


Recent Advances of MSCs in Renal IRI: From Injury to Renal Fibrosis

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Method
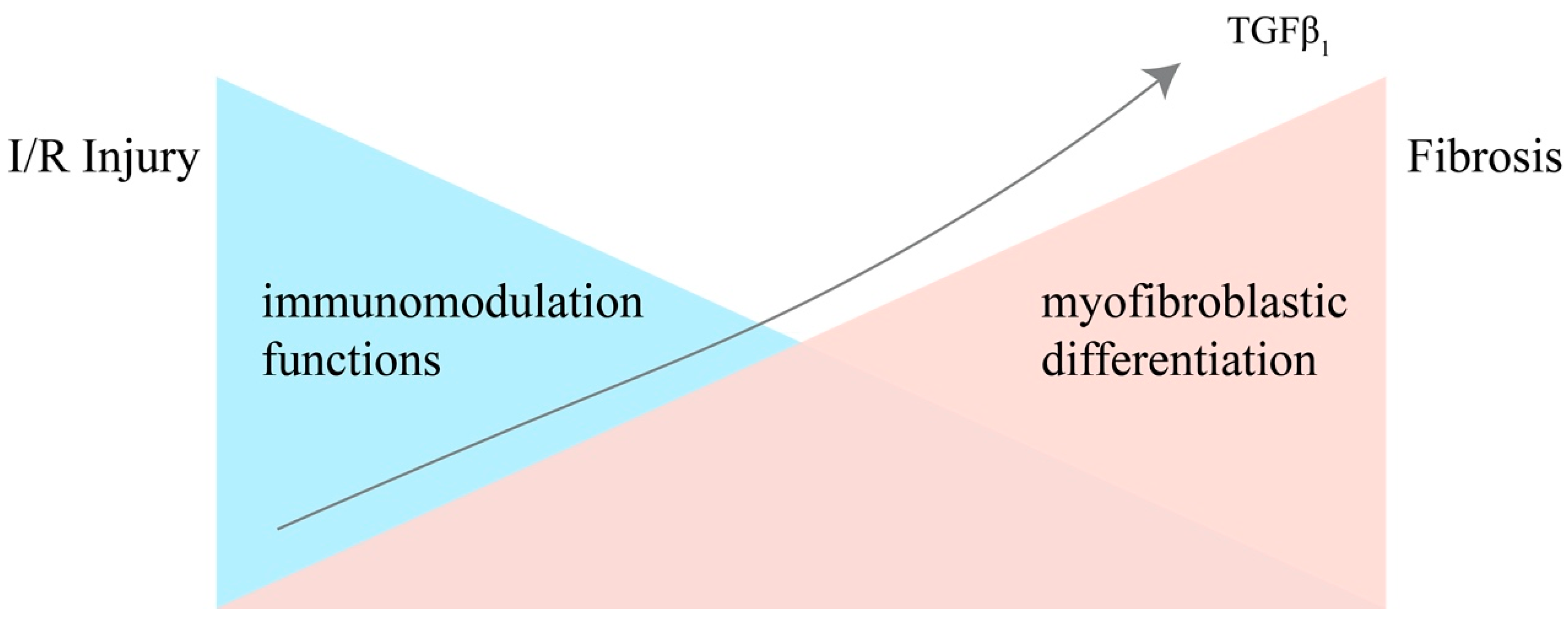
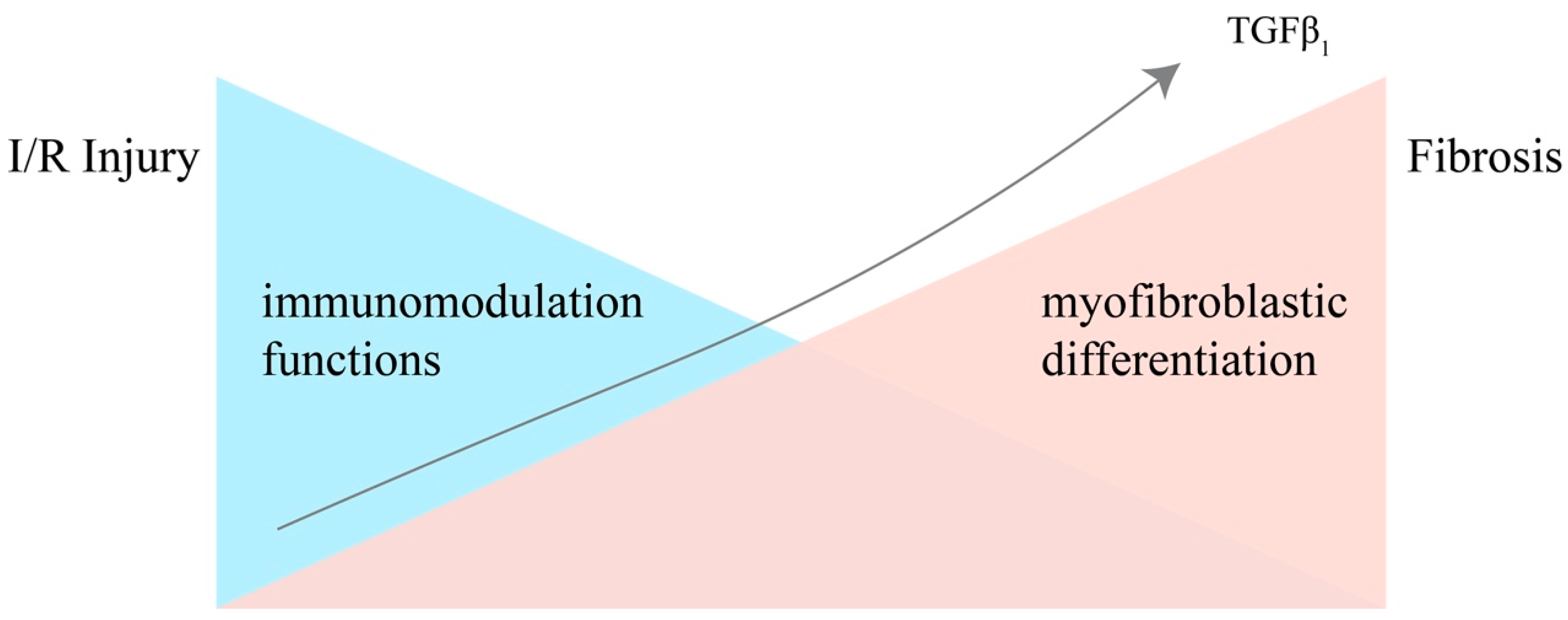
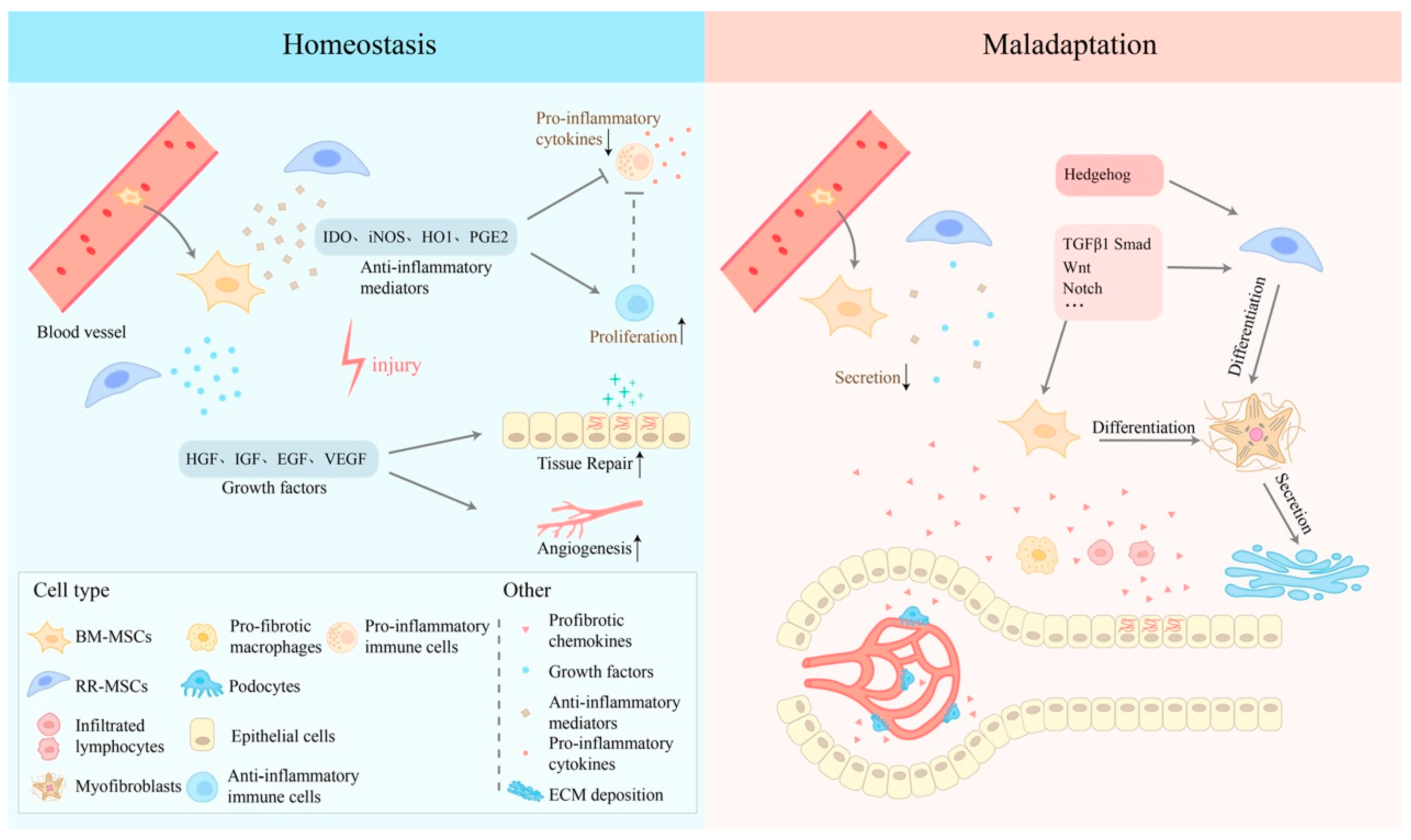
3. The Dual Nature of MSCs in Renal IRI
4. MSCs in the Early Stage of Injury
5. MSCs in the Progression of Renal Fibrosis
6. Signal Pathways
6.1. TGF-β1/Smad Pathway
6.2. Notch Signaling Pathway
6.3. Wnt/β-Catenin-Signaling Pathway
6.4. Hedgehog Pathway
7. Promising Targets
8. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lewington, A.J.P.; Cerdá, J.; Mehta, R.L. Raising Awareness of Acute Kidney Injury: A Global Perspective of a Silent Killer. Kidney Int. 2013, 84, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern. Med. 2017, 167, ITC66–ITC80. [Google Scholar] [CrossRef] [PubMed]
- Bon, D.; Chatauret, N.; Giraud, S.; Thuillier, R.; Favreau, F.; Hauet, T. New Strategies to Optimize Kidney Recovery and Preservation in Transplantation. Nat. Rev. Nephrol. 2012, 8, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Koza, Y. Acute Kidney Injury: Current Concepts and New Insights. J. Inj. Violence Res. 2016, 8, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Gazdic, M.; Simovic Markovic, B.; Jovicic, N.; Djonov, V.; Arsenijevic, N. Mesenchymal Stem Cell-Derived Factors: Immuno-Modulatory Effects and Therapeutic Potential. Biofactors 2017, 43, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yanagita, M. Immune Cells and Inflammation in AKI to CKD Progression. Am. J. Physiol. Renal Physiol. 2018, 315, F1501–F1512. [Google Scholar] [CrossRef]
- Mack, M.; Yanagita, M. Origin of Myofibroblasts and Cellular Events Triggering Fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Bochaton-Piallat, M.-L.; Gabbiani, G.; Hinz, B. The Myofibroblast in Wound Healing and Fibrosis: Answered and Unanswered Questions. F1000Research 2016, 5, F1000 Faculty Rev-752. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.M.; Pham, P.T.; Bach, T.Q.; Ngo, A.T.L.; Nguyen, Q.T.; Phan, T.T.K.; Nguyen, G.H.; Le, P.T.T.; Hoang, V.T.; Forsyth, N.R.; et al. Stem Cell-Based Therapy for Human Diseases. Signal Transduct. Target. Ther. 2022, 7, 272. [Google Scholar] [CrossRef]
- Najar, M.; Melki, R.; Khalife, F.; Lagneaux, L.; Bouhtit, F.; Moussa Agha, D.; Fahmi, H.; Lewalle, P.; Fayyad-Kazan, M.; Merimi, M. Therapeutic Mesenchymal Stem/Stromal Cells: Value, Challenges and Optimization. Front. Cell Dev. Biol. 2021, 9, 716853. [Google Scholar] [CrossRef]
- Zhou, J.; Shi, Y. Mesenchymal Stem/Stromal Cells (MSCs): Origin, Immune Regulation, and Clinical Applications. Cell. Mol. Immunol. 2023, 20, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Q.; Ma, R.; Yin, Y.; Lan, T.; Yu, M.; Ming, Y. Mesenchymal Stem Cells in Renal Fibrosis: The Flame of Cytotherapy. Stem Cells Int. 2019, 2019, 8387350. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Zhang, J.; Xin, H.; He, X.; Zhang, X. Current Perspectives on Role of MSC in Renal Pathophysiology. Front. Physiol. 2018, 9, 1323. [Google Scholar] [CrossRef] [PubMed]
- Basalova, N.; Sagaradze, G.; Arbatskiy, M.; Evtushenko, E.; Kulebyakin, K.; Grigorieva, O.; Akopyan, Z.; Kalinina, N.; Efimenko, A. Secretome of Mesenchymal Stromal Cells Prevents Myofibroblasts Differentiation by Transferring Fibrosis-Associated MicroRNAs within Extracellular Vesicles. Cells 2020, 9, 1272. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Jhao, P.-Y.; Hung, C.-T.; Wu, Y.-F.; Lin, S.-J.; Chiang, W.-C.; Lin, S.-L.; Yang, K.-C. Endoplasmic Reticulum Protein TXNDC5 Promotes Renal Fibrosis by Enforcing TGF-β Signaling in Kidney Fibroblasts. J. Clin. Investig. 2021, 131, e143645. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The Master Regulator of Fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A. International Society for Cellular Therapy Clarification of the Nomenclature for MSC: The International Society for Cellular Therapy Position Statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Du, Y.; Yuan, H.; Jiang, F.; Shen, M.; Wang, Y.; Wang, R. HAMSCs/HBMSCs Coculture System Ameliorates Osteogenesis and Angiogenesis against Glucolipotoxicity. Biochimie 2018, 152, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Domingues, A.; Ratajczak, J.; Ratajczak, M.Z. Potential Clinical Applications of Stem Cells in Regenerative Medicine. Adv. Exp. Med. Biol. 2019, 1201, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Xu, F.; Zhang, T.; Huang, J.; Guan, Q.; Wang, H.; Huang, Q. Inhibition of IL-18 Reduces Renal Fibrosis after Ischemia-Reperfusion. Biomed. Pharmacother. 2018, 106, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zou, X.; Miao, S.; Chen, J.; Du, T.; Zhong, L.; Ju, G.; Liu, G.; Zhu, Y. The Anti-Oxidative Role of Micro-Vesicles Derived from Human Wharton-Jelly Mesenchymal Stromal Cells through NOX2/Gp91(Phox) Suppression in Alleviating Renal Ischemia-Reperfusion Injury in Rats. PLoS ONE 2014, 9, e92129. [Google Scholar] [CrossRef] [PubMed]
- Rota, C.; Morigi, M.; Cerullo, D.; Introna, M.; Colpani, O.; Corna, D.; Capelli, C.; Rabelink, T.J.; Leuning, D.G.; Rottoli, D.; et al. Therapeutic Potential of Stromal Cells of Non-Renal or Renal Origin in Experimental Chronic Kidney Disease. Stem Cell Res. Ther. 2018, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Guo, J.; Zhang, P.; Cheuk, Y.C.; Jiang, Y.; Wang, J.; Xu, S.; Rong, R. Mesenchymal Stem Cell Protects Injured Renal Tubular Epithelial Cells by Regulating MTOR-Mediated Th17/Treg Axis. Front. Immunol. 2021, 12, 684197. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Su, J.; Roberts, A.I.; Shou, P.; Rabson, A.B.; Ren, G. How Mesenchymal Stem Cells Interact with Tissue Immune Responses. Trends Immunol. 2012, 33, 136–143. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and Function of Myofibroblasts in Kidney Fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ Progenitors Are Key Contributors to Injury-Induced Organ Fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Li, Q.; Liu, K.; Hou, J.; Shao, C.; Wang, Y. Immunoregulatory Mechanisms of Mesenchymal Stem and Stromal Cells in Inflammatory Diseases. Nat. Rev. Nephrol. 2018, 14, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, Q.; Lin, L.; Xu, C.; Zheng, C.; Chen, X.; Han, Y.; Li, M.; Cao, W.; Cao, K.; et al. Interleukin-17 Enhances Immunosuppression by Mesenchymal Stem Cells. Cell Death Differ. 2014, 21, 1758–1768. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.; Zhang, J.; Yuan, Z.; Ren, G.; Zhang, L.; Chen, X.; Rabson, A.B.; Roberts, A.I.; Wang, Y.; Shi, Y. Mesenchymal Stem Cells Use IDO to Regulate Immunity in Tumor Microenvironment. Cancer Res. 2014, 74, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Chen, X.; Huang, Y.; Li, W.; Li, J.; Cao, K.; Cao, G.; Zhang, L.; Li, F.; Roberts, A.I.; et al. Phylogenetic Distinction of INOS and IDO Function in Mesenchymal Stem Cell-Mediated Immunosuppression in Mammalian Species. Cell Death Differ. 2014, 21, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zheng, C.; Cao, J.; Cao, G.; Shou, P.; Lin, L.; Velletri, T.; Jiang, M.; Chen, Q.; Han, Y.; et al. Spermidine Alleviates Experimental Autoimmune Encephalomyelitis through Inducing Inhibitory Macrophages. Cell Death Differ. 2016, 23, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Maffioli, E.; Nonnis, S.; Angioni, R.; Santagata, F.; Calì, B.; Zanotti, L.; Negri, A.; Viola, A.; Tedeschi, G. Proteomic Analysis of the Secretome of Human Bone Marrow-Derived Mesenchymal Stem Cells Primed by pro-Inflammatory Cytokines. J. Proteom. 2017, 166, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yang, Y.; Wang, Z.; Liu, A.; Fang, L.; Wu, F.; Hong, J.; Shi, Y.; Leung, S.; Dong, C.; et al. Leukemia Inhibitory Factor Inhibits T Helper 17 Cell Differentiation and Confers Treatment Effects of Neural Progenitor Cell Therapy in Autoimmune Disease. Immunity 2011, 35, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Tiruppathi, C.; Nepal, S.; Zhao, Y.-Y.; Grzych, D.; Soni, D.; Prockop, D.J.; Malik, A.B. TNFα-Stimulated Gene-6 (TSG6) Activates Macrophage Phenotype Transition to Prevent Inflammatory Lung Injury. Proc. Natl. Acad. Sci. USA 2016, 113, E8151–E8158. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Xie, N.; Li, W.; Yuan, B.; Shi, Y.; Wang, Y. Immunobiology of Mesenchymal Stem Cells. Cell Death Differ. 2014, 21, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhang, G.; Cheng, Z.; Yin, D.; Du, T.; Ju, G.; Miao, S.; Liu, G.; Lu, M.; Zhu, Y. Microvesicles Derived from Human Wharton’s Jelly Mesenchymal Stromal Cells Ameliorate Renal Ischemia-Reperfusion Injury in Rats by Suppressing CX3CL1. Stem Cell Res. Ther. 2014, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Cahill, E.F.; Kennelly, H.; Carty, F.; Mahon, B.P.; English, K. Hepatocyte Growth Factor Is Required for Mesenchymal Stromal Cell Protection Against Bleomycin-Induced Pulmonary Fibrosis. Stem Cells Transl. Med. 2016, 5, 1307–1318. [Google Scholar] [CrossRef]
- Nimsanor, N.; Phetfong, J.; Kitiyanant, N.; Kamprom, W.; Supokawej, A. Overexpression of Anti-Fibrotic Factors Ameliorates Anti-Fibrotic Properties of Wharton’s Jelly Derived Mesenchymal Stem Cells under Oxidative Damage. Biosci. Trends 2019, 13, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Xiang, E.; Han, B.; Zhang, Q.; Rao, W.; Wang, Z.; Chang, C.; Zhang, Y.; Tu, C.; Li, C.; Wu, D. Human Umbilical Cord-Derived Mesenchymal Stem Cells Prevent the Progression of Early Diabetic Nephropathy through Inhibiting Inflammation and Fibrosis. Stem Cell Res. Ther. 2020, 11, 336. [Google Scholar] [CrossRef] [PubMed]
- El Agha, E.; Kramann, R.; Schneider, R.K.; Li, X.; Seeger, W.; Humphreys, B.D.; Bellusci, S. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem Cell 2017, 21, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Maione, A.S.; Stadiotti, I.; Pilato, C.A.; Perrucci, G.L.; Saverio, V.; Catto, V.; Vettor, G.; Casella, M.; Guarino, A.; Polvani, G.; et al. Excess TGF-Β1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 2673. [Google Scholar] [CrossRef]
- Zilberberg, L.; Todorovic, V.; Dabovic, B.; Horiguchi, M.; Couroussé, T.; Sakai, L.Y.; Rifkin, D.B. Specificity of Latent TGF-β Binding Protein (LTBP) Incorporation into Matrix: Role of Fibrillins and Fibronectin. J. Cell. Physiol. 2012, 227, 3828–3836. [Google Scholar] [CrossRef] [PubMed]
- Aragón, E.; Goerner, N.; Zaromytidou, A.-I.; Xi, Q.; Escobedo, A.; Massagué, J.; Macias, M.J. A Smad Action Turnover Switch Operated by WW Domain Readers of a Phosphoserine Code. Genes Dev. 2011, 25, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.; Zaromytidou, A.-I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs Drive Smad Transcriptional Activation and Turnover in BMP and TGF-Beta Pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Matsui, F.; Meldrum, K.K. The Role of the Janus Kinase Family/Signal Transducer and Activator of Transcription Signaling Pathway in Fibrotic Renal Disease. J. Surg. Res. 2012, 178, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Notch Signaling: Control of Cell Communication and Cell Fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental Signalling Pathways in Renal Fibrosis: The Roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Shrestha, R.; Qiu, C.; Kondo, A.; Huang, S.; Werth, M.; Li, M.; Barasch, J.; Suszták, K. Single-Cell Transcriptomics of the Mouse Kidney Reveals Potential Cellular Targets of Kidney Disease. Science 2018, 360, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H. Three Decades of Wnts: A Personal Perspective on How a Scientific Field Developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef] [PubMed]
- DiRocco, D.P.; Kobayashi, A.; Taketo, M.M.; McMahon, A.P.; Humphreys, B.D. Wnt4/β-Catenin Signaling in Medullary Kidney Myofibroblasts. J. Am. Soc. Nephrol. 2013, 24, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can We Safely Target the WNT Pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xiao, L.; Sun, L.; Liu, F. Wnt/Beta-Catenin Signaling: A Promising New Target for Fibrosis Diseases. Physiol. Res. 2012, 61, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Haegebarth, A.; Clevers, H. Wnt Signaling, Lgr5, and Stem Cells in the Intestine and Skin. Am. J. Pathol. 2009, 174, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Vooijs, M.; Liu, Z.; Kopan, R. Notch: Architect, Landscaper, and Guardian of the Intestine. Gastroenterology 2011, 141, 448–459. [Google Scholar] [CrossRef]
- Kuppe, C.; Kramann, R. Role of Mesenchymal Stem Cells in Kidney Injury and Fibrosis. Curr. Opin. Nephrol. Hypertens. 2016, 25, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Tan, R.J.; Liu, Y. Sonic Hedgehog Signaling in Kidney Fibrosis: A Master Communicator. Sci. China. Life Sci. 2016, 59, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; Nakano, Y.; Seger, C. Mechanisms and Functions of Hedgehog Signalling across the Metazoa. Nat. Rev. Genet. 2011, 12, 393–406. [Google Scholar] [CrossRef]
- Fabian, S.L.; Penchev, R.R.; St-Jacques, B.; Rao, A.N.; Sipilä, P.; West, K.A.; McMahon, A.P.; Humphreys, B.D. Hedgehog-Gli Pathway Activation during Kidney Fibrosis. Am. J. Pathol. 2012, 180, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Wongboonsin, J.; Chang-Panesso, M.; Machado, F.G.; Humphreys, B.D. Gli1+ Pericyte Loss Induces Capillary Rarefaction and Proximal Tubular Injury. J. Am. Soc. Nephrol. 2017, 28, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Miao, J.; Yang, Q.; Fang, L.; Fang, Y.; Ding, H.; Zhou, Y.; Jiang, L.; Dai, C.; Zen, K.; et al. Role of Pyruvate Kinase M2-Mediated Metabolic Reprogramming during Podocyte Differentiation. Cell Death Dis. 2020, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Drehmer, D.L.; de Aguiar, A.M.; Brandt, A.P.; Petiz, L.; Cadena, S.M.S.C.; Rebelatto, C.K.; Brofman, P.R.S.; Filipak Neto, F.; Dallagiovanna, B.; Abud, A.P.R. Metabolic Switches during the First Steps of Adipogenic Stem Cells Differentiation. Stem Cell Res. 2016, 17, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic Reprogramming during Neuronal Differentiation from Aerobic Glycolysis to Neuronal Oxidative Phosphorylation. Elife 2016, 5, e13374. [Google Scholar] [CrossRef]
- Haran, M.; Gross, A. Balancing Glycolysis and Mitochondrial OXPHOS: Lessons from the Hematopoietic System and Exercising Muscles. Mitochondrion 2014, 19 Pt A, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, T. Metabolic Regulation of Mesenchymal Stem Cell in Expansion and Therapeutic Application. Biotechnol. Prog. 2015, 31, 468–481. [Google Scholar] [CrossRef]
- Quaggin, S.E.; Kapus, A. Scar Wars: Mapping the Fate of Epithelial-Mesenchymal-Myofibroblast Transition. Kidney Int. 2011, 80, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Schrimpf, C.; Duffield, J.S. Mechanisms of Fibrosis: The Role of the Pericyte. Curr. Opin. Nephrol. Hypertens. 2011, 20, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Black, L.M.; Lever, J.M.; Agarwal, A. Renal Inflammation and Fibrosis: A Double-Edged Sword. J. Histochem. Cytochem. 2019, 67, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.H.; Li, G.; Liu, J.; Liu, L.; Wu, B.; Huang, W.; He, W.; Deng, C.; Wang, D.; Li, C.; et al. Nestin(+) Kidney Resident Mesenchymal Stem Cells for the Treatment of Acute Kidney Ischemia Injury. Biomaterials 2015, 50, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, G.; Pacilli, A.; Alviano, F.; Foroni, L.; Ricci, F.; Valente, S.; Orrico, C.; Lanzoni, G.; Buzzi, M.; Luigi Tazzari, P.; et al. Multidistrict Human Mesenchymal Vascular Cells: Pluripotency and Stemness Characteristics. Cytotherapy 2010, 12, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, Y.C.; Xu, S.; Zhu, D.; Luo, Y.; Chen, T.; Chen, J.; Li, J.; Shi, Y.; Zhang, Y.; Rong, R. Monocytic Myeloid-Derived Suppressor Cells Inhibit Myofibroblastic Differentiation in Mesenchymal Stem Cells Through IL-15 Secretion. Front. Cell Dev. Biol. 2022, 10, 817402. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, X.; Xu, X.; Xu, C.; Cheuk, Y.C.; Rong, R. Recent Advances of MSCs in Renal IRI: From Injury to Renal Fibrosis. Bioengineering 2024, 11, 432. https://doi.org/10.3390/bioengineering11050432
Niu X, Xu X, Xu C, Cheuk YC, Rong R. Recent Advances of MSCs in Renal IRI: From Injury to Renal Fibrosis. Bioengineering. 2024; 11(5):432. https://doi.org/10.3390/bioengineering11050432
Chicago/Turabian StyleNiu, Xinhao, Xiaoqing Xu, Cuidi Xu, Yin Celeste Cheuk, and Ruiming Rong. 2024. "Recent Advances of MSCs in Renal IRI: From Injury to Renal Fibrosis" Bioengineering 11, no. 5: 432. https://doi.org/10.3390/bioengineering11050432
APA StyleNiu, X., Xu, X., Xu, C., Cheuk, Y. C., & Rong, R. (2024). Recent Advances of MSCs in Renal IRI: From Injury to Renal Fibrosis. Bioengineering, 11(5), 432. https://doi.org/10.3390/bioengineering11050432