


Reverse Vaccinology Integrated with Biophysics Techniques for Designing a Peptide-Based Subunit Vaccine for Bourbon Virus
Abstract

1. Introduction
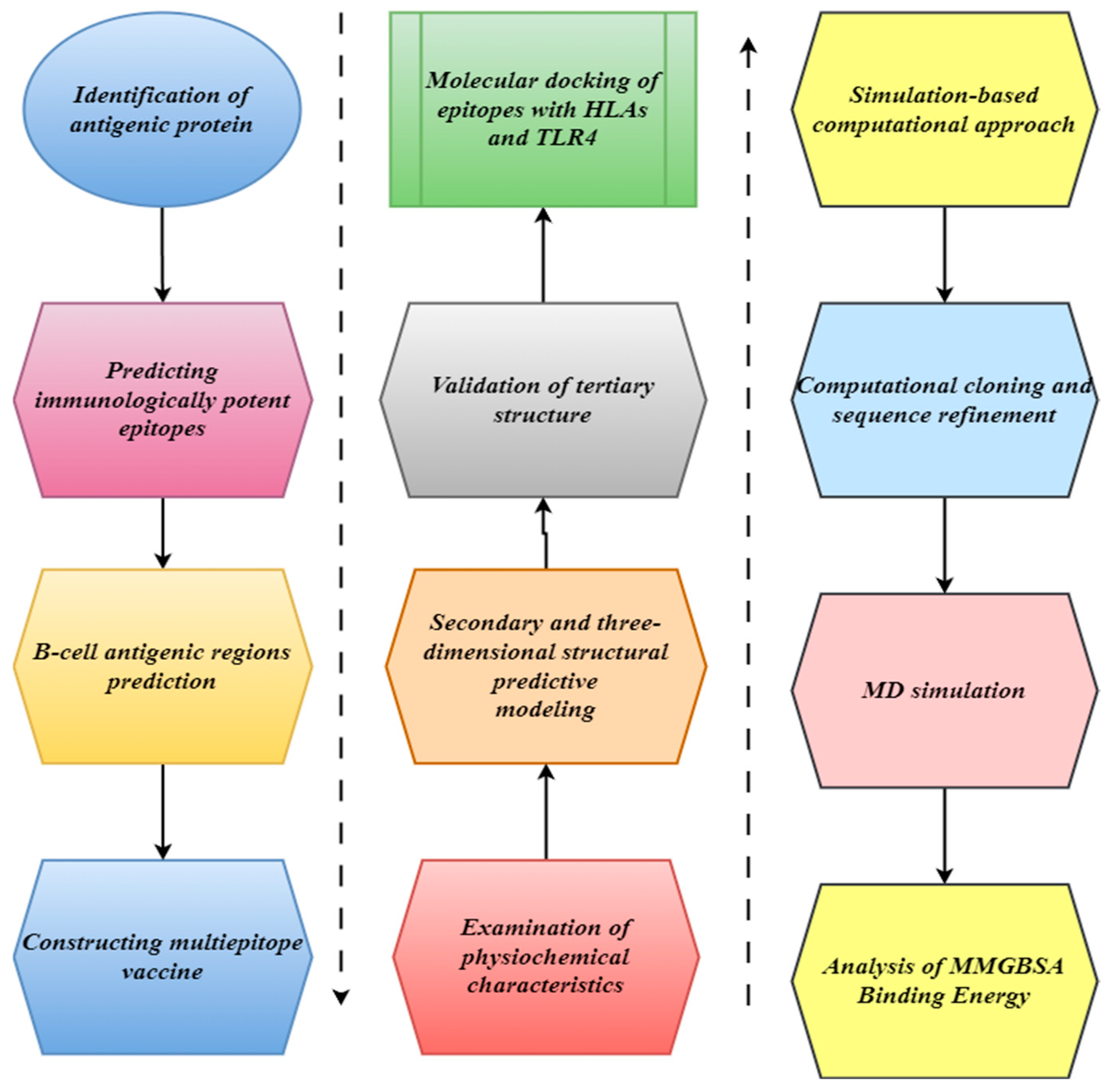
2. Materials and Methods
2.1. Identification of Antigenic Protein
2.2. Predicting Immunologically Potent Epitopes
2.3. B-Cell Antigenic Region Prediction
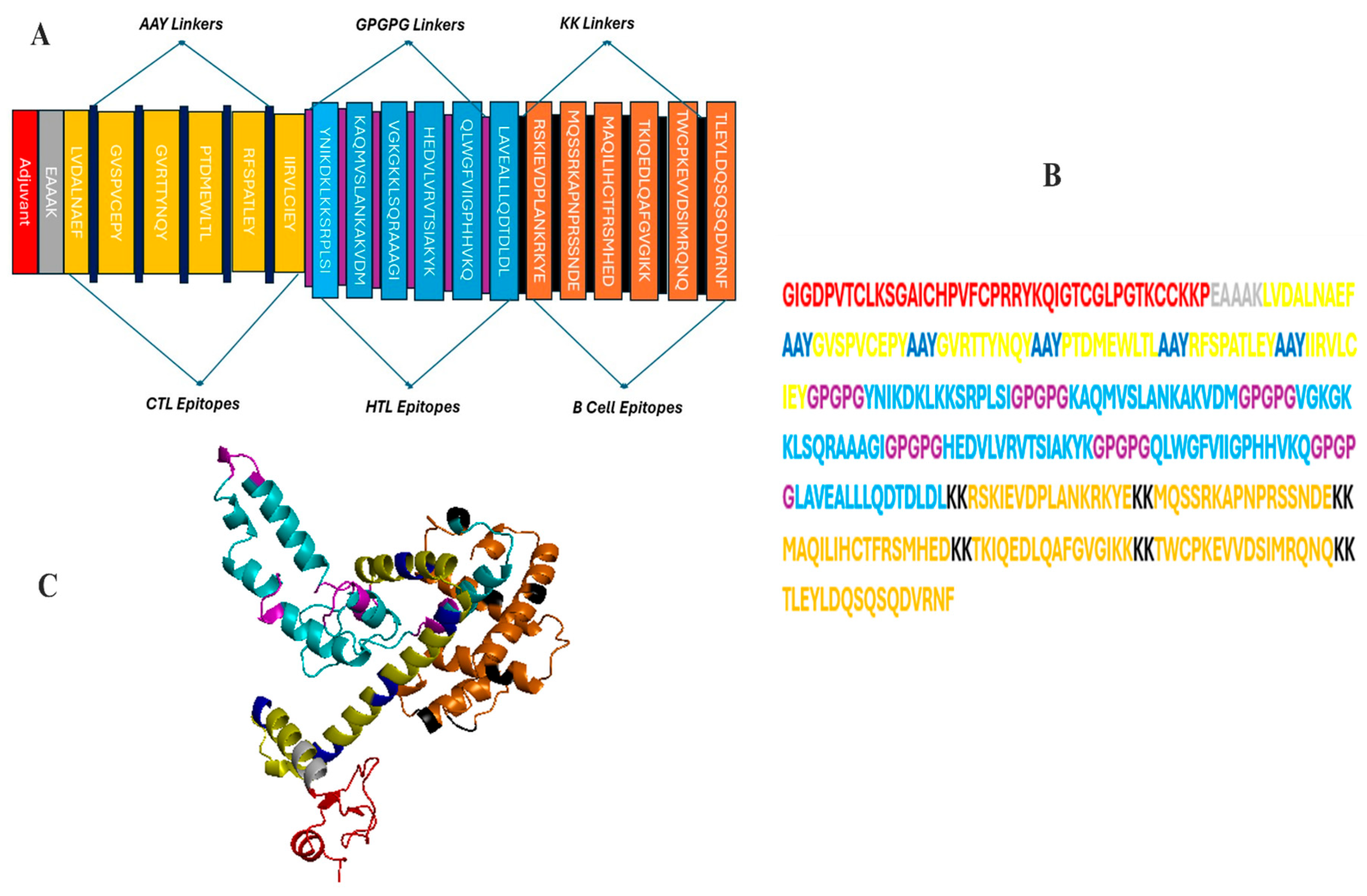
2.4. Constructing a Multi-Epitope Vaccine
2.5. Examination of Physiochemical Characteristics
2.6. Secondary and Three-Dimensional Structural Predictive Modeling
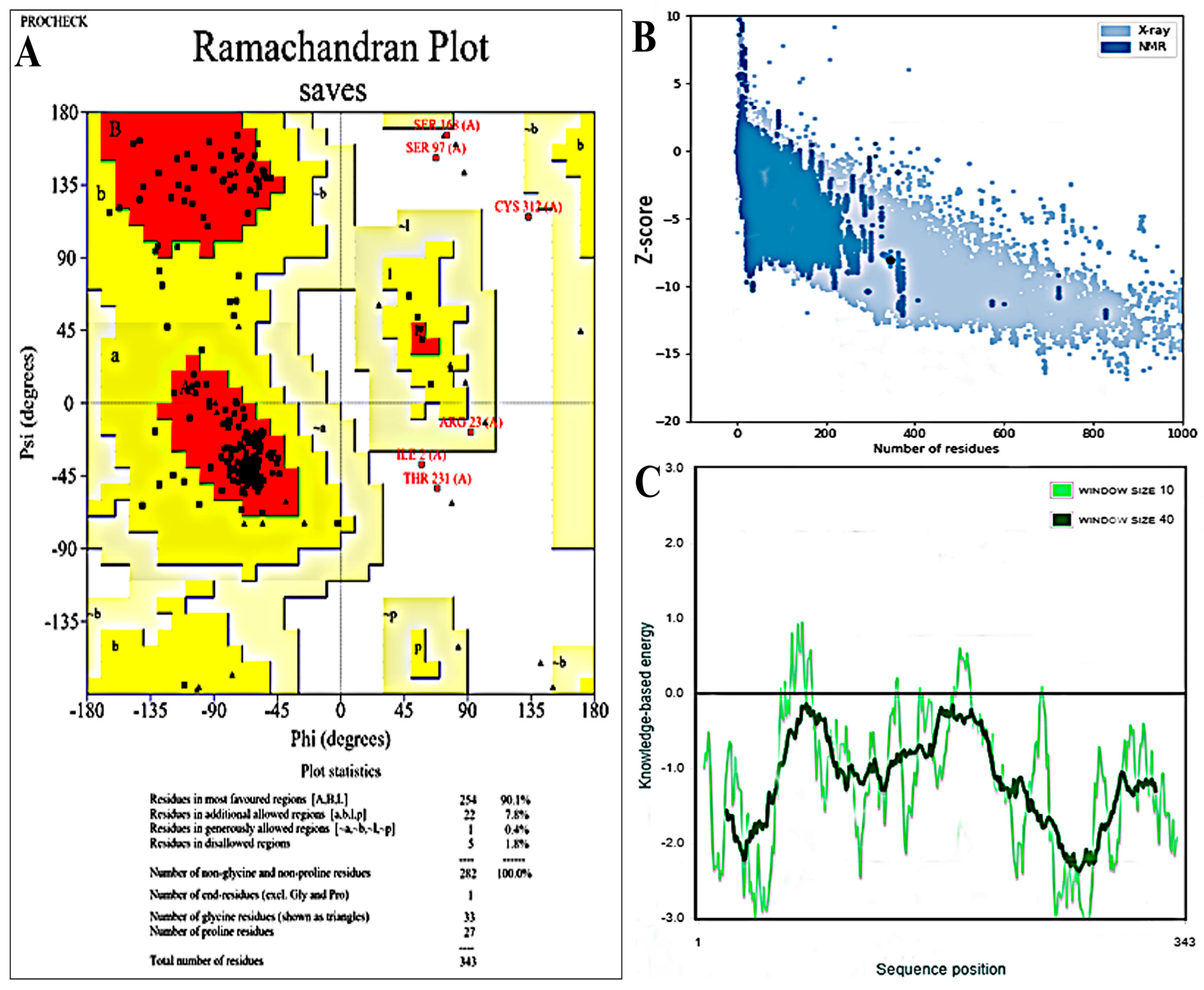
2.7. Validation of Tertiary Structure
2.8. Molecular Docking of Epitopes with HLAs and TLR4
2.9. Simulation-Based Computational Approach
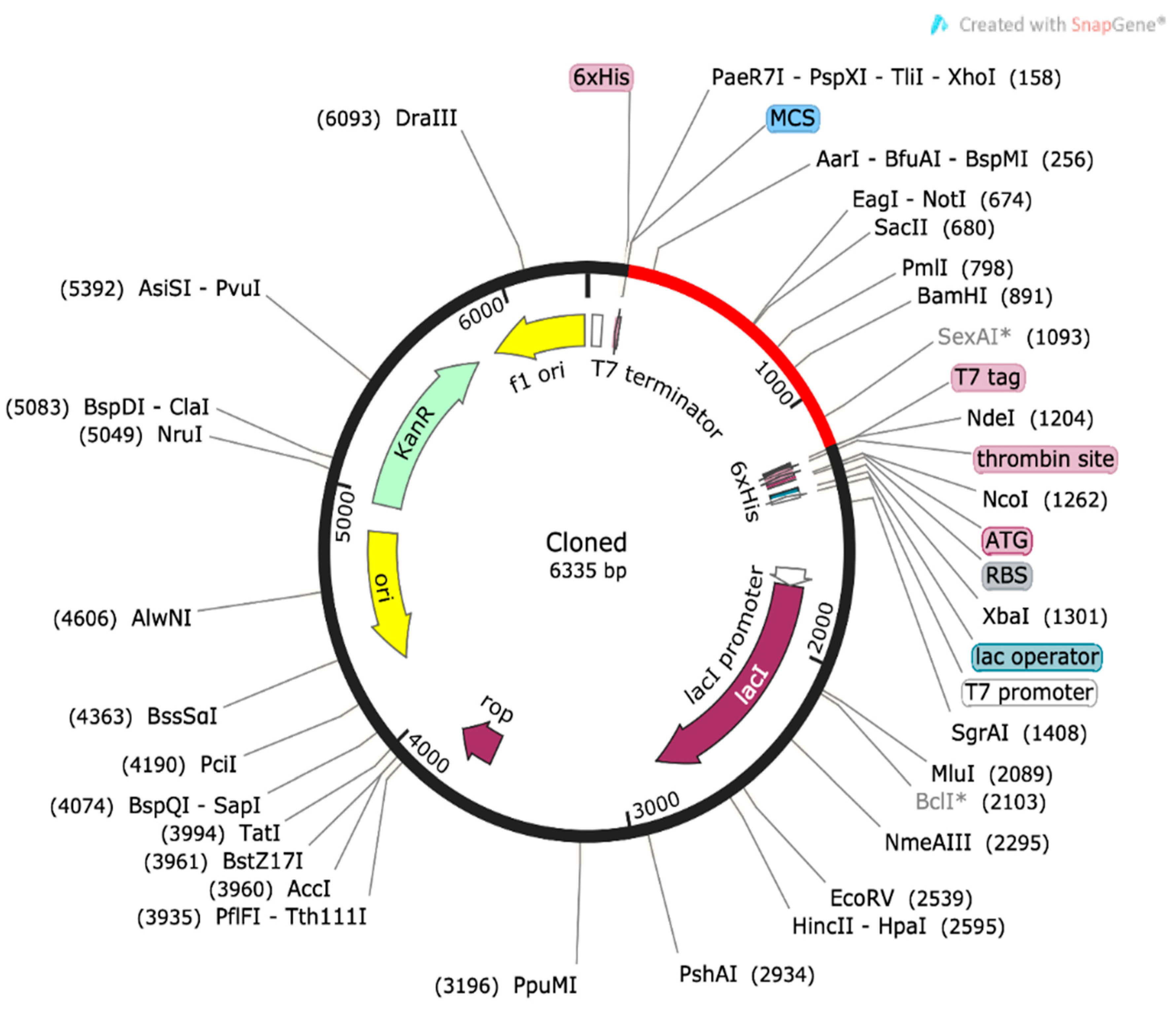
2.10. Computational Cloning and Sequence Refinement
2.11. MD Simulation
2.12. Analysis of MMGBSA Binding Energy
3. Results
3.1. Protein Retrieval
3.2. Immunogenic Epitope Profiling
3.3. Development of a MEVC Construct
3.4. Physicochemical Properties
3.5. Secondary and 3D Structural Modeling
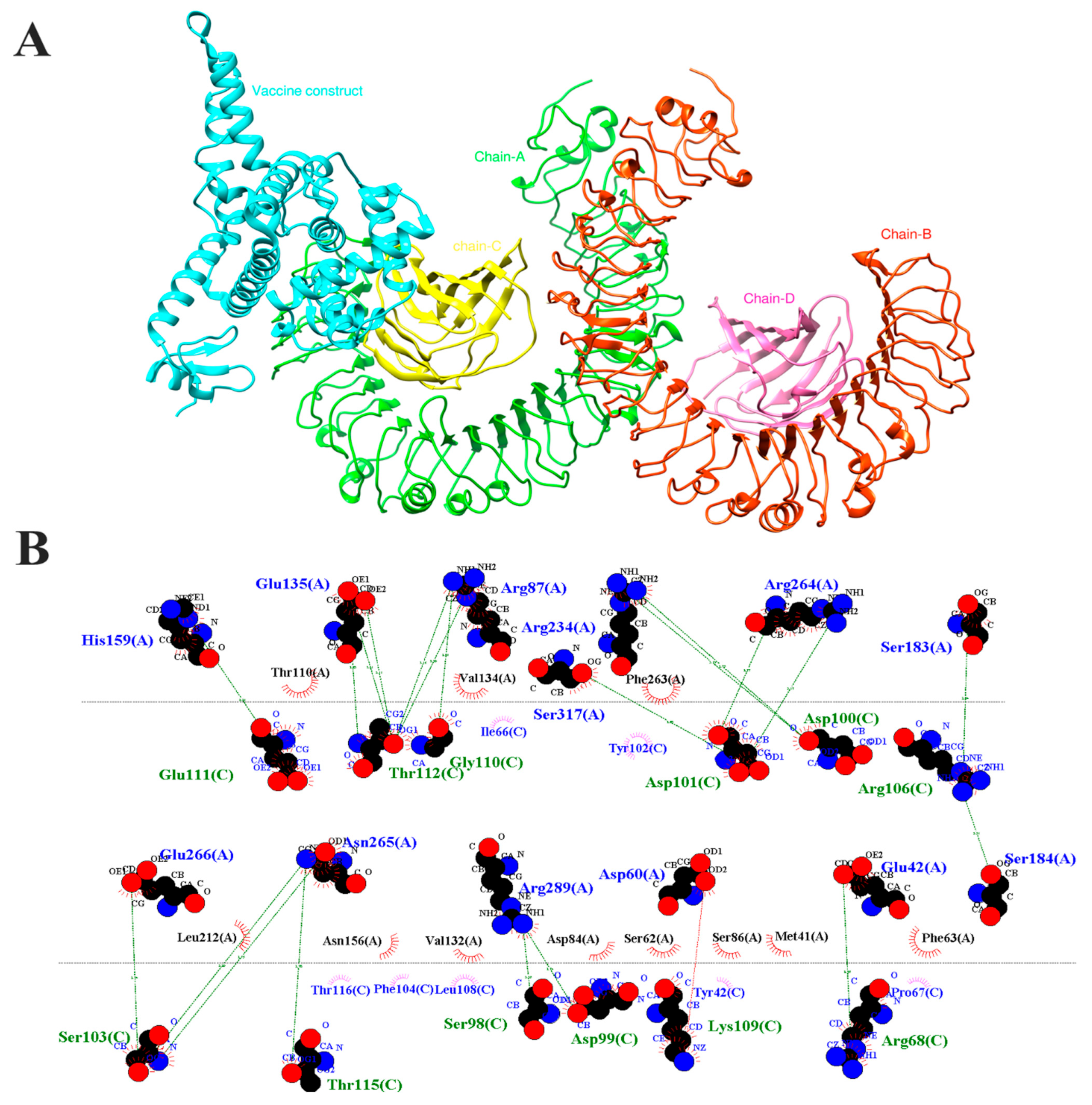
3.6. Docking Simulations Among Peptides and MEVC
3.7. Codon Optimization of the Comple Vaccine Construct
3.8. Immune Simulation
3.9. Molecular Docking
3.10. MD Simulation
3.11. Binding Free Energy Calculations
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Sayed, A.; Kamel, M. Climatic Changes and Their Role in Emergence and Re-Emergence of Diseases. Environ. Sci. Pollut. Res. Int. 2020, 27, 22336–22352. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.; Lindsey, N.P.; Fischer, M.; Gregory, C.J.; Hinckley, A.F.; Mead, P.S.; Paz-Bailey, G.; Waterman, S.H.; Drexler, N.A.; Kersh, G.J.; et al. Vital Signs: Trends in Reported Vectorborne Disease Cases—United States and Territories, 2004–2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Bush, L.M.; Vazquez-Pertejo, M.T. Tick Borne Illness-Lyme Disease. Dis. Mon. 2018, 64, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Rochlin, I.; Toledo, A. Emerging Tick-Borne Pathogens of Public Health Importance: A Mini-Review. J. Med. Microbiol. 2020, 69, 781–791. [Google Scholar] [CrossRef]
- Jackson, K.C.; Gidlewski, T.; Root, J.J.; Bosco-Lauth, A.M.; Lash, R.R.; Harmon, J.R.; Brault, A.C.; Panella, N.A.; Nicholson, W.L.; Komar, N. Bourbon Virus in Wild and Domestic Animals, Missouri, USA, 2012–2013. Emerg. Infect. Dis. 2019, 25, 1752–1753. [Google Scholar] [CrossRef]
- Kosoy, O.I.; Lambert, A.J.; Hawkinson, D.J.; Pastula, D.M.; Goldsmith, C.S.; Hunt, D.C.; Staples, J.E. Novel Thogotovirus Associated with Febrile Illness and Death, United States, 2014. Emerg. Infect. Dis. 2015, 21, 760–764. [Google Scholar] [CrossRef]
- Savage, H.M.; Burkhalter, K.L.; Godsey, M.S.; Panella, N.A.; Ashley, D.C.; Nicholson, W.L.; Lambert, A.J. Bourbon Virus in Field-Collected Ticks, Missouri, USA. Emerg. Infect. Dis. 2017, 23, 2017–2022. [Google Scholar] [CrossRef]
- Roe, M.K.; Huffman, E.R.; Batista, Y.S.; Papadeas, G.G.; Kastelitz, S.R.; Restivo, A.M.; Stobart, C.C. Comprehensive Review of Emergence and Virology of Tickborne Bourbon Virus in the United States. Emerg. Infect. Dis. 2023, 29, 1–7. [Google Scholar] [CrossRef]
- Garba, A.; Riley, J.; Lahmers, K.K.; Eastwood, G. Widespread Circulation of Tick-Borne Viruses in Virginia-Evidence of Exposure to Heartland, Bourbon, and Powassan Viruses in Wildlife and Livestock. Microorganisms 2024, 12, 899. [Google Scholar] [CrossRef]
- Shah, T.; Li, Q.; Wang, B.; Baloch, Z.; Xia, X. Geographical Distribution and Pathogenesis of Ticks and Tick-Borne Viral Diseases. Front. Microbiol. 2023, 14, 100903. [Google Scholar] [CrossRef]
- Fuchs, J.; Straub, T.; Seidl, M.; Kochs, G. Essential Role of Interferon Response in Containing Human Pathogenic Bourbon Virus. Emerg. Infect. Dis. 2019, 25, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Qi, J.; Wu, Y.; Wang, X.; Gao, G.F.; Peng, R.; Gao, F. Postfusion Structure of Human-Infecting Bourbon Virus Envelope Glycoprotein. J. Struct. Biol. 2019, 208, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Savage, H.M.; Godsey, M.S.; Tatman, J.; Burkhalter, K.L.; Hamm, A.; Panella, N.A.; Ghosh, A.; Raghavan, R.K. Surveillance for Heartland and Bourbon Viruses in Eastern Kansas, June 2016. J. Med. Entomol. 2018, 55, 1613–1616. [Google Scholar] [CrossRef] [PubMed]
- Cumbie, A.N.; Trimble, R.N.; Eastwood, G. Pathogen Spillover to an Invasive Tick Species: First Detection of Bourbon Virus in Haemaphysalis Longicornis in the United States. Pathogens 2022, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Moodley, A.; Fatoba, A.; Okpeku, M.; Emmanuel Chiliza, T.; Blessing Cedric Simelane, M.; Pooe, O.J. Reverse Vaccinology Approach to Design a Multi-Epitope Vaccine Construct Based on the Mycobacterium Tuberculosis Biomarker PE_PGRS17. Immunol. Res. 2022, 70, 501–517. [Google Scholar] [CrossRef]
- Patronov, A.; Doytchinova, I. T-Cell Epitope Vaccine Design by Immunoinformatics. Open Biol. 2021, 3, 120139. [Google Scholar] [CrossRef]
- Parihar, A.; Malviya, S.; Khan, R. Immunoinformatics and Reverse Vaccinomic Approaches for Effective Design. In Computational Approaches for Novel Therapeutic and Diagnostic Designing to Mitigate SARS-CoV-2 Infection; Academic Press: Cambridge, MA, USA, 2022; pp. 357–378. [Google Scholar] [CrossRef]
- Ong, E.; Wong, M.U.; Huffman, A.; He, Y. COVID-19 Coronavirus Vaccine Design Using Reverse Vaccinology and Machine Learning. bioRxiv 2020, 11, 1581. [Google Scholar] [CrossRef]
- Folaranmi, T.; Rubin, L.; Martin, S.W.; Patel, M.; MacNeil, J.R. Centers for Disease Control (CDC) Use of Serogroup B Meningococcal Vaccines in Persons Aged ≥10 Years at Increased Risk for Serogroup B Meningococcal Disease: Recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 608–612. [Google Scholar]
- Yang, H.-H.; Mascuch, S.J.; Madoff, L.C.; Paoletti, L.C. Recombinant Group B Streptococcus Alpha-Like Protein 3 Is an Effective Immunogen and Carrier Protein. Clin. Vaccine Immunol. 2008, 15, 1035–1041. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse Vaccinology: Developing Vaccines in the Era of Genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Huang, Y.; Li, Z.-S.; Lin, H.; Sui, Y.-F. Prediction and Analysis of HLA-A2/A24-Restricted Cytotoxic T-Lymphocyte Epitopes of the Tumor Antigen MAGE-n Using the Artificial Neural Networks Method on NetCTL1.2 Server. Oncol. Lett. 2010, 1, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Gartner, J.J.; Parkhurst, M.R.; Gros, A.; Tran, E.; Jafferji, M.S.; Copeland, A.; Hanada, K.-I.; Zacharakis, N.; Lalani, A.; Krishna, S.; et al. A Machine Learning Model for Ranking Candidate HLA Class I Neoantigens Based on Known Neoepitopes from Multiple Human Tumor Types. Nat. Cancer 2021, 2, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iTTCA-Hybrid: Improved and Robust Identification of Tumor T Cell Antigens by Utilizing Hybrid Feature Representation. Anal. Biochem. 2020, 599, 113747. [Google Scholar] [CrossRef] [PubMed]
- Prawiningrum, A.F.; Paramita, R.I.; Panigoro, S.S. Immunoinformatics Approach for Epitope-Based Vaccine Design: Key Steps for Breast Cancer Vaccine. Diagnostics 2022, 12, 2981. [Google Scholar] [CrossRef]
- Ayaz, H.; Almanaa, T.N.; Hassan Khan, U.; Ahmad, S.; Ahmad, F.; Irfan, M.; Waheed, Y. Multiepitope Subunit Vaccine against Colorado Tick Fever Virus by Using Reverse Vaccinology Approach. J. Mol. Liq. 2024, 402, 124725. [Google Scholar] [CrossRef]
- Alsubaiyel, A.M.; Bukhari, S.I. Computational Exploration and Design of a Multi-Epitopes Vaccine Construct against Chlamydia psittaci. J. Biomol. Struct. Dyn. 2023, 1–17. [Google Scholar] [CrossRef]
- Xu, Q.; Ma, X.; Wang, F.; Li, H.; Xiao, Y.; Zhao, X. Design and Construction of a Chimeric Multi-Epitope Gene as an Epitope-Vaccine Strategy against ALV-J. Protein Expr. Purif. 2015, 106, 18–24. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Kim, H. Discovering Peptides and Computational Investigations of a Multiepitope Vaccine Target Mycobacterium Tuberculosis. Synth. Syst. Biotechnol. 2024, 9, 391–405. [Google Scholar] [CrossRef]
- Yu, C.; Wu, Q.; Xin, J.; Yu, Q.; Ma, Z.; Xue, M.; Xu, Q.; Zheng, C. Designing a Smallpox B-Cell and T-Cell Multi-Epitope Subunit Vaccine Using a Comprehensive Immunoinformatics Approach. Microbiol. Spectr. 2024, 12, e0046524. [Google Scholar] [CrossRef]
- ProtParam, E. ExPASy-ProtParam Tool. 2017. Available online: https://web.expasy.org/protparam/ (accessed on 26 August 2024).
- Kim, D.E.; Chivian, D.; Baker, D. Protein Structure Prediction and Analysis Using the Robetta Server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Ghafoor, D.; Zeb, A.; Ali, S.S.; Ali, M.; Akbar, F.; Ud Din, Z.; Ur Rehman, S.; Suleman, M.; Khan, W. Immunoinformatic Based Designing of Potential Immunogenic Novel mRNA and Peptide-Based Prophylactic Vaccines against H5N1 and H7N9 Avian Influenza Viruses. J. Biomol. Struct. Dyn. 2024, 42, 3641–3658. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, Y.; Ningaraju, T.M.; Bhavani, P.; Kumar, B.; Peter, A. In silico analysis of lipase gene from Bacillus subtilis to predict the binding potential with industrial important compounds. J. Exp. Zool. India 2023, 26, 2369–2383. [Google Scholar] [CrossRef]
- Sura, K.; Rohilla, H.; Kumar, D.; Jakhar, R.; Ahlawat, V.; Kaushik, D.; Dangi, M.; Chhillar, A.K. Exploring Structural Antigens of Yellow Fever Virus to Design Multi-Epitope Subunit Vaccine Candidate by Utilizing an Immuno-Informatics Approach. J. Genet. Eng. Biotechnol. 2023, 21, 161. [Google Scholar] [CrossRef]
- Sarmadi, S.; Rahbar, M.R.; Najafi, H.; Chukwudozie, O.S.; Morowvat, M.H. In Silico Design and Evaluation of a Novel Therapeutic Agent Against the Spike Protein as a Novel Treatment Strategy for COVID-19 Treatment. Recent. Pat. Biotechnol. 2024, 18, 162–176. [Google Scholar] [CrossRef]
- Alam, M.M.; Saikat, A.S.M.; Uddin, M.E. Bioinformatics Approaches for Structural and Functional Annotation of an Uncharacterized Protein of Helicobacter Pylori. Eng. Proc. 2023, 37, 61. [Google Scholar] [CrossRef]
- Javid, M.T.; Rahim, F.; Taha, M.; Rehman, H.U.; Nawaz, M.; Wadood, A.; Imran, S.; Uddin, I.; Mosaddik, A.; Khan, K.M. Synthesis, in Vitro α-Glucosidase Inhibitory Potential and Molecular Docking Study of Thiadiazole Analogs. Bioorg Chem. 2018, 78, 201–209. [Google Scholar] [CrossRef]
- Mehrab, R.; Sedighian, H.; Sotoodehnejadnematalahi, F.; Halabian, R.; Fooladi, A.A.I. A Comparative Study of the Arazyme-Based Fusion Proteins with Various Ligands for More Effective Targeting Cancer Therapy: An in-Silico Analysis. Res. Pharm. Sci. 2023, 18, 159–176. [Google Scholar] [CrossRef]
- Nijanthi, P.; Santhi, S.; Munivelan, B. Molecular Dynamics Studies on the Arginine Kinase Protein of Aedes Sollicitans: Against the Natural Chemical Compound, Gedunin. Int. J. Mosq. Res. 2023, 10, 10–14. [Google Scholar] [CrossRef]
- Beikzadeh, B. Immunoinformatics Design of Multi-Epitope Vaccine Using OmpA, OmpD and Enterotoxin against Non-Typhoidal Salmonellosis. BMC Bioinform. 2023, 24, 63. [Google Scholar] [CrossRef]
- Mazumder, L.; Shahab, M.; Islam, S.; Begum, M.; Oliveira, J.I.N.; Begum, S.; Akter, S. An Immunoinformatics Approach to Epitope-Based Vaccine Design against PspA in Streptococcus Pneumoniae. J. Genet. Eng. Biotechnol. 2023, 21, 57. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.; Darden, T.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Götz, A.W.; Homeyer, N.; et al. Amber 16, University of California, San Francisco; University of California: San Francisco, CA, USA, 2016. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB Force Field. Amber 2014, 14, 29–31. [Google Scholar]
- Brice, A.R.; Dominy, B.N. Examining Electrostatic Influences on Base-Flipping: A Comparison of TIP3P and GB Solvent Models. Commun. Comput. Phys. 2013, 13, 223–237. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- 385 PDFs|Review Articles in XMGRACE. Available online: https://www.researchgate.net/topic/Xmgrace/publications (accessed on 31 December 2023).
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Rego, R.O.M.; Trentelman, J.J.A.; Anguita, J.; Nijhof, A.M.; Sprong, H.; Klempa, B.; Hajdusek, O.; Tomás-Cortázar, J.; Azagi, T.; Strnad, M.; et al. Counterattacking the Tick Bite: Towards a Rational Design of Anti-Tick Vaccines Targeting Pathogen Transmission. Parasites Vectors 2019, 12, 229. [Google Scholar] [CrossRef]
- Windhaber, S.; Xin, Q.; Lozach, P.-Y. Orthobunyaviruses: From Virus Binding to Penetration into Mammalian Host Cells. Viruses 2021, 13, 872. [Google Scholar] [CrossRef]
- Bloom, D.E. The Value of Vaccination. Adv. Exp. Med. Biol. 2011, 697, 1–8. [Google Scholar] [CrossRef]
- Gilbert, S.C. T-Cell-Inducing Vaccines—What’s the Future. Immunology 2012, 135, 19–26. [Google Scholar] [CrossRef]
- Thomas, S.J.; Nisalak, A.; Anderson, K.B.; Libraty, D.H.; Kalayanarooj, S.; Vaughn, D.W.; Putnak, R.; Gibbons, R.V.; Jarman, R.; Endy, T.P. Dengue Plaque Reduction Neutralization Test (PRNT) in Primary and Secondary Dengue Virus Infections: How Alterations in Assay Conditions Impact Performance. Am. J. Trop. Med. Hyg. 2009, 81, 825–833. [Google Scholar] [CrossRef]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-Presentation by Dendritic Cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Podojil, J.R.; Miller, S.D. Molecular Mechanisms of T-Cell Receptor and Costimulatory Molecule Ligation/Blockade in Autoimmune Disease Therapy. Immunol. Rev. 2009, 229, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.D.; Moss, P.A.; Goulder, P.J.; Barouch, D.H.; McHeyzer-Williams, M.G.; Bell, J.I.; McMichael, A.J.; Davis, M.M. Phenotypic Analysis of Antigen-Specific T Lymphocytes. Science 1996, 274, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Isosaka, T.; Matsuo, T.; Yamaguchi, T.; Funabiki, K.; Nakanishi, S.; Kobayakawa, R.; Kobayakawa, K. Htr2a-Expressing Cells in the Central Amygdala Control the Hierarchy between Innate and Learned Fear. Cell 2015, 163, 1153–1164. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||||
|---|---|---|---|---|---|---|
| Protein | Peptide | Affinity | Cleavage | Tap Score | Combined Score | |
| Nucleoprotein | LVDALNAEF | 0.2994 | 0.9286 | 2.5550 | 1.5382 | |
| GVSPVCEPY | 0.1263 | 0.9660 | 2.9280 | 0.8276 | ||
| GVRTTYNQY | 0.1057 | 0.9748 | 3.0650 | 0.7483 | ||
| Polymerase subunit PA | PTDMEWLTL | 0.2933 | 0.8914 | 0.4090 | 1.3995 | |
| RFSPATLEY | 0.1405 | 0.9767 | 3.3020 | 0.9083 | ||
| IIRVLCIEY | 0.0817 | 0.7837 | 3.0690 | 0.6178 | ||
| (B) | ||||||
| Protein | Allele | Peptide | Score | Allergenicity | IFN | |
| Nucleoprotein | HLA-DRB1*15:01 | YNIKDKLKKSRPLSI | 1.22 | Nill | Yes | |
| HLA-DRB1*15:01 | KAQMVSLANKAKVDM | 0.34 | Nill | Yes | ||
| HLA-DRB1*15:01 | VGKGKKLSQRAAAGI | 0.25 | Nill | Yes | ||
| Polymerase subunit PA | HLA-DRB1*15:01 | HEDVLVRVTSIAKYK | 0.53 | Nill | Yes | |
| HLA-DRB1*15:01 | QLWGFVIIGPHHVKQ | 0.67 | Nill | Yes | ||
| HLA-DRB1*15:01 | LAVEALLLQDTDLDL | 1.78 | Nill | Yes | ||
| (C) | ||||||
| Protein | Position | Peptide | Score | |||
| Nucleoprotein | 24 | RSKIEVDPLANKRKYE | 0.91 | |||
| 1 | MQSSRKAPNPRSSNDE | 0.91 | ||||
| 307 | MAQILIHCTFRSMHED | 0.85 | ||||
| Polymerase subunit PA | 295 | TKIQEDLQAFGVGIKK | 0.87 | |||
| 62 | TWCPKEVVDSIMRQNQ | 0.85 | ||||
| 132 | TLEYLDQSQSQDVRNF | 0.83 | ||||
| Energy Parameter | Vaccine–TLR4 Complex |
|---|---|
| Van der Waals Energy (kcal/mol) | −120.56 |
| Columbic Energy (kcal/mol) | −45.69 |
| Total Gas-Phase Energy (kcal/mol) | −166.25 |
| Total Solvation Energy (kcal/mol) | 31.42 |
| Net Energy (kcal/mol) | −134.83 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almanaa, T.N. Reverse Vaccinology Integrated with Biophysics Techniques for Designing a Peptide-Based Subunit Vaccine for Bourbon Virus. Bioengineering 2024, 11, 1056. https://doi.org/10.3390/bioengineering11111056
Almanaa TN. Reverse Vaccinology Integrated with Biophysics Techniques for Designing a Peptide-Based Subunit Vaccine for Bourbon Virus. Bioengineering. 2024; 11(11):1056. https://doi.org/10.3390/bioengineering11111056
Chicago/Turabian StyleAlmanaa, Taghreed N. 2024. "Reverse Vaccinology Integrated with Biophysics Techniques for Designing a Peptide-Based Subunit Vaccine for Bourbon Virus" Bioengineering 11, no. 11: 1056. https://doi.org/10.3390/bioengineering11111056
APA StyleAlmanaa, T. N. (2024). Reverse Vaccinology Integrated with Biophysics Techniques for Designing a Peptide-Based Subunit Vaccine for Bourbon Virus. Bioengineering, 11(11), 1056. https://doi.org/10.3390/bioengineering11111056