.; Pirri, F.; Audenino, A.; et al. A Methodological Approach for Interpreting and Comparing the Viscoelastic Behaviors of Soft Biological Tissues and Hydrogels at the Cell-Length Scale. Appl. Sci. 2024, 14, 1093. [Google Scholar] [CrossRef]
- Chang, Z.; Zhang, L.; Hang, J.-T.; Liu, W.; Xu, G.-K. Viscoelastic multiscale mechanical indexes for assessing liver fibrosis and treatment outcomes. Nano Lett. 2023, 23, 9618–9625. [Google Scholar] [CrossRef] [PubMed]
- Cherstvy, A.G.; Thapa, S.; Wagner, C.E.; Metzler, R. Non-Gaussian, non-ergodic, and non-Fickian diffusion of tracers in mucin hydrogels. Soft Matter 2019, 15, 2526–2551. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.E.; Turner, B.S.; Rubinstein, M.; McKinley, G.H.; Ribbeck, K. A Rheological Study of the Association and Dynamics of MUC5AC Gels. Biomacromolecules 2017, 18, 3654–3664. [Google Scholar] [CrossRef]
- Thapa, S.; Lomholt, M.A.; Krog, J.; Cherstvy, A.G.; Metzler, R. Bayesian analysis of single-particle tracking data using the nested-sampling algorithm: Maximum-likelihood model selection applied to stochastic-diffusivity data. Phys. Chem. Chem. Phys. PCCP 2018, 20, 29018–29037. [Google Scholar] [CrossRef]
- Dose, V. Bayesian inference in physics: Case studies. Rep. Prog. Phys. 2003, 66, 1421. [Google Scholar] [CrossRef]
- Yu, J. Paradoxical response of pulmonary slowly adapting units during constant pressure lung inflation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 321, R220–R227. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Peng, Y.; Mansy, H.A.; Sandler, R.H.; Royston, T.J. A model of lung parenchyma stress relaxation using fractional viscoelasticity. Med. Eng. Phys. 2015, 37, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Monjezi, M.; Saidi, M.S. Fluid-structure interaction analysis of air flow in pulmonary alveoli during normal breathing in healthy humans. Sci. Iran. 2016, 23, 1826–1836. [Google Scholar] [CrossRef]
- Francis, I.; Saha, S.C. Surface tension effects on flow dynamics and alveolar mechanics in the acinar region of human lung. Heliyon 2022, 8, e11026. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Ikegami, M.; Cherniack, R.M.; Mason, R.J. Increased surface tension of the lung and surfactant in bleomycin-induced pulmonary fibrosis in rats. Am. J. Respir. Crit. Care Med. 1996, 154, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Osanai, K.; Takahashi, K.; Sato, S.; Iwabuchi, K.; Ohtake, K.; Sata, M.; Yasui, S.; Gwinn, W.M.; Kapita, M.C.; Wang, P.M.; et al. Changes of lung surfactant and pressure-volume curve in bleomycin-induced pulmonary fibrosis. J. Appl. Physiol. 1991, 70, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Tlatelpa-Romero, B.; Cázares-Ordoñez, V.; Oyarzábal, L.F.; Vázquez-De-Lara, L.G. The Role of Pulmonary Surfactant Phospholipids in Fibrotic Lung Diseases. Int. J. Mol. Sci. 2022, 24, 326. [Google Scholar] [CrossRef] [PubMed]
- Günther, A.; Schmidt, R.; Nix, F.; Yabut-Perez, M.; Guth, C.; Rosseau, S.; Siebert, C.; Grimminger, F.; Morr, H.; Velcovsky, H.; et al. Surfactant abnormalities in idiopathic pulmonary fibrosis, hypersensitivity pneumonitis and sarcoidosis. Eur. Respir. J. 1999, 14, 565–573. [Google Scholar] [CrossRef]
- Correll, K.A.; Edeen, K.E.; Zemans, R.L.; Redente, E.F.; Mikels-Vigdal, A.; Mason, R.J. TGF beta inhibits expression of SP-A, SP-B, SP-C, but not SP-D in human alveolar type II cells. Biochem. Biophys. Res. Commun. 2018, 499, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, E.; Boden, C.; Echaide, M.; Perez-Gil, J.; Kolb, M.; Gauldie, J.; Maus, U.A.; Ochs, M.; Knudsen, L. Surfactant dysfunction during overexpression of TGF-β1 precedes profibrotic lung remodeling in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L1260–L1271. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.; Wang, M.; Du, C.; Zhang, W.; Jiang, Y.; Zhang, W.; Jiang, X.; Ren, D.; Wang, H.; et al. Pulmonary Surfactant Homeostasis Dysfunction Mediates Multiwalled Carbon Nanotubes Induced Lung Fibrosis via Elevating Surface Tension. ACS Nano 2023, 18, 2828–2840. [Google Scholar] [CrossRef] [PubMed]
- Possmayer, F.; Zuo, Y.Y.; Veldhuizen, R.A.; Petersen, N.O. Pulmonary Surfactant: A Mighty Thin Film. Chem. Rev. 2023, 123, 13209–13290. [Google Scholar] [CrossRef] [PubMed]
- Ciutara, C.O.; Iasella, S.V.; Huang, B.; Barman, S.; Zasadzinski, J.A. Evolution of interfacial mechanics of lung surfactant mimics progression of acute respiratory distress syndrome. Proc. Natl. Acad. Sci. USA 2023, 120, e2309900120. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tao, W.; Ji, W.; Lu, Y.; Zhao, X. Effects of pulmonary fibrosis and surface tension on alveolar sac mechanics in diffuse alveolar damage. J. Biomech. Eng. 2021, 143, 081013. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Z.; Ji, G.; Wang, S.; Mo, C.; Ding, B. Lung regeneration: Diverse cell types and the therapeutic potential. Medcomm 2024, 5, e494. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V.; Sheetz, M. Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Guillot, L.; Nathan, N.; Tabary, O.; Thouvenin, G.; Le Rouzic, P.; Corvol, H.; Amselem, S.; Clement, A. Alveolar epithelial cells: Master regulators of lung homeostasis. Int. J. Biochem. Cell Biol. 2013, 45, 2568–2573. [Google Scholar] [CrossRef]
- Jansing, N.L.; McClendon, J.; Henson, P.M.; Tuder, R.M.; Hyde, D.M.; Zemans, R.L. Unbiased Quantitation of Alveolar Type II to Alveolar Type I Cell Transdifferentiation during Repair after Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 57, 519–526. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Markov, N.S.; Lu, Z.; Aillon, R.P.; Soberanes, S.; Runyan, C.E.; Ren, Z.; Grant, R.A.; Maciel, M.; Abdala-Valencia, H.; et al. Resetting proteostasis with ISRIB promotes epithelial differentiation to attenuate pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2101100118. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Benítez, N.E.; Parotto, M.; Post, M.; Han, B.; Spieth, P.M.; Cheng, W.-E.; Valladares, F.; Villar, J.; Liu, M.; Sato, M.; et al. Mechanical stress induces lung fibrosis by epithelial–mesenchymal transition. Crit. Care Med. 2012, 40, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Marchioni, A.; Tonelli, R.; Cerri, S.; Castaniere, I.; Andrisani, D.; Gozzi, F.; Bruzzi, G.; Manicardi, L.; Moretti, A.; Demurtas, J.; et al. Pulmonary Stretch and Lung Mechanotransduction: Implications for Progression in the Fibrotic Lung. Int. J. Mol. Sci. 2021, 22, 6443. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-L.; Li, J.-Y.; Chen, X.; Liu, J.-W.; Zhang, Q.; Liu, J.-Y.; Wen, J.; Wang, N.; Lei, M.; Wei, J.-P.; et al. Mechanosensitive channels TMEM63A and TMEM63B mediate lung inflation-induced surfactant secretion. J. Clin. Investig. 2024, 134, e174508. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Saito, Y.; Matsuda, K.; Kamio, K.; Abe, S.; Kubota, K.; Azuma, A.; Gemma, A. Cyclic mechanical stretch-induced oxidative stress occurs via a NOX-dependent mechanism in type II alveolar epithelial cells. Respir. Physiol. Neurobiol. 2017, 242, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Valentine, M.S.; Link, P.A.; Herbert, J.A.; Gninzeko, F.J.K.; Schneck, M.B.; Shankar, K.; Nkwocha, J.; Reynolds, A.M.; Heise, R.L. Inflammation and Monocyte Recruitment Due to Aging and Mechanical Stretch in Alveolar Epithelium are Inhibited by the Molecular Chaperone 4-Phenylbutyrate. Cell. Mol. Bioeng. 2018, 11, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Xu, L.; Yang, T.; Wang, F. High-mobility group box-1 and its role in angiogenesis. J. Leukoc. Biol. 2014, 95, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Zabini, D.; Crnkovic, S.; Xu, H.; Tscherner, M.; Ghanim, B.; Klepetko, W.; Olschewski, A.; Kwapiszewska, G.; Marsh, L.M. High-mobility group box-1 induces vascular remodelling processes via c-Jun activation. J. Cell. Mol. Med. 2015, 19, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as master regulators in cancer: Modulation, function and therapeutic approaches. Nat. Cancer 2022, 4, 9–26. [Google Scholar] [CrossRef] [PubMed]
- LaCanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Investig. 2019, 129, 2107–2122. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Gokey, J.J.; Sridharan, A.; Xu, Y.; Green, J.; Carraro, G.; Stripp, B.R.; Perl, A.-K.T.; Whitsett, J.A. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. J. Clin. Investig. 2018, 3, e98738. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, J.C. From the library of the Netherlands Journal of Medicine. Rudolf Virchow: Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre; 1858. Ned. Tijdschr. Voor Geneeskd. 2003, 147, 2236–2244. [Google Scholar]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, V.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.F.; Desjardins-Park, H.E.; Mascharak, S.; Borrelli, M.R.; Longaker, M.T. Understanding the impact of fibroblast heterogeneity on skin fibrosis. Dis. Model. Mech. 2020, 13, dmm044164. [Google Scholar] [CrossRef] [PubMed]
- Talbott, H.E.; Mascharak, S.; Griffin, M.; Wan, D.C.; Longaker, M.T. Wound healing, fibroblast heterogeneity, and fibrosis. Cell Stem Cell 2022, 29, 1161–1180. [Google Scholar] [CrossRef] [PubMed]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Valenzi, E.; Bulik, M.; Tabib, T.; Morse, C.; Sembrat, J.; Bittar, H.T.; Rojas, M.; Lafyatis, R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 2019, 78, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-F.; Liao, S.-K.; Huang, C.-C.; Hung, M.-J.; A Quinn, D. Serine/threonine kinase-protein kinase B and extracellular signal-regulated kinase regulate ventilator-induced pulmonary fibrosis after bleomycin-induced acute lung injury: A prospective, controlled animal experiment. Crit. Care 2008, 12, R103. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Mei, S.; Xu, Q.; Feng, J.; Zhou, Y.; Xing, S.; He, Z.; Gao, Y. ASK1-ER stress pathway-mediated fibrotic-EV release contributes to the interaction of alveolar epithelial cells and lung fibroblasts to promote mechanical ventilation-induced pulmonary fibrosis. Exp. Mol. Med. 2022, 54, 2162–2174. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Wang, Y.; Liu, Y.-J.; Mao, Y.-F.; Dong, W.-W.; Ding, Z.-N.; Meng, G.-X.; Jiang, L.; Zhu, X.-Y. NLRP3 Inflammasome Activation Contributes to Mechanical Stretch–Induced Endothelial-Mesenchymal Transition and Pulmonary Fibrosis. Crit. Care Med. 2018, 46, e49–e58. [Google Scholar] [CrossRef] [PubMed]
- Haak, A.J.; Kostallari, E.; Sicard, D.; Ligresti, G.; Choi, K.M.; Caporarello, N.; Jones, D.L.; Tan, Q.; Meridew, J.; Espinosa, A.M.D.; et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 2019, 11, eaau6296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; He, X.; Caldwell, L.; Goru, S.K.; Severino, L.U.; Tolosa, M.F.; Misra, P.S.; McEvoy, C.M.; Christova, T.; Liu, Y.; et al. NUAK1 promotes organ fibrosis via YAP and TGF-β/SMAD signaling. Sci. Transl. Med. 2022, 14, eaaz4028. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.; Sun, H.; Gulati, M.; Herazo-Maya, J.D.; Chen, Y.; Osafo-Addo, A.; Brandsdorfer, C.; Winkler, J.; Blaul, C.; Faunce, J.; et al. Extracellular Mitochondrial DNA Is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, C.; Zhang, Y.; He, X.; Chen, X.; Zeng, X.; Liu, F.; Chen, Y.; Chen, J. Targeting Mechanics-Induced Fibroblast Activation through CD44-RhoA-YAP Pathway Ameliorates Crystalline Silica-Induced Silicosis. Theranostics 2019, 9, 4993. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Das, A.M.; Seideman, J.; Griswold, D.; Afuh, C.N.; Kobayashi, T.; Abe, S.; Fang, Q.; Hashimoto, M.; Rennard, S.I. The CC chemokine ligand 2 (CCL2) mediates fibroblast survival through IL-6. Am. J. Respir. Cell Mol. Biol. 2007, 37, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Y.; Zhu, W.-W.; Wang, M.-Y.; Zhai, R.-D.; Wang, Q.; Shen, W.-L.; Liu, L.-K. Cancer-associated fibroblasts promote oral squamous cell carcinoma progression through LOX-mediated matrix stiffness. J. Transl. Med. 2021, 19, 513. [Google Scholar] [CrossRef]
- Meli, V.S.; Atcha, H.; Veerasubramanian, P.K.; Nagalla, R.R.; Luu, T.U.; Chen, E.Y.; Guerrero-Juarez, C.F.; Yamaga, K.; Pandori, W.; Hsieh, J.Y.; et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Sci. Adv. 2020, 6, eabb8471. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, Y.; Zhou, P.; Liu, X.; Zhao, H.; Zhou, X.; Zhou, Q.; Gu, B.; Li, X.; Shi, Q. Substrate stiffness modulates bone marrow-derived macrophage polarization through NF-κB signaling pathway. Bioact. Mater. 2020, 5, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Taufalele, P.V.; Wang, W.; Simmons, A.J.; Southard-Smith, A.N.; Chen, B.; Greenlee, J.D.; King, M.R.; Lau, K.S.; Hassane, D.C.; Bordeleau, F.; et al. Matrix stiffness enhances cancer-macrophage interactions and M2-like macrophage accumulation in the breast tumor microenvironment. Acta Biomater. 2023, 163, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Zhang, N.; Xia, J.; Zhan, Q.; Wang, C. Potential role of M2 macrophage polarization in ventilator-induced lung fibrosis. Int. Immunopharmacol. 2019, 75, 105795. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Xiao, R.; Zhao, J.; Zhao, Q.; Luo, M.; Li, F.; Zhang, W.; Wu, M. Matrix stiffness affects tumor-associated macrophage functional polarization and its potential in tumor therapy. J. Transl. Med. 2024, 22, 85. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Brown, K.K.; Collard, H.R.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Martinez, F.J.; Noble, P.W.; Song, J.W.; et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: A randomised, double-blind, controlled, phase 2 trial. Lancet Respir. Med. 2016, 5, 22–32. [Google Scholar] [CrossRef]
- Hutchinson, J.H.; Rowbottom, M.W.; Lonergan, D.; Darlington, J.; Prodanovich, P.; King, C.D.; Evans, J.F.; Bain, G. Small Molecule Lysyl Oxidase-like 2 (LOXL2) Inhibitors: The Identification of an Inhibitor Selective for LOXL2 over LOX. ACS Med. Chem. Lett. 2017, 8, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lucas, M.; Leonte, L.E.; Garcia-Montolio, M.; Singh, L.B.; Findlay, A.D.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Timpson, P.; et al. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017, 8, 26066–26078. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.-J.; Hinz, B. The single-molecule mechanics of the latent TGF-β1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Glaser, K.J.; Manduca, A.; Mounajjed, T.; Malhi, H.; Simonetto, D.A.; Wang, R.; Yang, L.; Mao, S.A.; Glorioso, J.M.; et al. Distinguishing between Hepatic Inflammation and Fibrosis with MR Elastography. Radiology 2017, 284, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanchez, W.; Callstrom, M.R.; Gorman, B.; Lewis, J.T.; Sanderson, S.O.; Greenleaf, J.F.; Xie, H.; Shi, Y.; Pashley, M.; et al. Assessment of liver viscoelasticity by using shear waves induced by ultrasound radiation force. Radiology 2013, 266, 964–970. [Google Scholar] [CrossRef]
- Sundarakrishnan, A.; Chen, Y.; Black, L.D.; Aldridge, B.B.; Kaplan, D.L. Engineered cell and tissue models of pulmonary fibrosis. Adv. Drug Deliv. Rev. 2017, 129, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Phogat, S.; Thiam, F.; Al Yazeedi, S.; Abokor, F.A.; Osei, E.T. 3D in vitro hydrogel models to study the human lung extracellular matrix and fibroblast function. Respir. Res. 2023, 24, 242. [Google Scholar] [CrossRef] [PubMed]
- Giménez, A.; Duch, P.; Puig, M.; Gabasa, M.; Xaubet, A.; Alcaraz, J. Dysregulated Collagen Homeostasis by Matrix Stiffening and TGF-β1 in Fibroblasts from Idiopathic Pulmonary Fibrosis Patients: Role of FAK/Akt. Int. J. Mol. Sci. 2017, 18, 2431. [Google Scholar] [CrossRef] [PubMed]
- Cacopardo, L.; Ahluwalia, A. Engineering and Monitoring 3D Cell Constructs with Time-Evolving Viscoelasticity for the Study of Liver Fibrosis In Vitro. Bioengineering 2021, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Major, G.; Ahn, M.; Cho, W.-W.; Santos, M.; Wise, J.; Phillips, E.; Wise, S.G.; Jang, J.; Rnjak-Kovacina, J.; Woodfield, T.; et al. Programming temporal stiffness cues within extracellular matrix hydrogels for modelling cancer niches. Mater. Today Bio 2024, 25, 101004. [Google Scholar] [CrossRef]
- Daly, A.C.; Davidson, M.D.; Burdick, J.A. 3D bioprinting of high cell-density heterogeneous tissue models through spheroid fusion within self-healing hydrogels. Nat. Commun. 2021, 12, 753. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.M.; Brunel, L.G.; Heilshorn, S.C. 3D Bioprinting of Cell-Laden Hydrogels for Improved Biological Functionality. Adv. Mater. 2021, 34, 2103691. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, B.; Paulsen, S.J.; Corbett, D.C.; Sazer, D.W.; Fortin, C.L.; Zaita, A.J.; Greenfield, P.T.; Calafat, N.J.; Gounley, J.P.; Ta, A.H.; et al. Multivascular networks and functional intravascular topologies within biocompatible hydrogels. Science 2019, 364, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Park, J.A.; Kim, W.; Kim, S.; Lee, H.; Kim, W.; Yoo, J.; Jung, S. All-Inkjet-Printed 3D Alveolar Barrier Model with Physiologically Relevant Microarchitecture. Adv. Sci. 2021, 8, 2004990. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, A.; He, L.; Chen, T.; Li, X.; Cao, C. Biomechanical Properties and Cellular Responses in Pulmonary Fibrosis. Bioengineering 2024, 11, 747. https://doi.org/10.3390/bioengineering11080747
He A, He L, Chen T, Li X, Cao C. Biomechanical Properties and Cellular Responses in Pulmonary Fibrosis. Bioengineering. 2024; 11(8):747. https://doi.org/10.3390/bioengineering11080747
Chicago/Turabian StyleHe, Andong, Lizhe He, Tianwei Chen, Xuejin Li, and Chao Cao. 2024. "Biomechanical Properties and Cellular Responses in Pulmonary Fibrosis" Bioengineering 11, no. 8: 747. https://doi.org/10.3390/bioengineering11080747
APA StyleHe, A., He, L., Chen, T., Li, X., & Cao, C. (2024). Biomechanical Properties and Cellular Responses in Pulmonary Fibrosis. Bioengineering, 11(8), 747. https://doi.org/10.3390/bioengineering11080747