

Identification of a Novel Signature Based on Ferritinophagy-Related Genes to Predict Prognosis in Lung Adenocarcinoma: Focus on AHNAK2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Analysis of Ferritinophagy-Related Genes
2.3. Subtype Analysis
2.4. Model Construction and Evaluation
2.5. Functional Enrichment Analysis
2.6. Immune-Related Analysis
2.7. Somatic Mutation, MSI and TIDE Analysis
2.8. Analysis of Drug Sensitivity
2.9. Single Cell Sequencing Analysis
2.10. Cell Culture and Quantitative Real-Time PCR (RT-qPCR)
2.11. CCK8, Colony Formation, Transwell, and Wound-Healing Assay
2.12. Fe2+, ROS, GSH, and MDA Assay
2.13. Transmission Electron Microscopy
2.14. Statistical Analysis
3. Results
3.1. Landscape of Ferritinophagy-Related Genes in LUAD
3.2. WGCNA
3.3. Construction of Prognostic Model
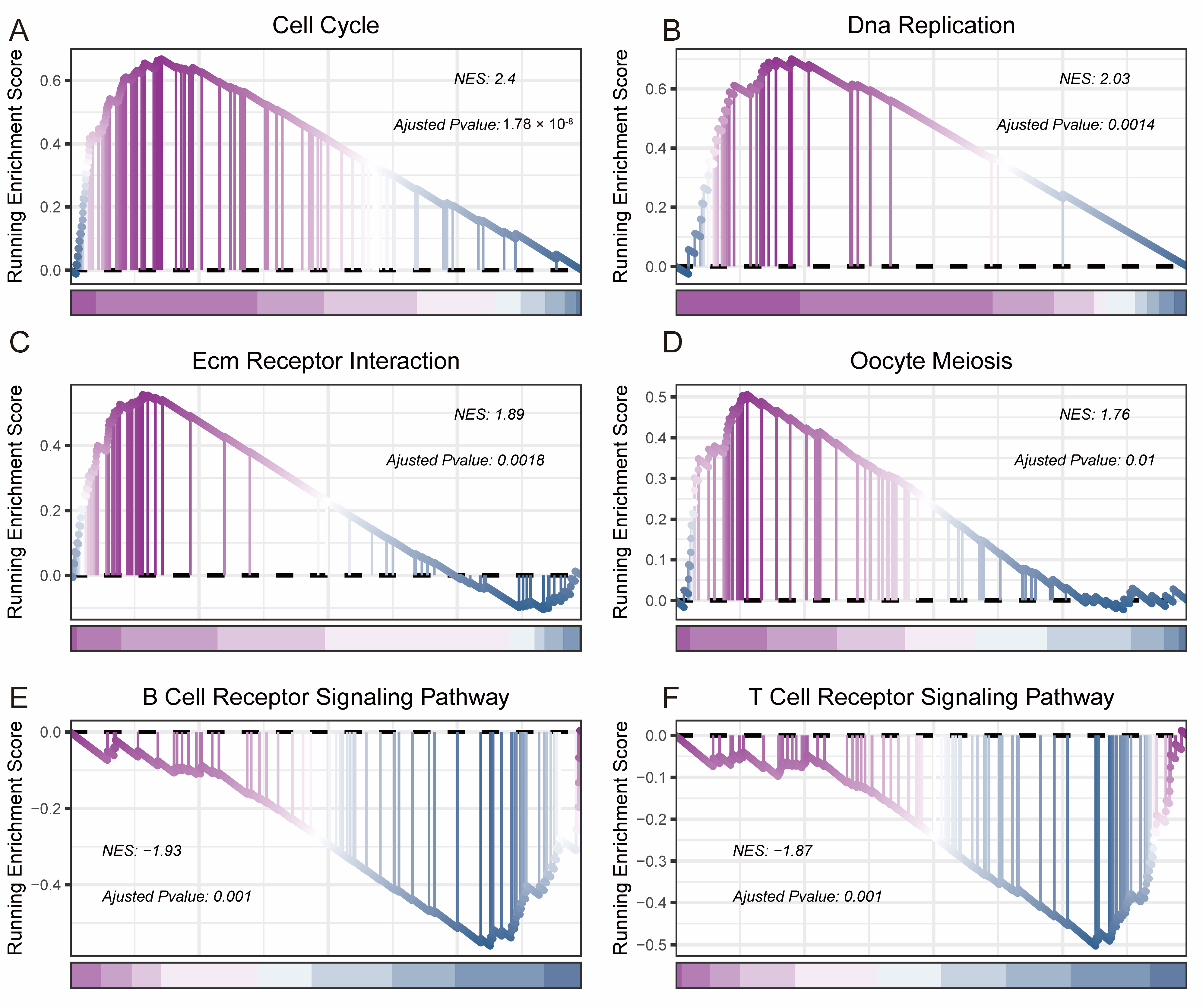
3.4. Biological Functional Analysis
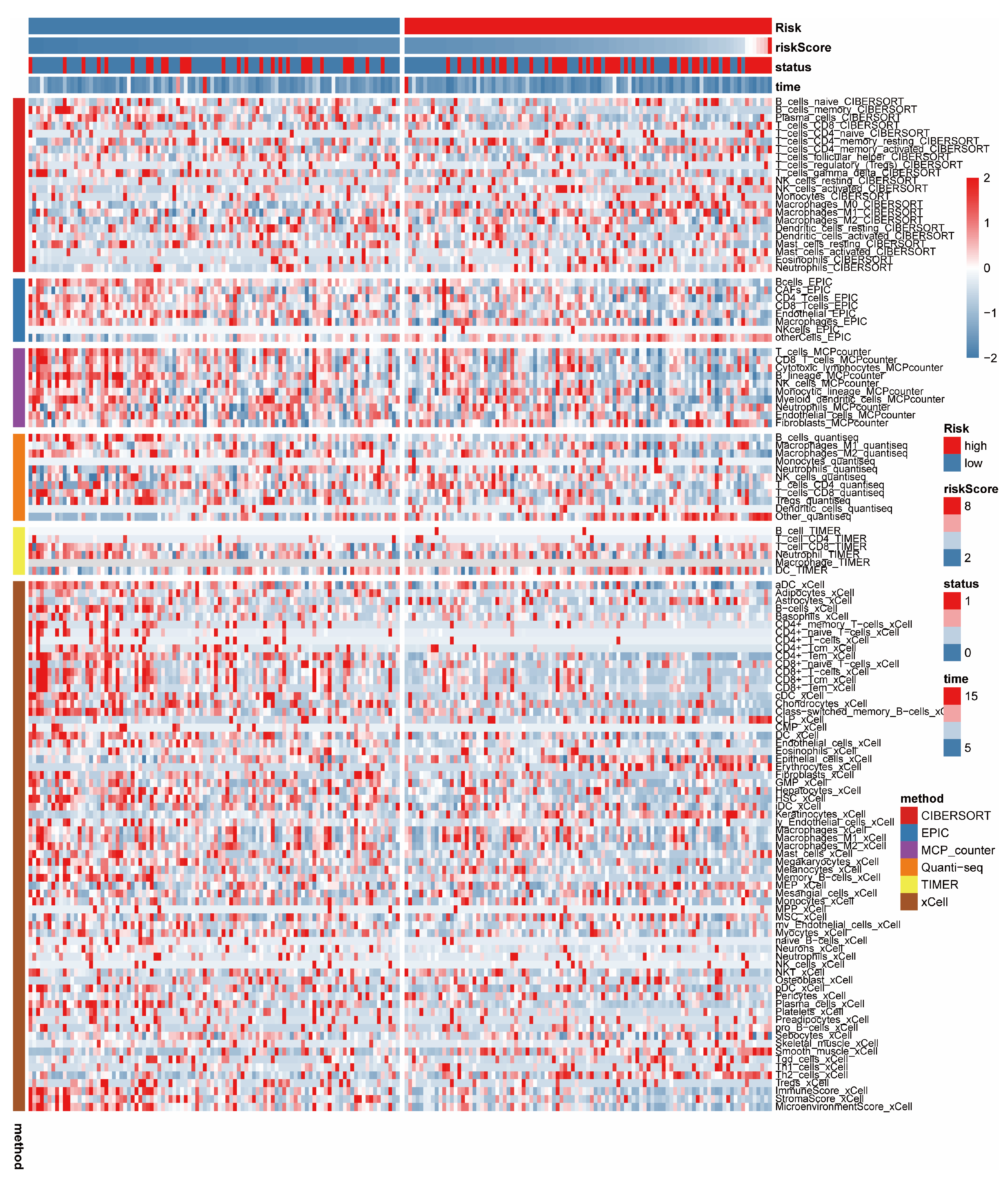
3.5. Immune-Related Analysis
3.6. Multi-Omics Analysis
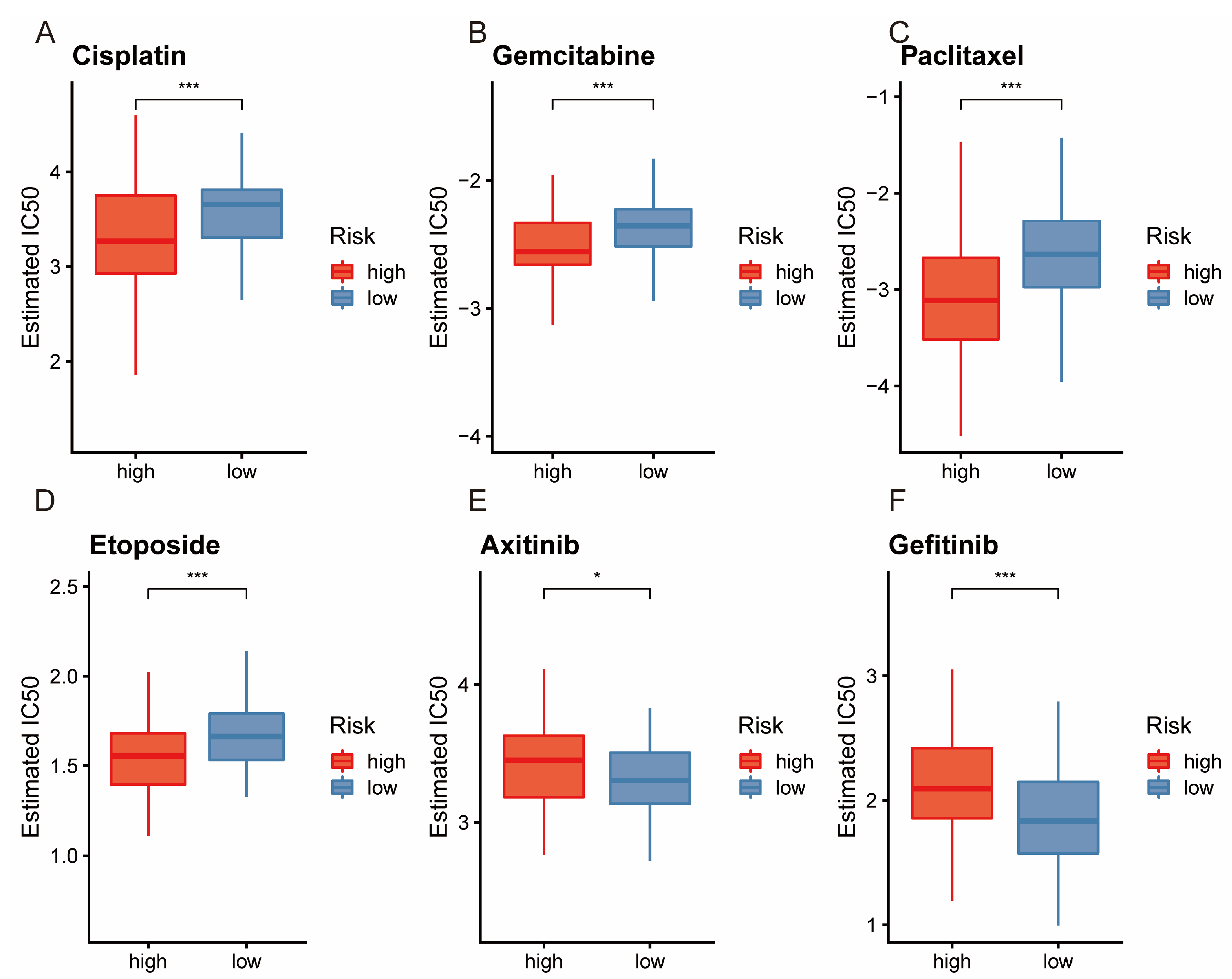
3.7. Analysis of Drug Sensitivity
3.8. Independent Prognosis Analysis and Constructing the Nomogram and Calibration Curves
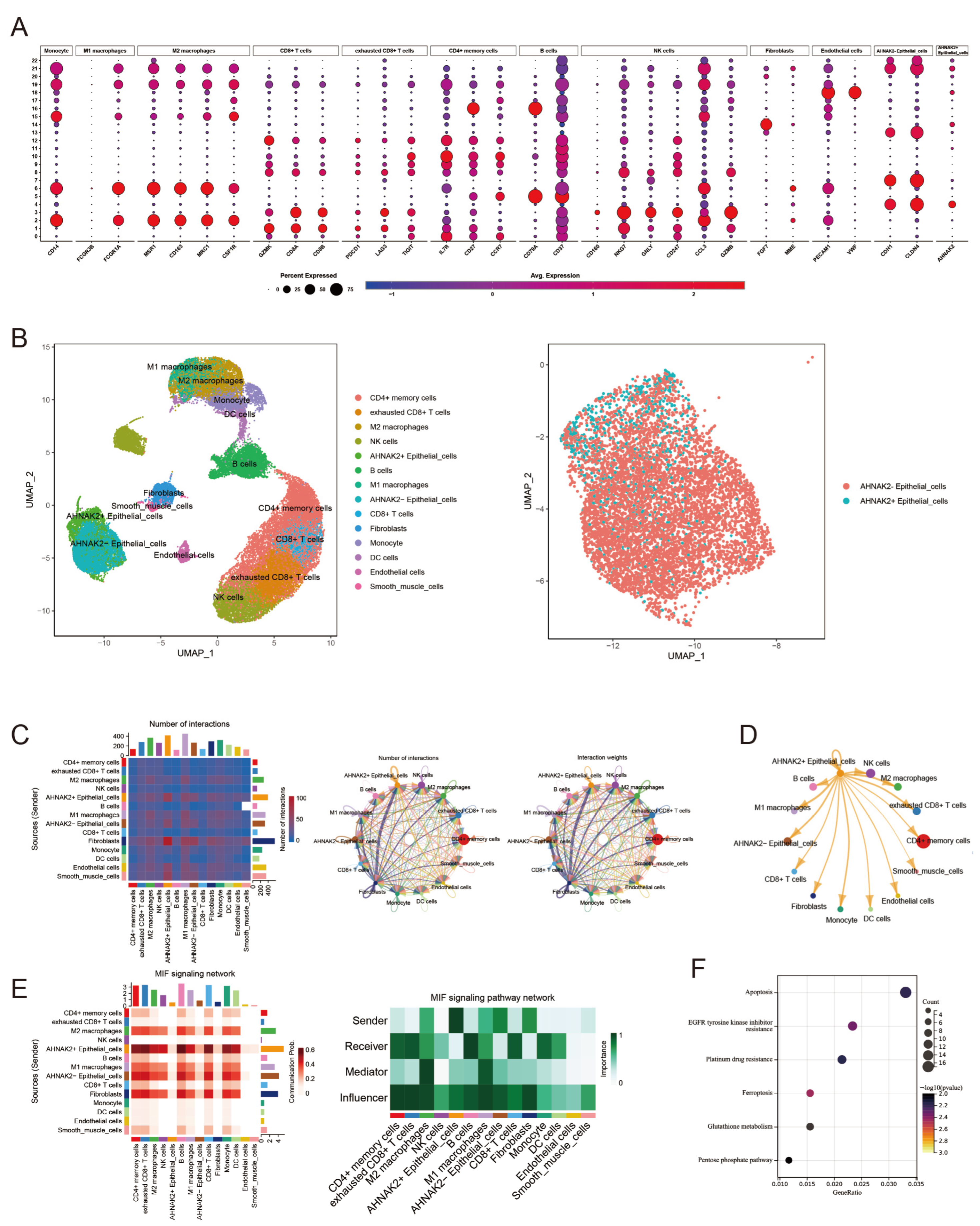
3.9. Validation of Model Genes with Single-Cell Sequencing
3.10. Identification of AHNAK2+ Epithelial Cells Subtype
3.11. AHNAK2 Was Significantly Up-Regulated in LUAD Tissues and Cells
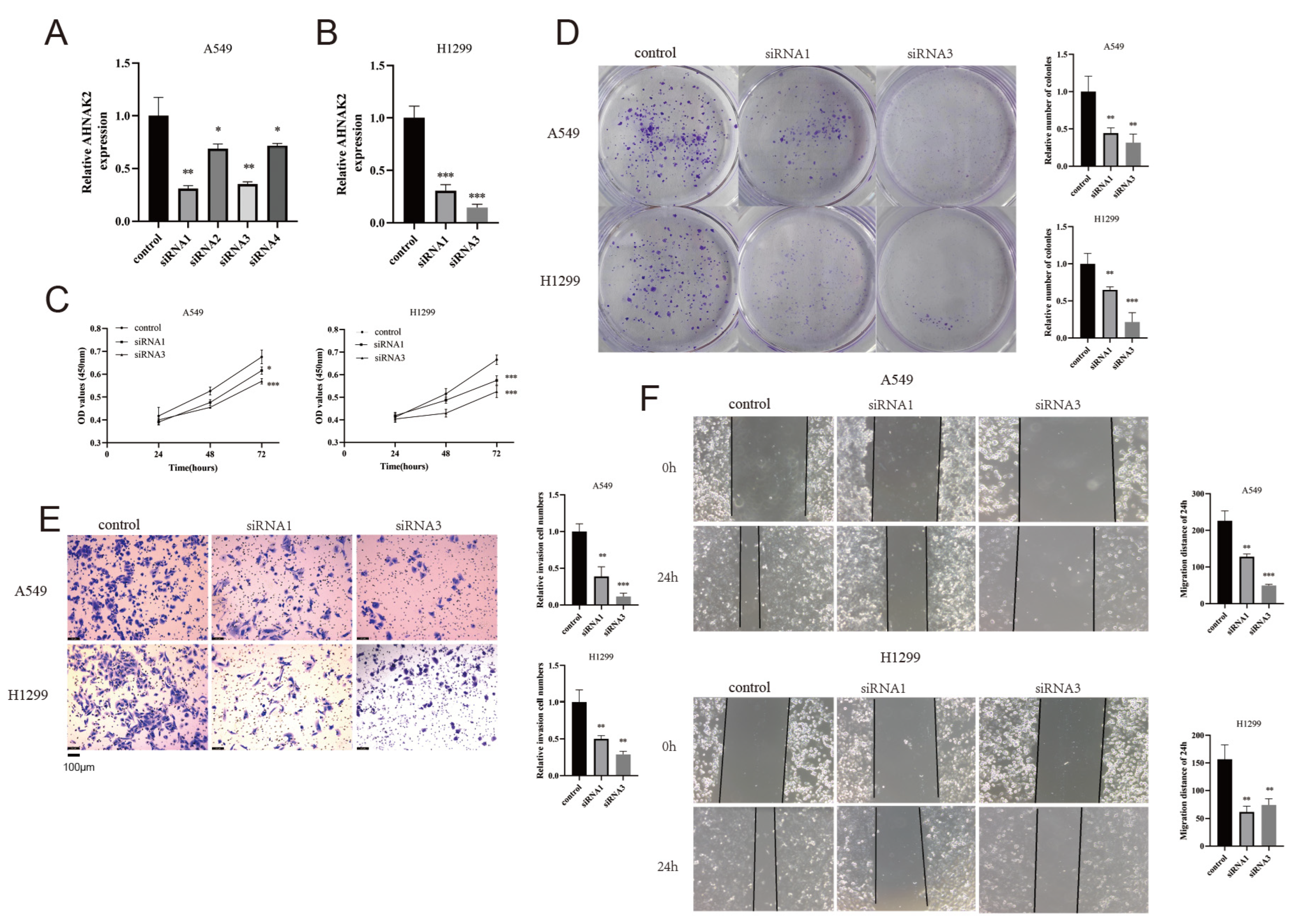
3.12. Silencing AHNAK2 Suppresses the Cell Proliferation, Invasion, and Migration of LUAD Cells
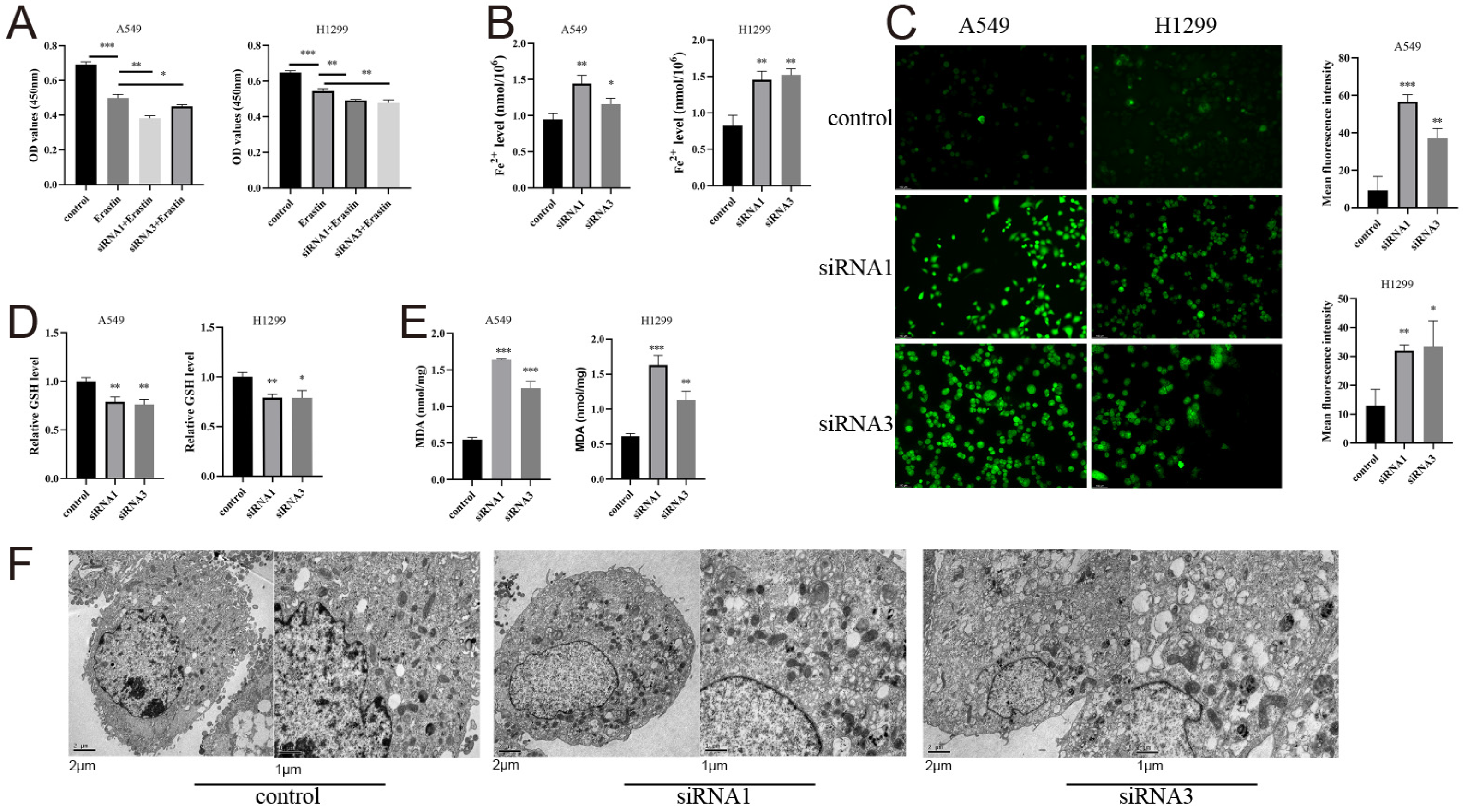
3.13. AHNAK2 Attenuates Ferroptosis of LUAD Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Li, C.; Liao, S.; Yao, X.; Ouyang, Y.; Liu, Y.; Wang, Z.; Li, Z.; Yao, F. Ferritinophagy, a form of autophagic ferroptosis: New insights into cancer treatment. Front. Pharmacol. 2022, 13, 1043344. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Jiang, C.; Zou, Z.; Huang, H.; Li, X.; Xu, S.; Tan, R. Ferritinophagy in the etiopathogenic mechanism of related diseases. J. Nutr. Biochem. 2023, 117, 109339. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, J.; Zhu, L.; Zhou, Z.; Ma, Z.; Zhang, H.; Yang, Y.; Niu, Q.; Wang, X. Identification of hepatocellular carcinoma-related subtypes and development of a prognostic model: A study based on ferritinophagy-related genes. Discover. Oncol. 2023, 14, 147. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Tian, Y.; Lu, J.; Hao, X.; Li, H.; Zhang, G.; Liu, X.; Li, X.; Zhao, C.; Kuang, W.; Chen, D.; et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef]
- Zardab, M.; Stasinos, K.; Grose, R.P.; Kocher, H.M. The Obscure Potential of AHNAK2. Cancers 2022, 14, 528. [Google Scholar] [CrossRef]
- Wang, D.W.; Zheng, H.Z.; Cha, N.; Zhang, X.J.; Zheng, M.; Chen, M.M.; Tian, L.X. Down-Regulation of AHNAK2 Inhibits Cell Proliferation, Migration and Invasion Through Inactivating the MAPK Pathway in Lung Adenocarcinoma. Technol. Cancer Res. Treat. 2020, 19, 1533033820957006. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, H.; Shao, M.M.; Miao, J.; Fu, Y.; Hu, B. Downregulation of AHNAK2 inhibits cell cycle of lung adenocarcinoma cells by interacting with RUVBL1. Thorac. Cancer 2023, 14, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Guo, Z.; Zhang, Q.; Liu, Z.; Zhu, D. AHNAK2 Promotes Migration, Invasion, and Epithelial-Mesenchymal Transition in Lung Adenocarcinoma Cells via the TGF-β/Smad3 Pathway. OncoTargets Ther. 2020, 13, 12893–12903. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liu, X.; Wu, Y.; Liang, X.; Dai, J.; Zhang, Z.; Guo, R. Deleterious AHNAK2 Mutation as a Novel Biomarker for Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer. Front. Oncol. 2022, 12, 798401. [Google Scholar] [CrossRef] [PubMed]
- Okayama, H.; Kohno, T.; Ishii, Y.; Shimada, Y.; Shiraishi, K.; Iwakawa, R.; Furuta, K.; Tsuta, K.; Shibata, T.; Yamamoto, S.; et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012, 72, 100–111. [Google Scholar] [CrossRef]
- Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; Misek, D.E.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar]
- Schabath, M.B.; Welsh, E.A.; Fulp, W.J.; Chen, L.; Teer, J.K.; Thompson, Z.J.; Engel, B.E.; Xie, M.; Berglund, A.E.; Creelan, B.C.; et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016, 35, 3209–3216. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.I.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Blanche, P.; Dartigues, J.F.; Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013, 32, 5381–5397. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Ye, Z.; Shen, R.; Yu, G.; Wu, J.; Xiong, Y.; Zhou, R.; Qiu, W.; Huang, N.; Sun, L.; et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 2021, 12, 687975. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 2017, 6, e26476. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef]
- Lee, S.; Hwang, N.; Seok, B.G.; Lee, S.; Lee, S.J.; Chung, S.W. Autophagy mediates an amplification loop during ferroptosis. Cell Death Dis. 2023, 14, 464. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, J.; Bian, T.; Liu, L.; Sun, H.; Zhou, H.; Zhao, C.; Yang, Z.; Shi, J.; Liu, Y. Correlation between prognostic indicator AHNAK2 and immune infiltrates in lung adenocarcinoma. Int. Immunopharmacol. 2021, 90, 107134. [Google Scholar] [CrossRef]
- Zheng, L.; Li, S.; Zheng, X.; Guo, R.; Qu, W. AHNAK2 is a novel prognostic marker and correlates with immune infiltration in papillary thyroid cancer: Evidence from integrated analysis. Int. Immunopharmacol. 2021, 90, 107185. [Google Scholar] [CrossRef]
- Koguchi, D.; Matsumoto, K.; Shimizu, Y.; Kobayashi, M.; Hirano, S.; Ikeda, M.; Sato, Y.; Iwamura, M. Prognostic Impact of AHNAK2 Expression in Patients Treated with Radical Cystectomy. Cancers 2021, 13, 1748. [Google Scholar] [CrossRef]
- Ye, R.; Liu, D.; Guan, H.; AiErken, N.; Fang, Z.; Shi, Y.; Zhang, Y.; Wang, S. AHNAK2 promotes thyroid carcinoma progression by activating the NF-κB pathway. Life Sci. 2021, 286, 120032. [Google Scholar] [CrossRef]
- Xu, M.; Wen, J.; Xu, Q.; Li, H.; Lin, B.; Bhandari, A.; Qu, J. AHNAK2 promotes the progression of differentiated thyroid cancer through PI3K/AKT signaling pathway. Curr. Cancer Drug Targets 2022, 24, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.Y.; Qi, Q.L.; Hou, S.; Chen, Z.; Jiang, N.; Zhang, L.; Lin, C.H. Silencing of AHNAK2 restricts thyroid carcinoma progression by inhibiting the Wnt/β-catenin pathway. Neoplasma 2021, 68, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, T.; Xue, W.; Fang, Y.; Chen, X.; Xu, L.; Zhang, L.; Guan, K.; Pan, J.; Zheng, L.; et al. ARNTL2 promotes pancreatic ductal adenocarcinoma progression through TGF/BETA pathway and is regulated by miR-26a-5p. Cell Death Dis. 2020, 11, 692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shan, G.; Jin, X.; Yu, X.; Bi, G.; Feng, M.; Wang, H.; Lin, M.; Zhan, C.; Wang, Q.; et al. ARNTL2 is an indicator of poor prognosis, promotes epithelial-to-mesenchymal transition and inhibits ferroptosis in lung adenocarcinoma. Transl. Oncol. 2022, 26, 101562. [Google Scholar] [CrossRef]
- Wang, T.; Wang, K.; Zhu, X.; Chen, N. ARNTL2 upregulation of ACOT7 promotes NSCLC cell proliferation through inhibition of apoptosis and ferroptosis. BMC Mol. Cell Biol. 2023, 24, 14. [Google Scholar] [CrossRef]
- Buchan, S.L.; Rogel, A.; Al-Shamkhani, A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 2018, 131, 39–48. [Google Scholar] [CrossRef]
- Starzer, A.M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: CD27 (TNFRSF7). ESMO Open 2020, 4, e000629. [Google Scholar] [CrossRef]
- Borelli, A.; Irla, M. Lymphotoxin: From the physiology to the regeneration of the thymic function. Cell Death Differ. 2021, 28, 2305–2314. [Google Scholar] [CrossRef]
- Legut, M.; Gajic, Z.; Guarino, M.; Daniloski, Z.; Rahman, J.A.; Xue, X.; Lu, C.; Lu, L.; Mimitou, E.P.; Hao, S.; et al. A genome-scale screen for synthetic drivers of T cell proliferation. Nature 2022, 603, 728–735. [Google Scholar] [CrossRef]
- Wu, P.; Guo, Y. Susceptibility Loci in SLC15A1, UGT1A3, and CWC27 Genes Associated with Bladder Cancer in the Northeast Chinese Population. BioMed Res. Int. 2022, 2022, 2988159. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, Q.; Guo, Y.; Zhu, K. Eight-gene signature predicts recurrence in lung adenocarcinoma. Cancer Biomark. Sect. A Dis. Markers 2020, 28, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, C.; Gan, G.; Zhang, J.; Wu, J.; Miao, Y.; Zhang, M.; Li, B.; Mi, J. Cancer-associated fibroblasts enhance tumor (18)F-FDG uptake and contribute to the intratumor heterogeneity of PET-CT. Theranostics 2018, 8, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Xuan, B.; Yan, T.; Ma, Y.; Xu, P.; Tian, X.; Zhang, X.; Cao, Y.; Ma, D.; Zhu, X.; et al. m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol. Cancer 2020, 19, 72. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Yang, J.W.; Zeng, J.; Chen, X.Q.; Gao, Y.; Kui, X.Y.; Liu, X.Y.; Zhang, Y.; Zhang, Y.H.; Pei, Z.J. SLC2A1 is a Diagnostic Biomarker Involved in Immune Infiltration of Colorectal Cancer and Associated With m6A Modification and ceRNA. Front. Cell Dev. Biol. 2022, 10, 853596. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zeng, X.; Wu, B.; Zhao, J.; Pan, Y. RNA-Seq analysis of peripheral blood mononuclear cells reveals unique transcriptional signatures associated with radiotherapy response of nasopharyngeal carcinoma and prognosis of head and neck cancer. Cancer Biol. Ther. 2020, 21, 139–146. [Google Scholar] [CrossRef]
- Evans, C.S.; Ruhl, D.A.; Chapman, E.R. An Engineered Metal Sensor Tunes the Kinetics of Synaptic Transmission. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 11769–11779. [Google Scholar] [CrossRef]
- Li, L.; Liu, H.; Krout, M.; Richmond, J.E.; Wang, Y.; Bai, J.; Weeratunga, S.; Collins, B.M.; Ventimiglia, D.; Yu, Y.; et al. A novel dual Ca2+ sensor system regulates Ca2+-dependent neurotransmitter release. J. Cell Biol. 2021, 220, e202008121. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, L.; Ma, H. Identification of a Novel Signature Based on Ferritinophagy-Related Genes to Predict Prognosis in Lung Adenocarcinoma: Focus on AHNAK2. Bioengineering 2024, 11, 1070. https://doi.org/10.3390/bioengineering11111070
Xia L, Ma H. Identification of a Novel Signature Based on Ferritinophagy-Related Genes to Predict Prognosis in Lung Adenocarcinoma: Focus on AHNAK2. Bioengineering. 2024; 11(11):1070. https://doi.org/10.3390/bioengineering11111070
Chicago/Turabian StyleXia, Liangjiang, and Haitao Ma. 2024. "Identification of a Novel Signature Based on Ferritinophagy-Related Genes to Predict Prognosis in Lung Adenocarcinoma: Focus on AHNAK2" Bioengineering 11, no. 11: 1070. https://doi.org/10.3390/bioengineering11111070
APA StyleXia, L., & Ma, H. (2024). Identification of a Novel Signature Based on Ferritinophagy-Related Genes to Predict Prognosis in Lung Adenocarcinoma: Focus on AHNAK2. Bioengineering, 11(11), 1070. https://doi.org/10.3390/bioengineering11111070