


Isolation and Biological Evaluation of Alfa-Mangostin as Potential Therapeutic Agents against Liver Fibrosis

 , , ,
, , ,
Abstract
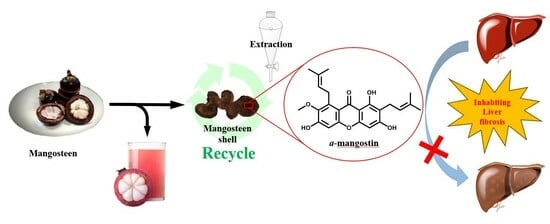
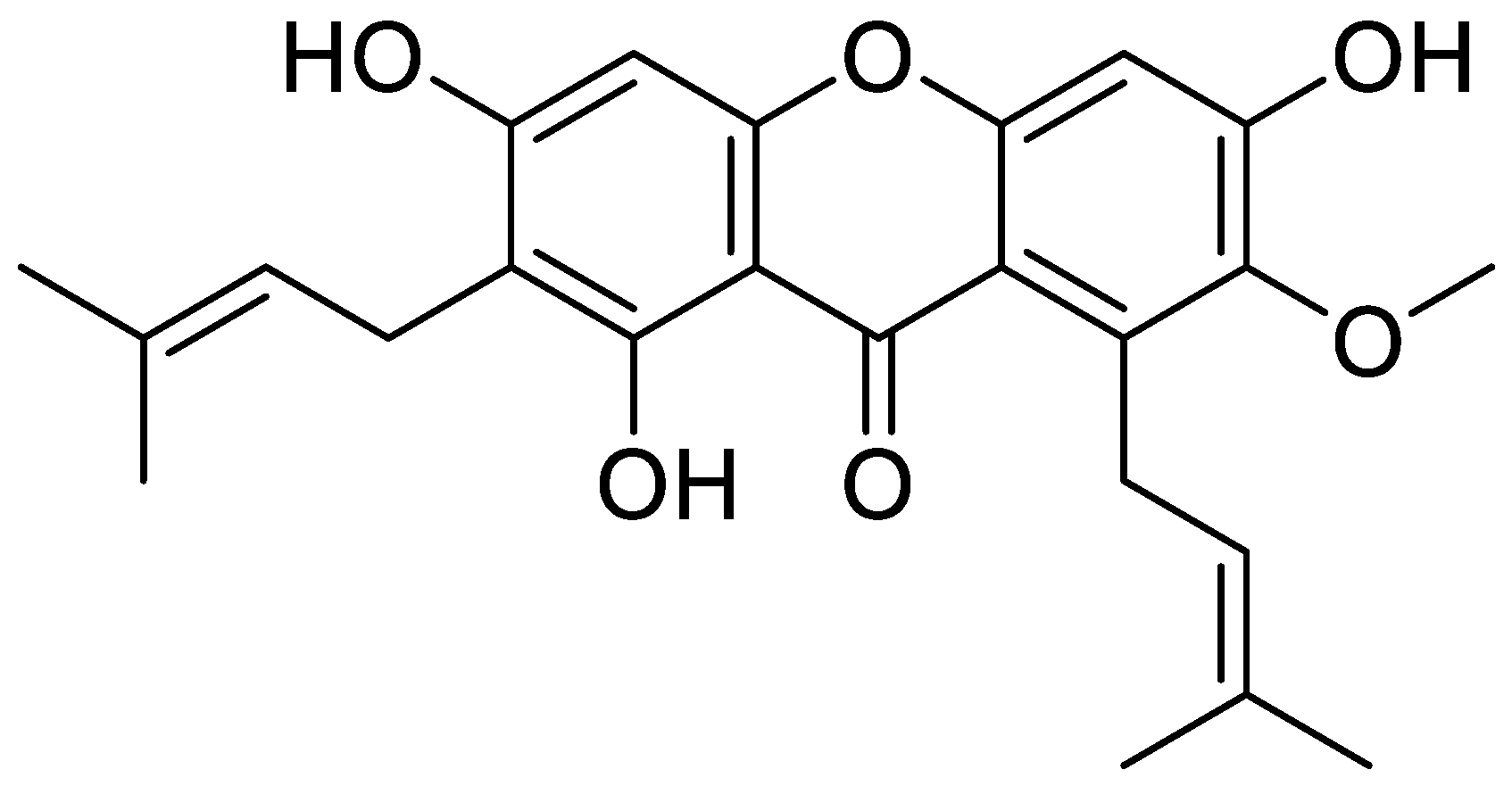
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
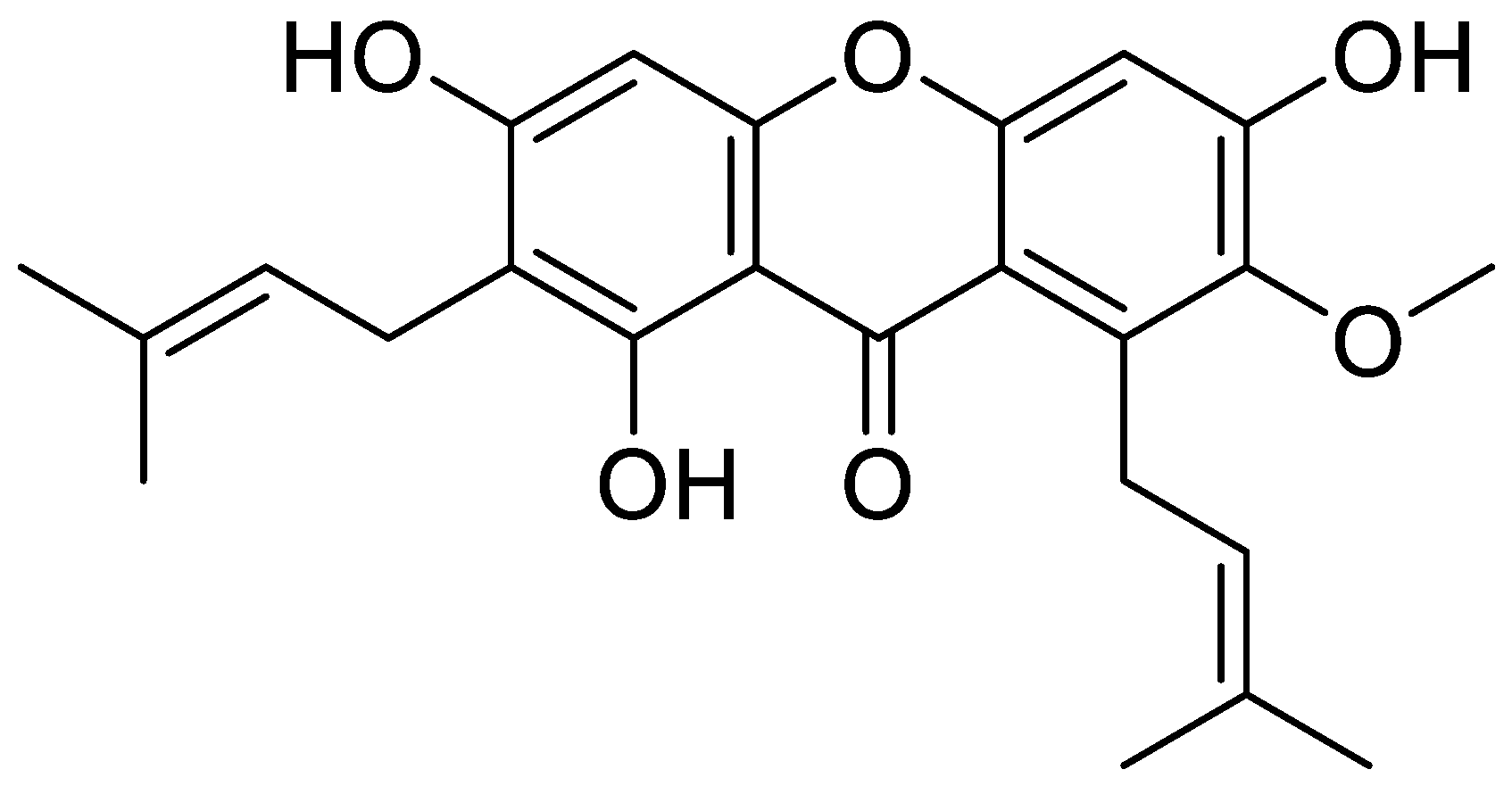
2.2. Isolation and Purification of α-Mangostin from Mangosteen Pericarp
2.3. Cell Culture
2.4. TGF-β1, LPS, and PDGF-BB Treatments
2.5. Cell Viability Assay
2.6. Animals and Experimental Design
2.7. Western Blot and IHC Staining
2.8. Real-Time PCR
2.9. Seahorse Assay
2.10. Statistical Analyses
3. Results
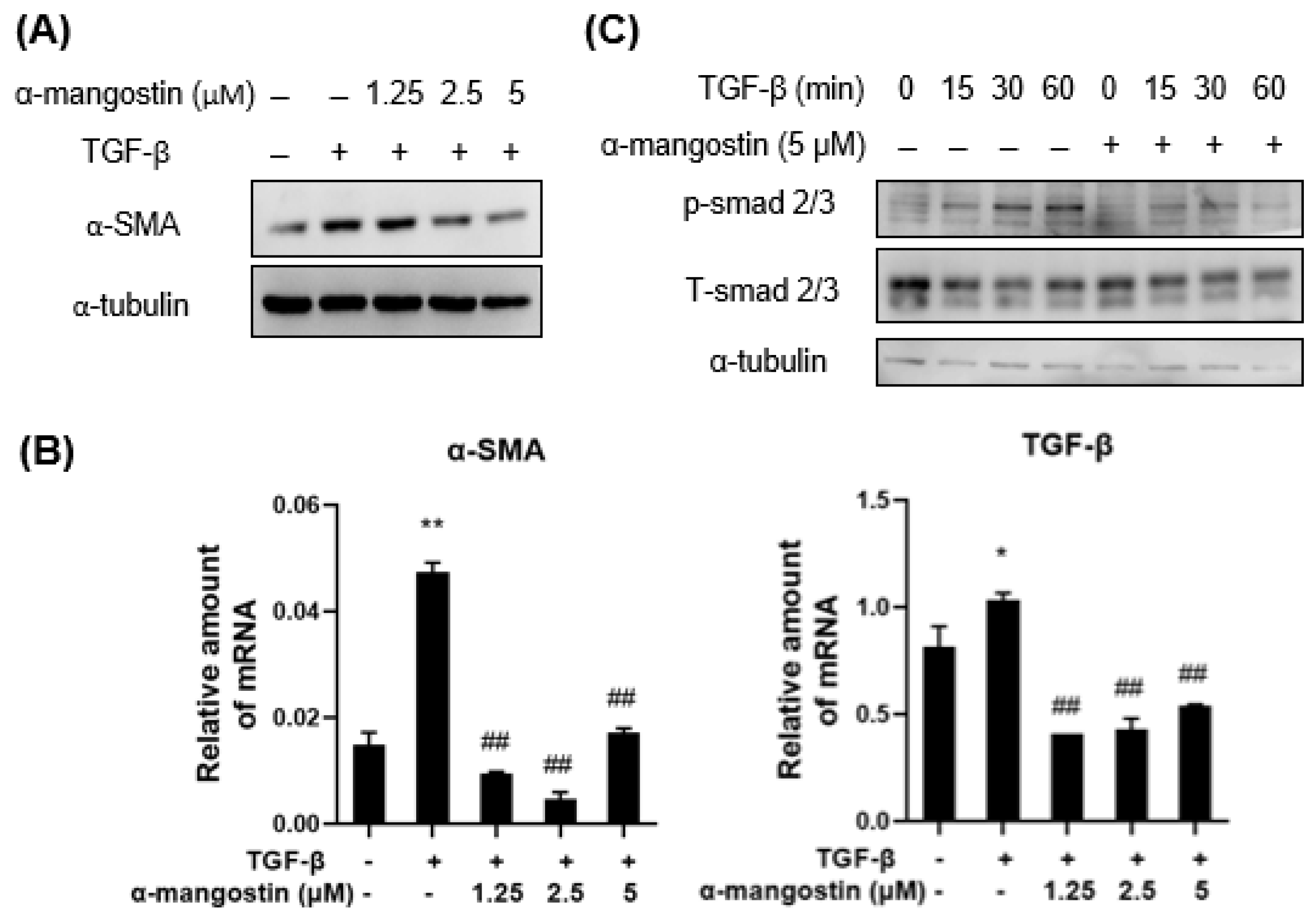
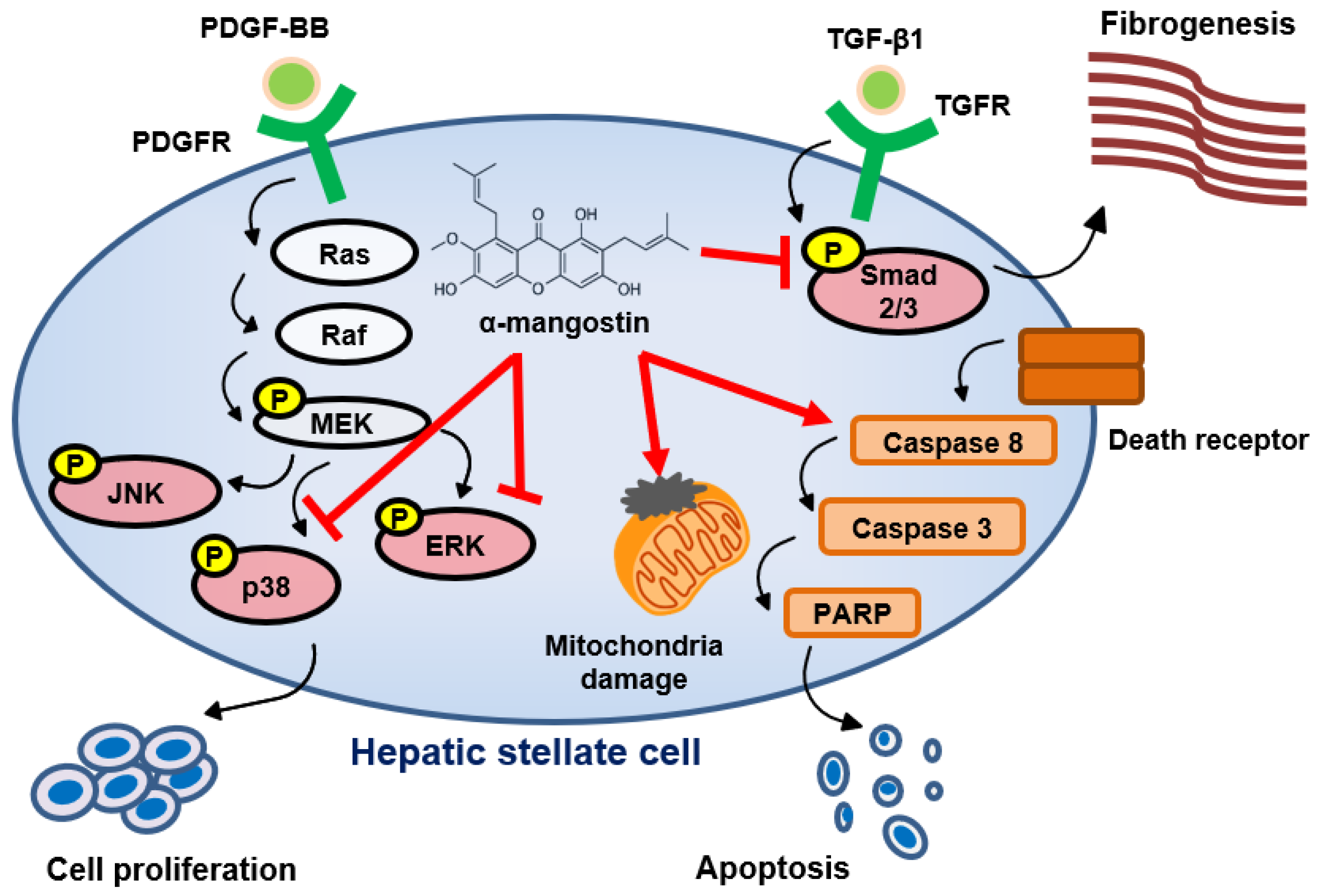
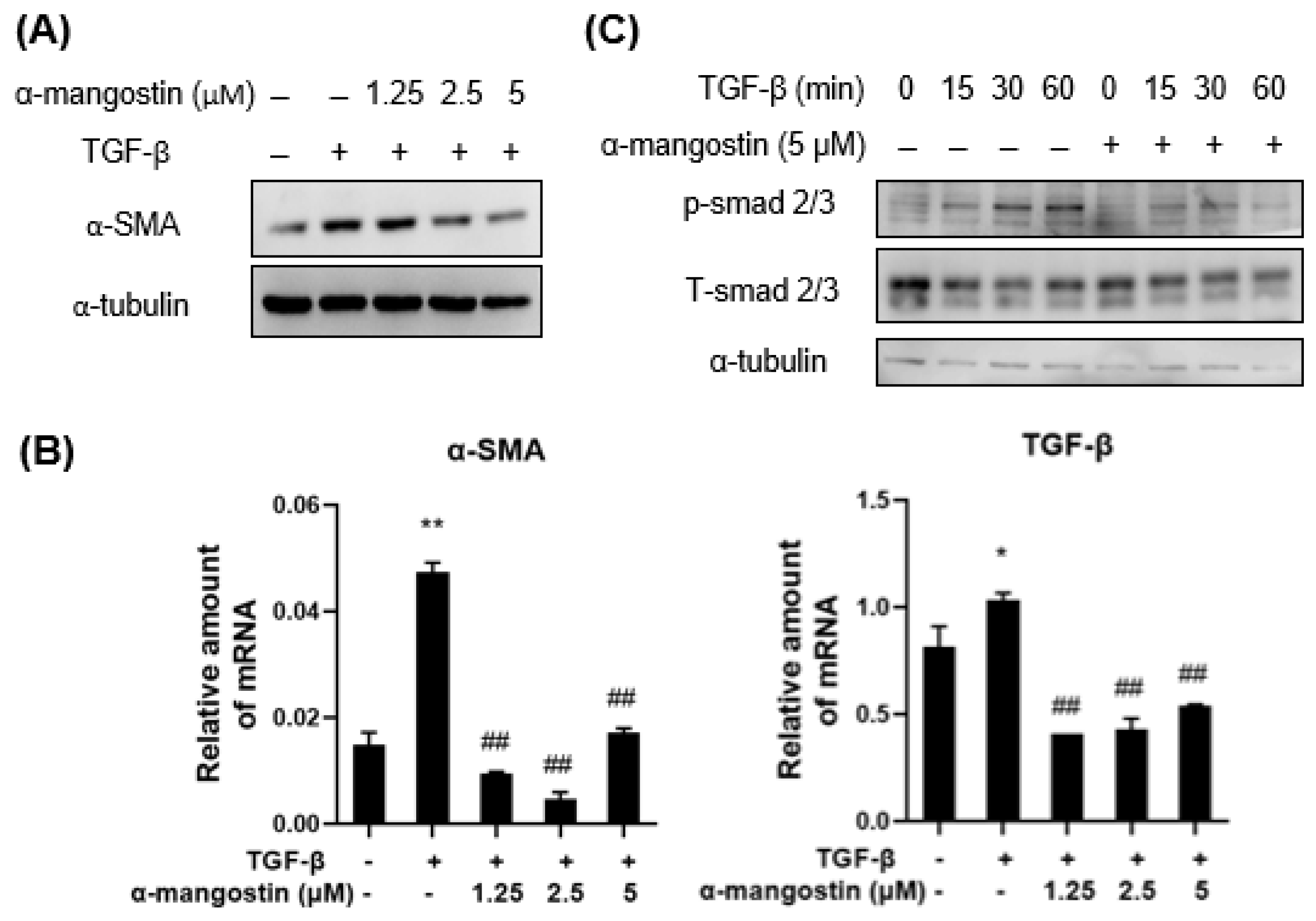
3.1. α-Mangostin Inhibits TGF-β1-Induced HSCs Activation
3.2. α-Mangostin Inhibits HSCs Proliferation through Blocking PDGF-BB-Induced ERK and p38 Signaling
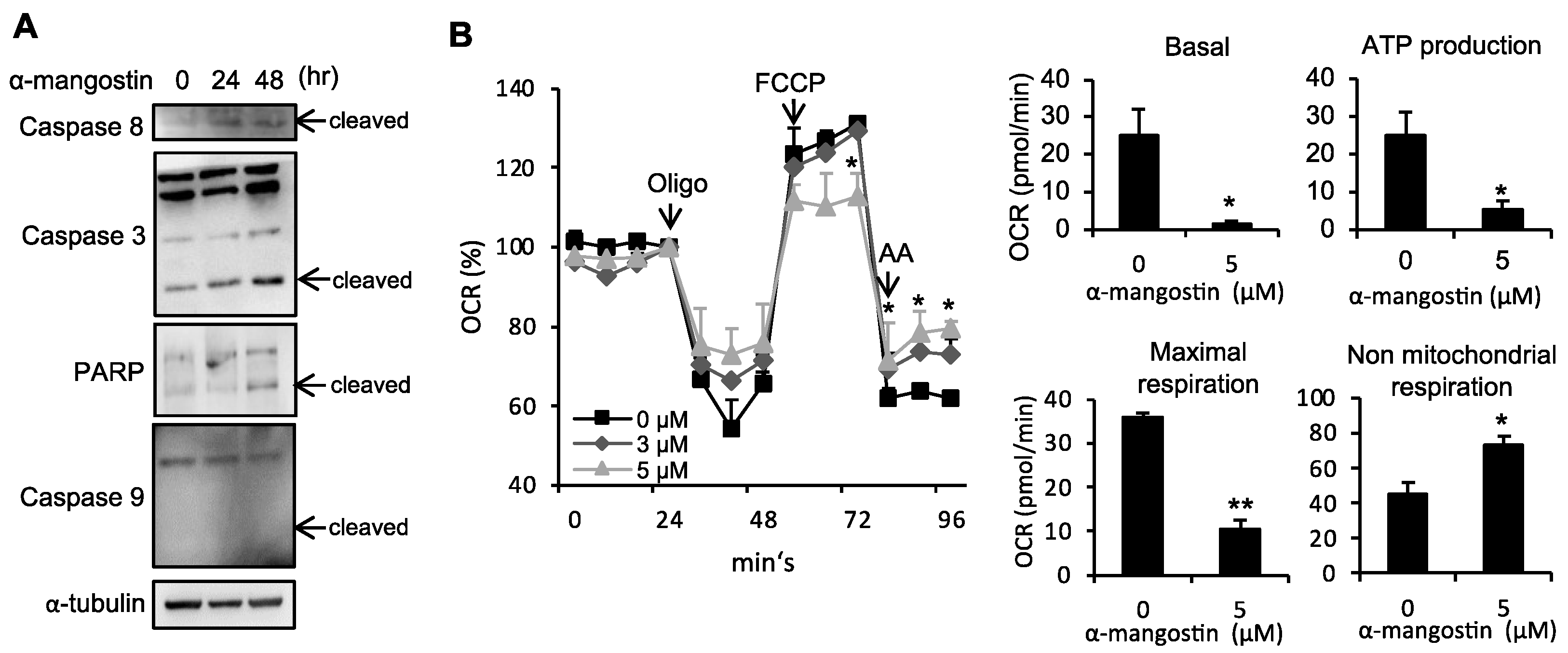
3.3. α-Mangostin Induces Apoptosis in HSCs
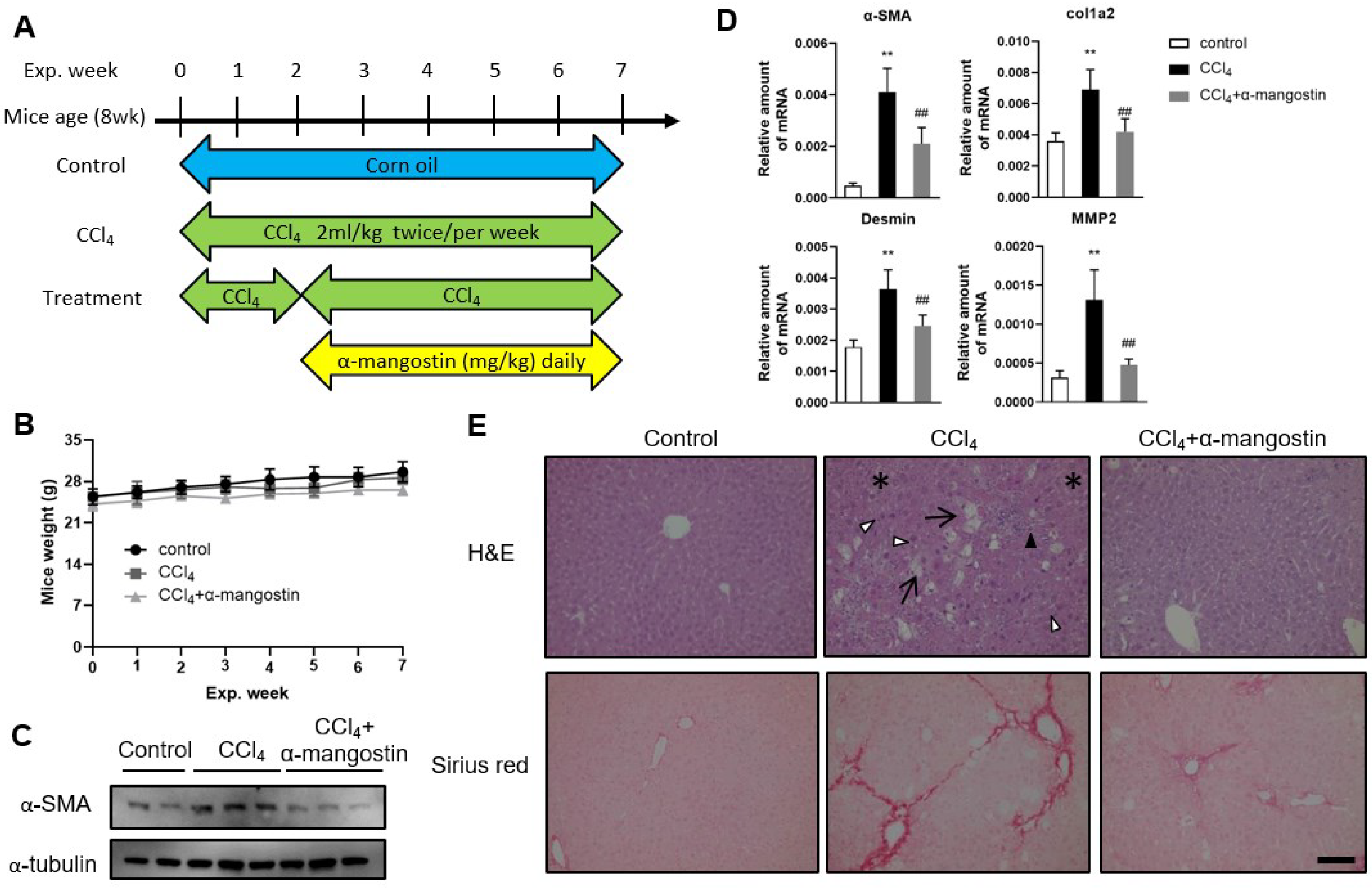
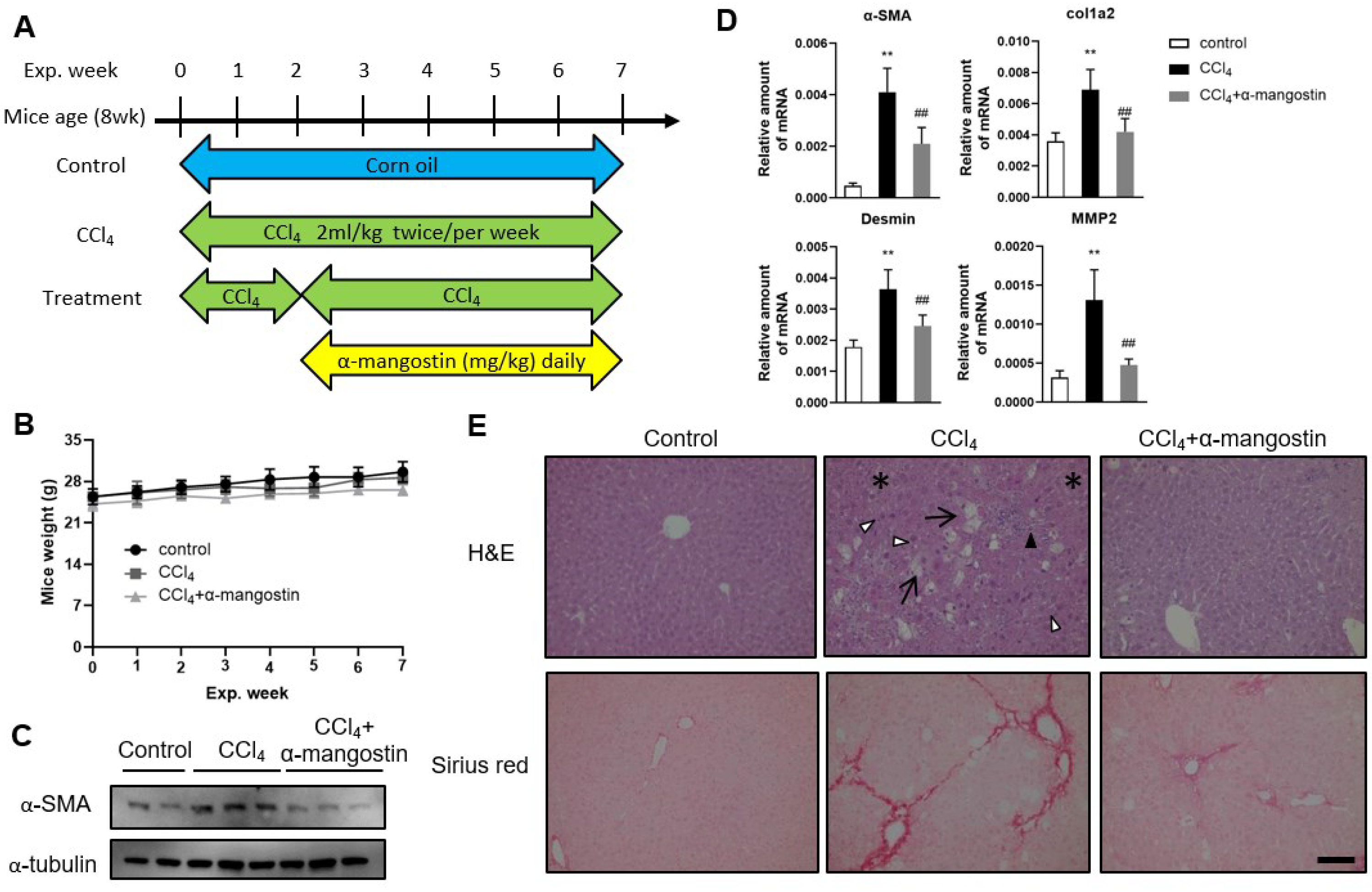
3.4. α-Mangostin Ameliorates CCl4-Induced Liver Fibrosis in Mice
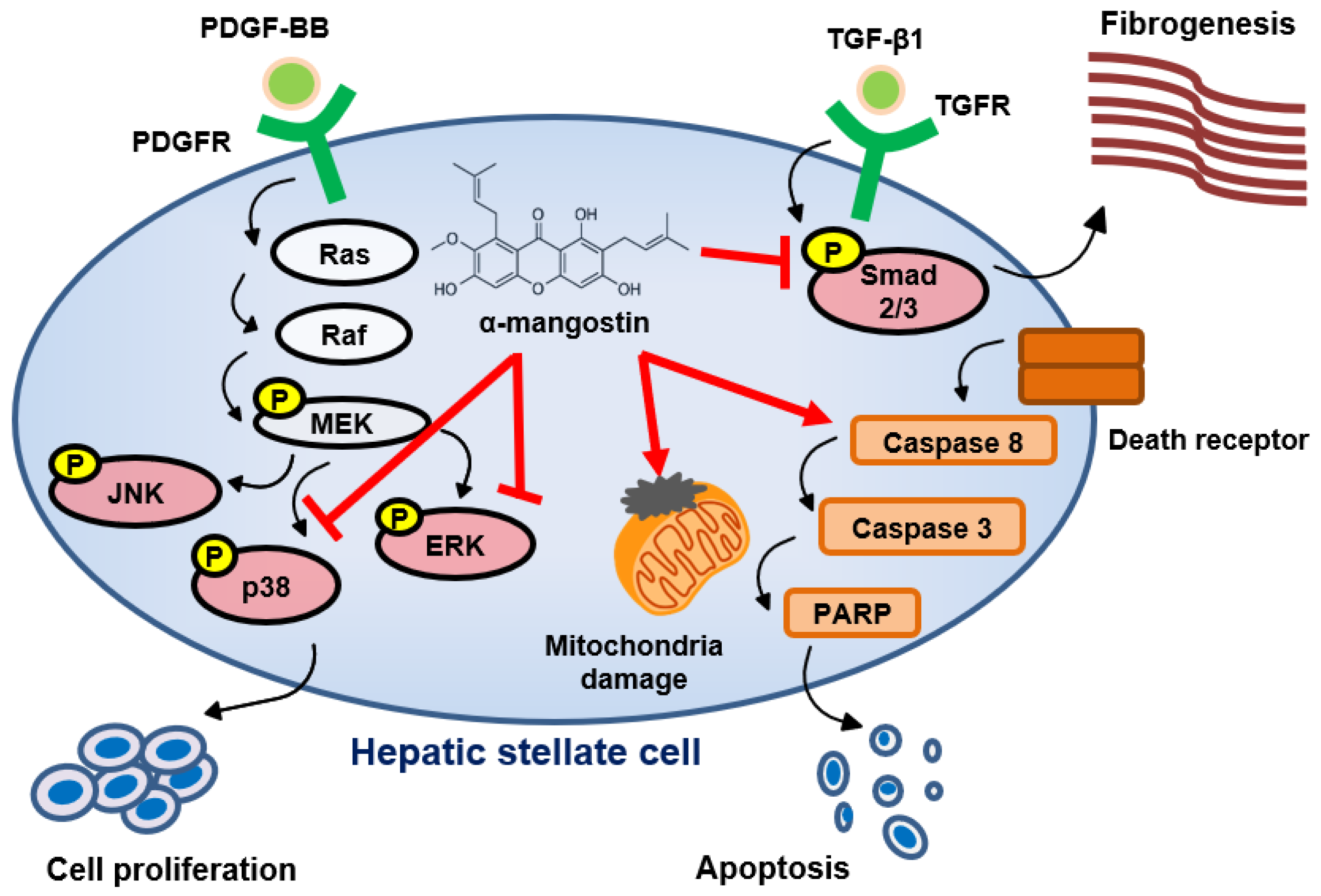
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Niu, Y.; Li, Q.; Tu, C.; Li, N.; Gao, L.; Lin, H.; Wang, Z.; Zhou, Z.; Li, L. Hypouricemic Actions of the Pericarp of Mangosteen In Vitro and In Vivo. J. Nat. Prod. 2023, 86, 24–33. [Google Scholar] [CrossRef]
- Asyifah, M.R.; Lu, K.; Ting, H.L.; Zhang, D. Hidden potential of tropical fruit waste components as a useful source of remedy for obesity. J. Agric. Food Chem. 2014, 62, 3505–3516. [Google Scholar] [CrossRef]
- Sunil Richard, A.; Verma, R.S. Antioxidant α-Mangostin Coated Woven Polycaprolactone Nanofibrous Yarn Scaffold for Cardiac Tissue Repair. ACS Appl. Nano Mater. 2022, 5, 5075–5086. [Google Scholar] [CrossRef]
- Chen, L.-G.; Yang, L.-L.; Wang, C.-C. Anti-inflammatory activity of mangostins from Garcinia mangostana. Food Chem. Toxicol. 2008, 46, 688–693. [Google Scholar] [CrossRef]
- Matsumoto, K.; Akao, Y.; Kobayashi, E.; Ohguchi, K.; Ito, T.; Tanaka, T.; Iinuma, M.; Nozawa, Y. Induction of apoptosis by xanthones from mangosteen in human leukemia cell lines. J. Nat. Prod. 2003, 66, 1124–1127. [Google Scholar] [CrossRef]
- Wu, C.-P.; Hsiao, S.-H.; Murakami, M.; Lu, Y.-J.; Li, Y.-Q.; Huang, Y.-H.; Hung, T.-H.; Ambudkar, S.V.; Wu, Y.-S. Alpha-mangostin reverses multidrug resistance by attenuating the function of the multidrug resistance-linked ABCG2 transporter. Mol. Pharm. 2017, 14, 2805–2814. [Google Scholar] [CrossRef]
- Koh, J.-J.; Qiu, S.; Zou, H.; Lakshminarayanan, R.; Li, J.; Zhou, X.; Tang, C.; Saraswathi, P.; Verma, C.; Tan, D.T.H.; et al. Rapid bactericidal action of alpha-mangostin against MRSA as an outcome of membrane targeting. Biochim. Biophys. Acta BBA Biomembr. 2013, 1828, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Sin, W.L.W.; Koh, J.-J.; Lim, F.; Wang, L.; Cao, D.; Beuerman, R.W.; Ren, L.; Liu, S. Semisynthesis and biological evaluation of xanthone amphiphilics as selective, highly potent antifungal agents to combat fungal resistance. J. Med. Chem. 2017, 60, 10135–10150. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Huang, Y.-Y.; Zhou, Q.; Gao, Y.; Li, Z.; Wu, D.; Yu, S.; Guo, L.; Chen, Z.; Huang, L. Discovery and optimization of α-mangostin derivatives as novel PDE4 inhibitors for the treatment of vascular dementia. J. Med. Chem. 2020, 63, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bian, Y.; Wang, J.-W.; Gong, T.-T.; Ying, Y.-M.; Ma, L.-F.; Shan, W.-G.; Xie, X.-Q.; Zhan, Z.-J. Effects of α-mangostin derivatives on the Alzheimer’s disease model of rats and their mechanism: A combination of experimental study and computational systems pharmacology analysis. ACS Omega 2020, 5, 9846–9863. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38 (Suppl. 1), S38–S53. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P.; Thompson, A.; Henderson, N.C. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim. Biophys. Acta 2013, 1832, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Investig. 2007, 117, 539–548. [Google Scholar] [CrossRef]
- Forbes, S.J.; Parola, M. Liver fibrogenic cells. Best Pract. Research. Clin. Gastroenterol. 2011, 25, 207–217. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic fibrosis—Overview. Toxicology 2008, 254, 120–129. [Google Scholar] [CrossRef]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Majumdar, A.; Curley, S.A.; Wu, X.; Brown, P.; Hwang, J.P.; Shetty, K.; Yao, Z.X.; He, A.R.; Li, S.; Katz, L.; et al. Hepatic stem cells and transforming growth factor beta in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Okazaki, I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair. Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Fallowfield, J.A. Therapeutic targets in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G709–G715. [Google Scholar] [CrossRef]
- Yata, Y.; Gotwals, P.; Koteliansky, V.; Rockey, D.C. Dose-dependent inhibition of hepatic fibrosis in mice by a TGF-beta soluble receptor: Implications for antifibrotic therapy. Hepatology 2002, 35, 1022–1030. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef]
- Liu, Y.; Wen, X.M.; Lui, E.L.; Friedman, S.L.; Cui, W.; Ho, N.P.; Li, L.; Ye, T.; Fan, S.T.; Zhang, H. Therapeutic targeting of the PDGF and TGF-beta-signaling pathways in hepatic stellate cells by PTK787/ZK22258. Lab. Investig. 2009, 89, 1152–1160. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Xu, W.; Wang, S.; Hu, Z.; Zhang, Q.; Deng, X.; Wang, J.; Zhang, J.; Guo, C. Antifibrotic effects of luteolin on hepatic stellate cells and liver fibrosis by targeting AKT/mTOR/p70S6K and TGFbeta/Smad signalling pathways. Liver Int. 2015, 35, 1222–1233. [Google Scholar] [CrossRef]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Aoyama, T.; Paik, Y.H.; Seki, E. Toll-like receptor signaling and liver fibrosis. Gastroenterol. Res. Pract. 2010, 2010, 192543. [Google Scholar] [CrossRef]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr161. [Google Scholar] [CrossRef]
- Poelstra, K.; Schuppan, D. Targeted therapy of liver fibrosis/cirrhosis and its complications. J. Hepatol. 2011, 55, 726–728. [Google Scholar] [CrossRef]
- Elsharkawy, A.M.; Oakley, F.; Mann, D.A. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis Int. J. Program. Cell Death 2005, 10, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Zheng, W.D.; Shi, M.N.; Wang, X.Z. Effects of interleukin-10 on activation and apoptosis of hepatic stellate cells in fibrotic rat liver. World J. Gastroenterol. 2006, 12, 1918–1923. [Google Scholar] [CrossRef]
- He, L.; Hou, X.; Fan, F.; Wu, H. Quercetin stimulates mitochondrial apoptosis dependent on activation of endoplasmic reticulum stress in hepatic stellate cells. Pharm. Biol. 2016, 54, 3237–3243. [Google Scholar] [CrossRef]
- Lee, P.J.; Woo, S.J.; Jee, J.G.; Sung, S.H.; Kim, H.P. Bisdemethoxycurcumin Induces apoptosis in activated hepatic stellate cells via cannabinoid receptor 2. Molecules 2015, 20, 1277–1292. [Google Scholar] [CrossRef]
- Twu, Y.C.; Lee, T.S.; Lin, Y.L.; Hsu, S.M.; Wang, Y.H.; Liao, C.Y.; Wang, C.K.; Liang, Y.C.; Liao, Y.J. Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation. Int. J. Mol. Sci. 2016, 17, 1122. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef]
- Mason, E.F.; Rathmell, J.C. Cell metabolism: An essential link between cell growth and apoptosis. Biochim. Biophys. Acta 2011, 1813, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Neff, G.W.; Duncan, C.W.; Schiff, E.R. The current economic burden of cirrhosis. Gastroenterol. Hepatol. 2011, 7, 661–671. [Google Scholar]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Gabele, E.; Reif, S.; Tsukada, S.; Bataller, R.; Yata, Y.; Morris, T.; Schrum, L.W.; Brenner, D.A.; Rippe, R.A. The role of p70S6K in hepatic stellate cell collagen gene expression and cell proliferation. J. Biol. Chem. 2005, 280, 13374–13382. [Google Scholar] [CrossRef] [PubMed]
- Reif, S.; Lang, A.; Lindquist, J.N.; Yata, Y.; Gabele, E.; Scanga, A.; Brenner, D.A.; Rippe, R.A. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J. Biol. Chem. 2003, 278, 8083–8090. [Google Scholar] [CrossRef]
- Marra, F.; Arrighi, M.C.; Fazi, M.; Caligiuri, A.; Pinzani, M.; Romanelli, R.G.; Efsen, E.; Laffi, G.; Gentilini, P. Extracellular signal-regulated kinase activation differentially regulates platelet-derived growth factor’s actions in hepatic stellate cells, and is induced by in vivo liver injury in the rat. Hepatology 1999, 30, 951–958. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, H.; Wang, N.; Ouyang, Y.; Yi, Y.; Liao, L.; Shen, H.; Hu, G.; Wang, Z.; Tao, L. Fluorofenidone attenuates hepatic fibrosis by suppressing the proliferation and activation of hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G253–G263. [Google Scholar] [CrossRef]
- Son, M.K.; Ryu, Y.L.; Jung, K.H.; Lee, H.; Lee, H.S.; Yan, H.H.; Park, H.J.; Ryu, J.K.; Suh, J.K.; Hong, S.; et al. HS-173, a novel PI3K inhibitor, attenuates the activation of hepatic stellate cells in liver fibrosis. Sci. Rep. 2013, 3, 3470. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Chen, M.F.; Huang, S.J.; Huang, C.C.; Liu, P.S.; Lin, K.I.; Liu, C.W.; Hsieh, W.C.; Shiu, L.Y.; Chen, C.H. Saikosaponin d induces cell death through caspase-3-dependent, caspase-3-independent and mitochondrial pathways in mammalian hepatic stellate cells. BMC Cancer 2016, 16, 532. [Google Scholar] [CrossRef]
- Pan, Q.; Luo, Y.; Xia, Q.; He, K. Ferroptosis and Liver Fibrosis. Int. J. Med. Sci. 2021, 18, 3361–3366. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Liu, R.; Cheng, Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 2019, 109, 2043–2053. [Google Scholar] [CrossRef]
- Shan, L.; Wang, F.; Zhai, D.; Meng, X.; Liu, J.; Lv, X. New Drugs for Hepatic Fibrosis. Front. Pharmacol. 2022, 13, 874408. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Han, X.; Hao, M.; Yang, Q.; Lyu, Q.; Tang, D.; Shen, Z.; Wang, K.; Kuang, H.; Cao, G.; et al. Nanodrug rescues liver fibrosis via synergistic therapy with H2O2 depletion and Saikosaponin b1 sustained release. Commun. Biol. 2023, 6, 184. [Google Scholar] [CrossRef] [PubMed]
- Wathoni, N.; Rusdin, A.; Motoyama, K.; Joni, I.M.; Lesmana, R.; Muchtaridi, M. Nanoparticle drug delivery systems for mangostin. Nanotechnol. Sci. Appl. 2020, 13, 23–26. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, Y.-J.; Lee, C.-Y.; Twu, Y.-C.; Suk, F.-M.; Lai, T.-C.; Chang, Y.-C.; Lai, Y.-C.; Yuan, J.-W.; Jhuang, H.-M.; Jian, H.-R.; et al. Isolation and Biological Evaluation of Alfa-Mangostin as Potential Therapeutic Agents against Liver Fibrosis. Bioengineering 2023, 10, 1075. https://doi.org/10.3390/bioengineering10091075
Liao Y-J, Lee C-Y, Twu Y-C, Suk F-M, Lai T-C, Chang Y-C, Lai Y-C, Yuan J-W, Jhuang H-M, Jian H-R, et al. Isolation and Biological Evaluation of Alfa-Mangostin as Potential Therapeutic Agents against Liver Fibrosis. Bioengineering. 2023; 10(9):1075. https://doi.org/10.3390/bioengineering10091075
Chicago/Turabian StyleLiao, Yi-Jen, Chun-Ya Lee, Yuh-Ching Twu, Fat-Moon Suk, Tzu-Chieh Lai, Ya-Ching Chang, Yi-Cheng Lai, Jing-Wei Yuan, Hong-Ming Jhuang, Huei-Ruei Jian, and et al. 2023. "Isolation and Biological Evaluation of Alfa-Mangostin as Potential Therapeutic Agents against Liver Fibrosis" Bioengineering 10, no. 9: 1075. https://doi.org/10.3390/bioengineering10091075
APA StyleLiao, Y.-J., Lee, C.-Y., Twu, Y.-C., Suk, F.-M., Lai, T.-C., Chang, Y.-C., Lai, Y.-C., Yuan, J.-W., Jhuang, H.-M., Jian, H.-R., Huang, L.-C., Chen, K.-P., & Hsu, M.-H. (2023). Isolation and Biological Evaluation of Alfa-Mangostin as Potential Therapeutic Agents against Liver Fibrosis. Bioengineering, 10(9), 1075. https://doi.org/10.3390/bioengineering10091075