
MiMultiCat: A Unified Cloud Platform for the Analysis of Microbiome Data with Multi-Categorical Responses


Abstract
1. Introduction
2. Materials and Methods
2.1. Association Testing
2.2. Prediction Modeling
2.3. Web Server Architecture
2.4. Data Availability
3. Results
3.1. Data Processing
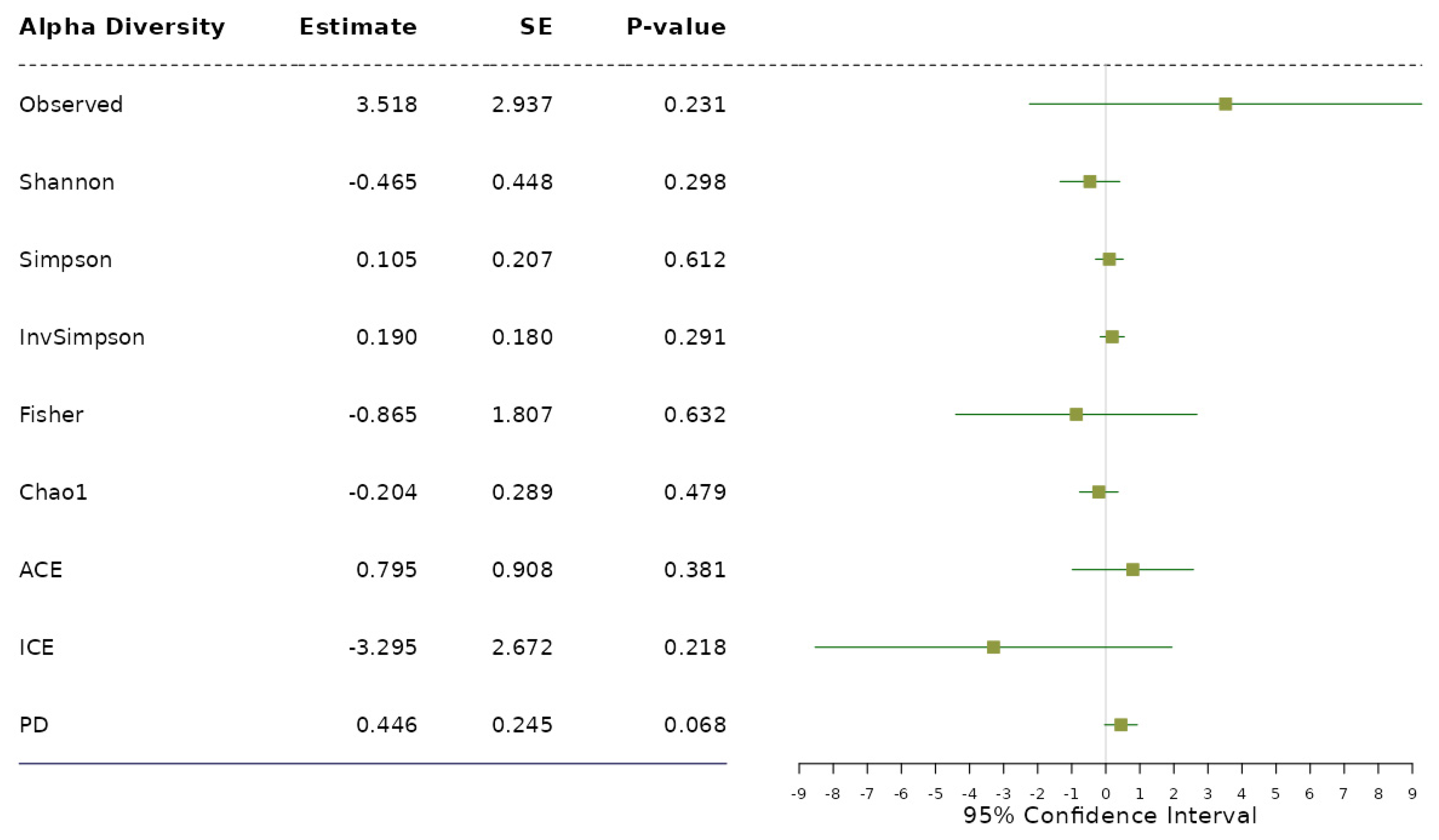
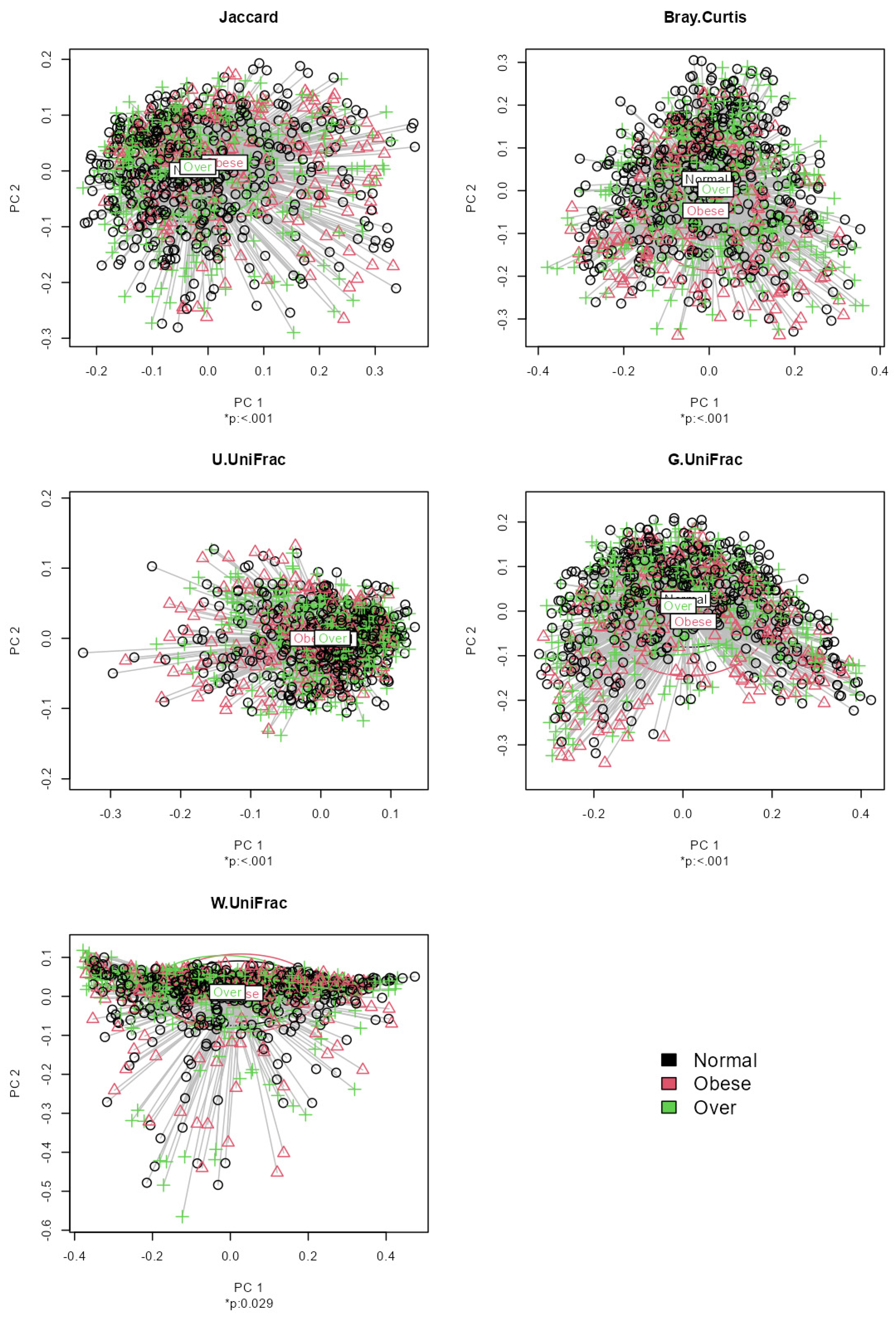
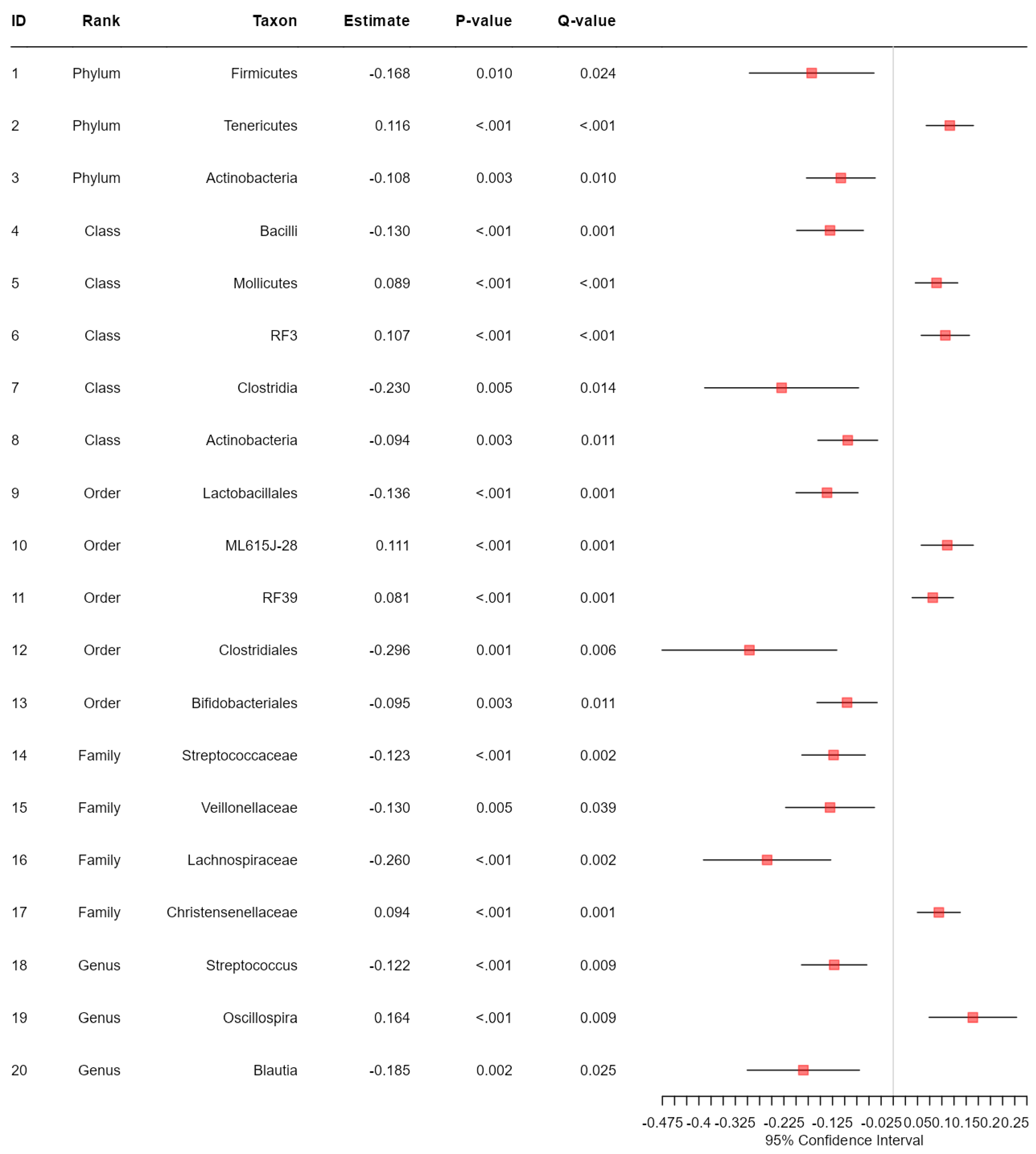
3.2. Data Analysis: Association
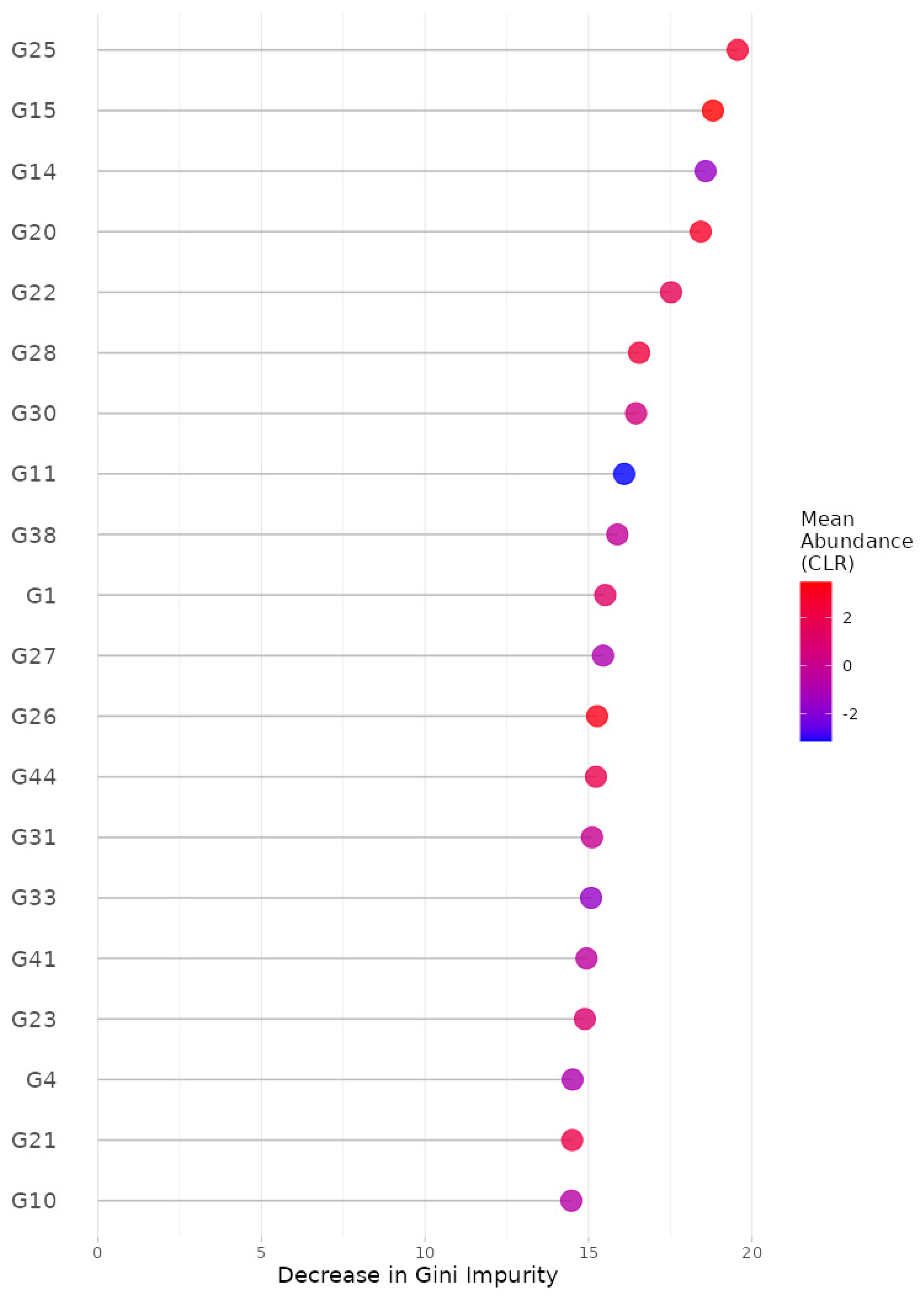
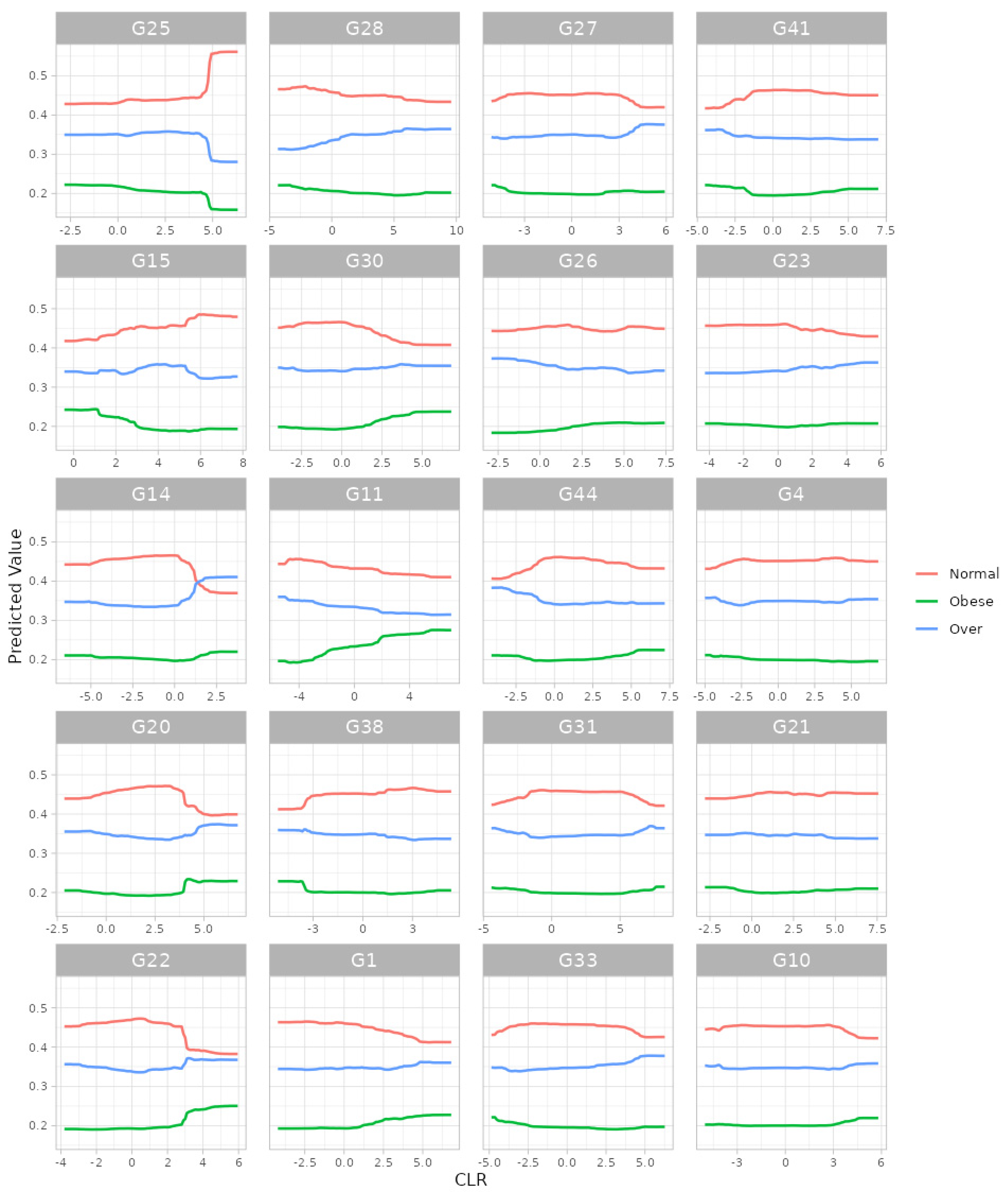
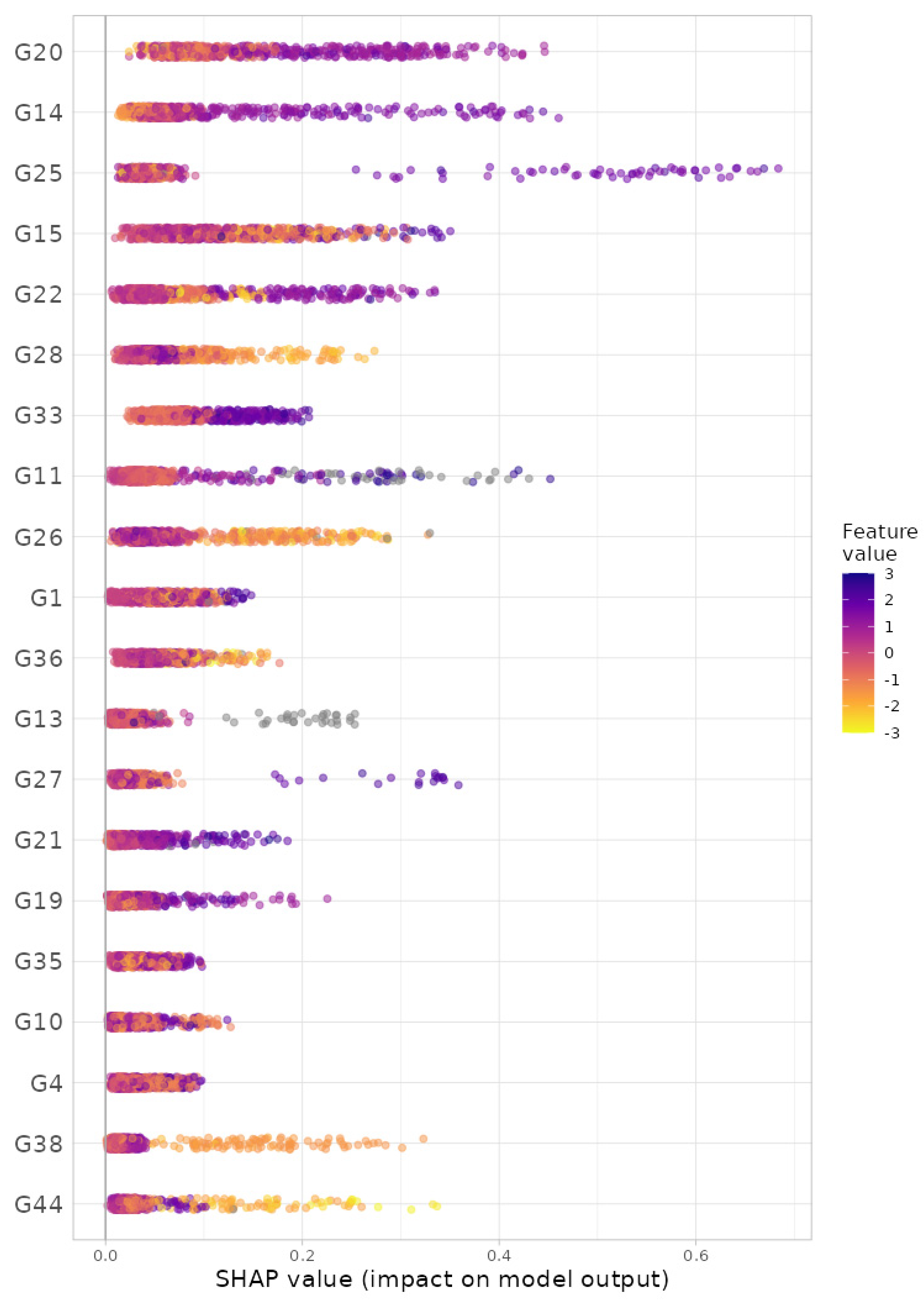
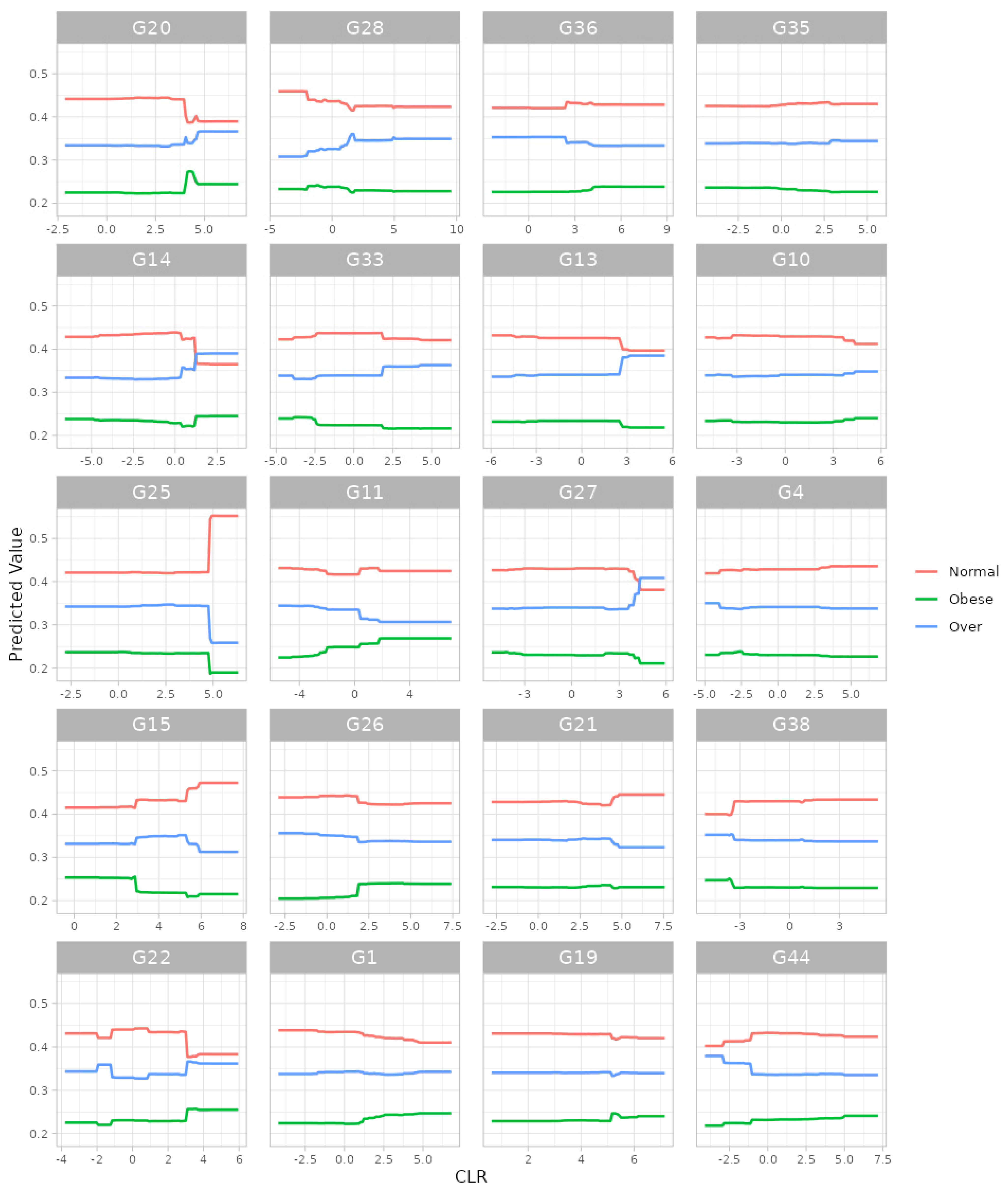
3.3. Data Analysis: Prediction
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woese, C.R.; Fox, G.E. Phylogenetic Structure of the Prokaryotic Domain: The Primary Kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a Natural System of Organisms: Proposal for the Domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A Guide from Sampling to Data Analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The Commensal Microbiome Is Associated with Anti–PD-1 Efficacy in Metastatic Melanoma Patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti–PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Limeta, A.; Ji, B.; Levin, M.; Gatto, F.; Nielsen, J. Meta-Analysis of the Gut Microbiota in Predicting Response to Cancer Immunotherapy in Metastatic Melanoma. JCI Insight 2020, 5, e140940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Li, J.; Krautkramer, K.A.; Badri, M.; Battaglia, T.; Borbet, T.C.; Koh, H.; Ng, S.; Sibley, R.A.; Li, Y. Antibiotic-Induced Acceleration of Type 1 Diabetes Alters Maturation of Innate Intestinal Immunity. eLife 2018, 7, e37816. [Google Scholar] [CrossRef] [PubMed]
- Livanos, A.E.; Greiner, T.U.; Vangay, P.; Pathmasiri, W.; Stewart, D.; McRitchie, S.; Li, H.; Chung, J.; Sohn, J.; Kim, S. Antibiotic-Mediated Gut Microbiome Perturbation Accelerates Development of Type 1 Diabetes in Mice. Nat. Microbiol. 2016, 1, 16140. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef]
- De Palma, G.; Lynch, M.D.J.; Lu, J.; Dang, V.T.; Deng, Y.; Jury, J.; Umeh, G.; Miranda, P.M.; Pigrau Pastor, M.; Sidani, S.; et al. Transplantation of Fecal Microbiota from Patients with Irritable Bowel Syndrome Alters Gut Function and Behavior in Recipient Mice. Sci. Transl. Med. 2017, 9, eaaf6397. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, P.H.; Hilpüsch, F.; Cavanagh, J.P.; Leikanger, I.S.; Kolstad, C.; Valle, P.C.; Goll, R. Faecal Microbiota Transplantation versus Placebo for Moderate-to-Severe Irritable Bowel Syndrome: A Double-Blind, Randomised, Placebo-Controlled, Parallel-Group, Single-Centre Trial. Lancet Gastroenterol. Hepatol. 2018, 3, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Koh, H.; Patatanian, M.; Reyes-Caballero, H.; Zhao, N.; Meinert, J.; Holbrook, J.T.; Leinbach, L.I.; Biswal, S. The Mediating Roles of the Oral Microbiome in Saliva and Subgingival Sites between E-Cigarette Smoking and Gingival Inflammation. BMC Microbiol. 2023, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Khamash, D.F.; Koh, H.; Voskertchian, A.; Egbert, E.; Mongodin, E.F.; White, J.R.; Hittle, L.; Colantuoni, E.; Milstone, A.M. Low Diversity in Nasal Microbiome Associated with Staphylococcus Aureus Colonization and Bloodstream Infections in Hospitalized Neonates. Open Forum Infect Dis 2021, 8, ofab475. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Chen, J.; Carroll, I.M.; Ringel-Kulka, T.; Epstein, M.P.; Zhou, H.; Zhou, J.J.; Ringel, Y.; Li, H.; Wu, M.C. Testing in Microbiome-Profiling Studies with MiRKAT, the Microbiome Regression-Based Kernel Association Test. Am. J. Hum. Genet. 2015, 96, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.; Blaser, M.J.; Li, H. A Powerful Microbiome-Based Association Test and a Microbial Taxa Discovery Framework for Comprehensive Association Mapping. Microbiome 2017, 5, 45. [Google Scholar] [CrossRef]
- Koh, H. An Adaptive Microbiome α-Diversity-Based Association Analysis Method. Sci Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Koh, H.; Zhao, N. A Powerful Microbial Group Association Test Based on the Higher Criticism Analysis for Sparse Microbial Association Signals. Microbiome 2020, 8, 63. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of Composition of Microbiomes: A Novel Method for Studying Microbial Composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A Web-Based Tool for Comprehensive Statistical, Visual and Meta-Analysis of Microbiome Data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Xia, J.; Liu, Y.; Zhou, Y.; Guo, A.C.; Cruz, J.A.; Sinelnikov, I.; Budwill, K.; Nesbø, C.L.; Wishart, D.S. METAGENassist: A Comprehensive Web Server for Comparative Metagenomics. Nucleic Acids Res. 2012, 40, W88–W95. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S rRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.; Liou, D.; Dommer, J.; MacMenamin, P.; Quiñones, M.; Misner, I.; Oler, A.J.; Wan, J.; Kim, L.; McCarthy, M.C.; et al. Nephele: A Cloud Platform for Simplified, Standardized and Reproducible Microbiome Data Analysis. Bioinformatics 2018, 34, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Navas-Molina, J.A.; Kosciolek, T.; McDonald, D.; Vázquez-Baeza, Y.; Ackermann, G.; DeReus, J.; Janssen, S.; Swafford, A.D.; Orchanian, S.B.; et al. Qiita: Rapid, Web-Enabled Microbiome Meta-Analysis. Nat. Methods 2018, 15, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; Ronas, J.; Dao, C.; Freise, A.C.; Mangul, S.; Shapiro, C.; Moberg-Parker, J. PUMAA: A Platform for Accessible Microbiome Analysis in the Undergraduate Classroom. Front. Microbiol. 2020, 11, 584699. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Moon, J.; Chisina, C.; Kang, B.; Park, T.; Koh, H. MiCloud: A Unified Web Platform for Comprehensive Microbiome Data Analysis. PLoS ONE 2022, 17, e0272354. [Google Scholar] [CrossRef]
- Jang, H.; Park, S.; Koh, H. Comprehensive Microbiome Causal Mediation Analysis Using MiMed on User-Friendly Web Interfaces. Biol. Methods Protoc. 2023, 8, bpad023. [Google Scholar] [CrossRef]
- Kim, J.; Koh, H. MiTree: A Unified Web Cloud Analytic Platform for User-Friendly and Interpretable Microbiome Data Mining Using Tree-Based Methods. Microorganisms 2023, 11, 2816. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; ACM: San Francisco, CA, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Kruskal, W.H.; Wallis, W.A. Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Dunn, O.J. Multiple Comparisons among Means. J. Am. Stat. Assoc. 1961, 56, 52–64. [Google Scholar] [CrossRef]
- McCullagh, P. Regression Models for Ordinal Data. J. R. Stat. Soc. Ser. B Methodol. 1980, 42, 109–127. [Google Scholar] [CrossRef]
- Jiang, Z.; He, M.; Chen, J.; Zhao, N.; Zhan, X. MiRKAT-MC: A Distance-Based Microbiome Kernel Association Test with Multi-Categorical Outcomes. Front. Genet. 2022, 13, 841764. [Google Scholar] [CrossRef]
- Anderson, M.J. A New Method for Non-parametric Multivariate Analysis of Variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- McArdle, B.H.; Anderson, M.J. Fitting Multivariate Models to Community Data: A Comment on Distance-Based Redundancy Analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Fisher, R.A.; Corbet, A.S.; Williams, C.B. The Relation between the Number of Species and the Number of Individuals in a Random Sample of an Animal Population. J. Anim. Ecol. 1943, 12, 42–58. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Chao, A.; Lee, S.-M. Estimating the Number of Classes via Sample Coverage. J. Am. Stat. Assoc. 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Lee, S.M.; Chao, A. Estimating Population Size via Sample Coverage for Closed Capture-Recapture Models. Biometrics 1994, 50, 88–97. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Jaccard, P. The Distribution of the Flora in the Alpine zone. New Phytol 1912, 11, 37–50. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating Microbiome Composition with Environmental Covariates Using Generalized UniFrac Distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and Qualitative β Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, J. The Statistical Analysis of Compositional Data. J. R. Stat. Soc. Ser. B Methodol. 1982, 44, 139–160. [Google Scholar] [CrossRef]
- Sanders, H.L. Marine Benthic Diversity: A Comparative Study. Am. Nat. 1968, 102, 243–282. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Variable | Ordinal Variable | |
|---|---|---|
| Covariate-adjusted analysis | Multinomial logistic regression | Proportional odds model |
| Unadjusted analysis | ANOVA F-test and Tukey’s test, Kruskal–Wallis and Dunn’s test, Multinomial logistic regression | Proportional odds model |
| Nominal Variable | Ordinal Variable | |
|---|---|---|
| Covariate-adjusted analysis | MiRKAT-MC | MiRKAT-MC |
| Unadjusted analysis | PERMANOVA MiRKAT-MC | MiRKAT-MC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Jang, H.; Koh, H. MiMultiCat: A Unified Cloud Platform for the Analysis of Microbiome Data with Multi-Categorical Responses. Bioengineering 2024, 11, 60. https://doi.org/10.3390/bioengineering11010060
Kim D, Jang H, Koh H. MiMultiCat: A Unified Cloud Platform for the Analysis of Microbiome Data with Multi-Categorical Responses. Bioengineering. 2024; 11(1):60. https://doi.org/10.3390/bioengineering11010060
Chicago/Turabian StyleKim, Donghwan, Hyojung Jang, and Hyunwook Koh. 2024. "MiMultiCat: A Unified Cloud Platform for the Analysis of Microbiome Data with Multi-Categorical Responses" Bioengineering 11, no. 1: 60. https://doi.org/10.3390/bioengineering11010060
APA StyleKim, D., Jang, H., & Koh, H. (2024). MiMultiCat: A Unified Cloud Platform for the Analysis of Microbiome Data with Multi-Categorical Responses. Bioengineering, 11(1), 60. https://doi.org/10.3390/bioengineering11010060