
Discovery and Characteristics of a Novel Antitumor Cyclopeptide Derived from Shark

 , , and
, , and
Abstract

1. Introduction
2. Results
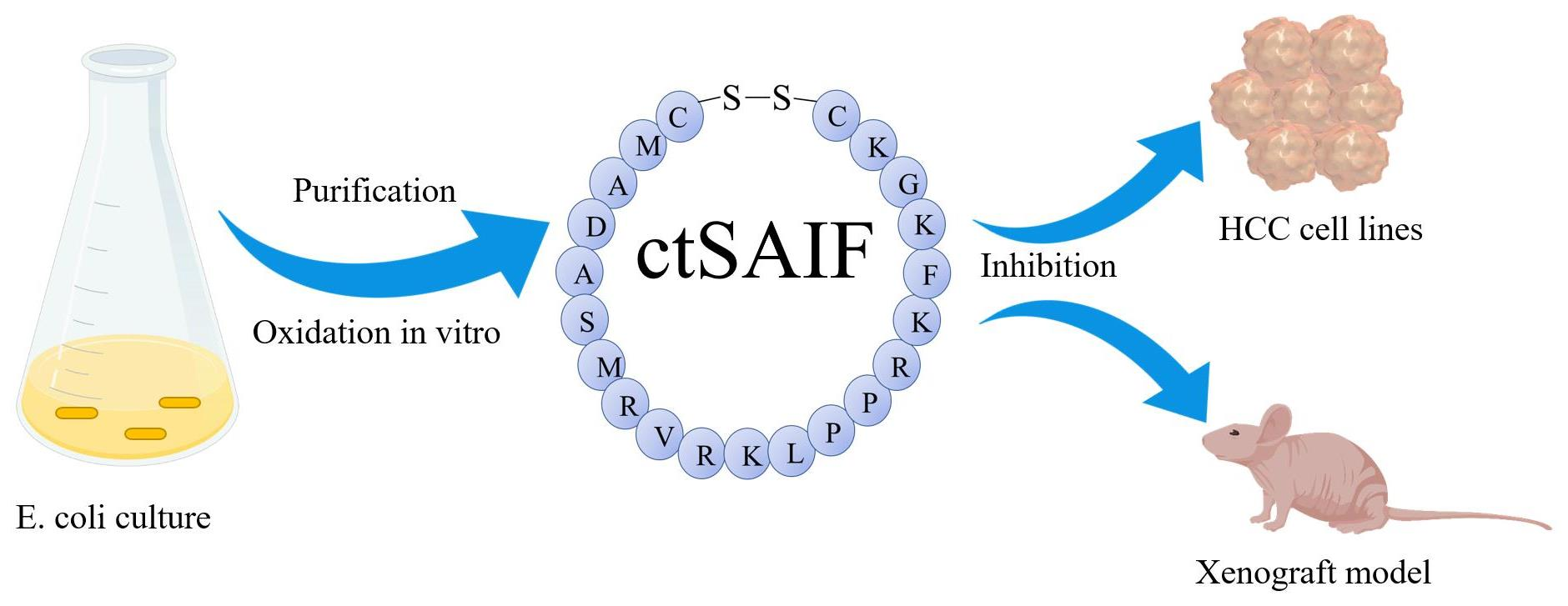
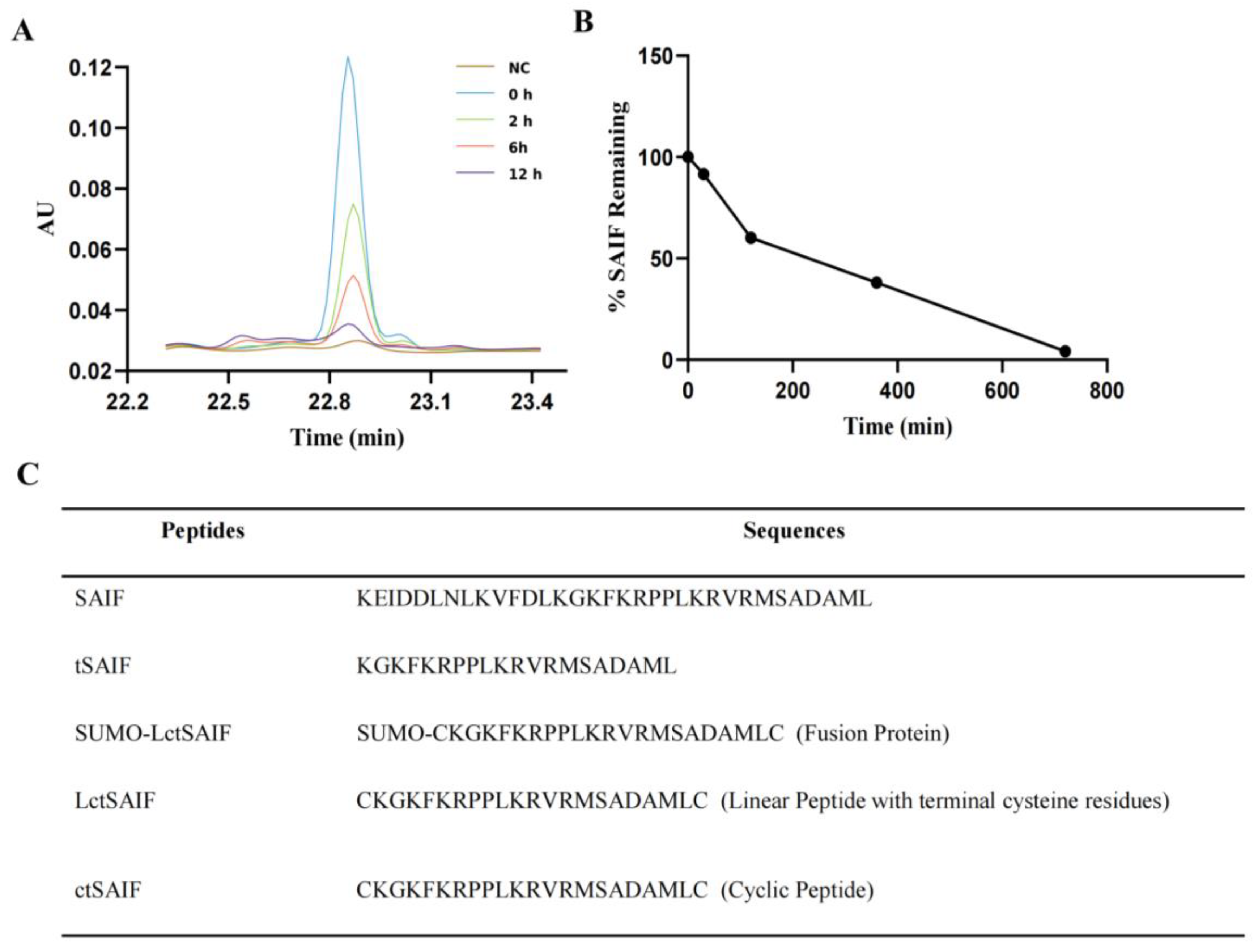
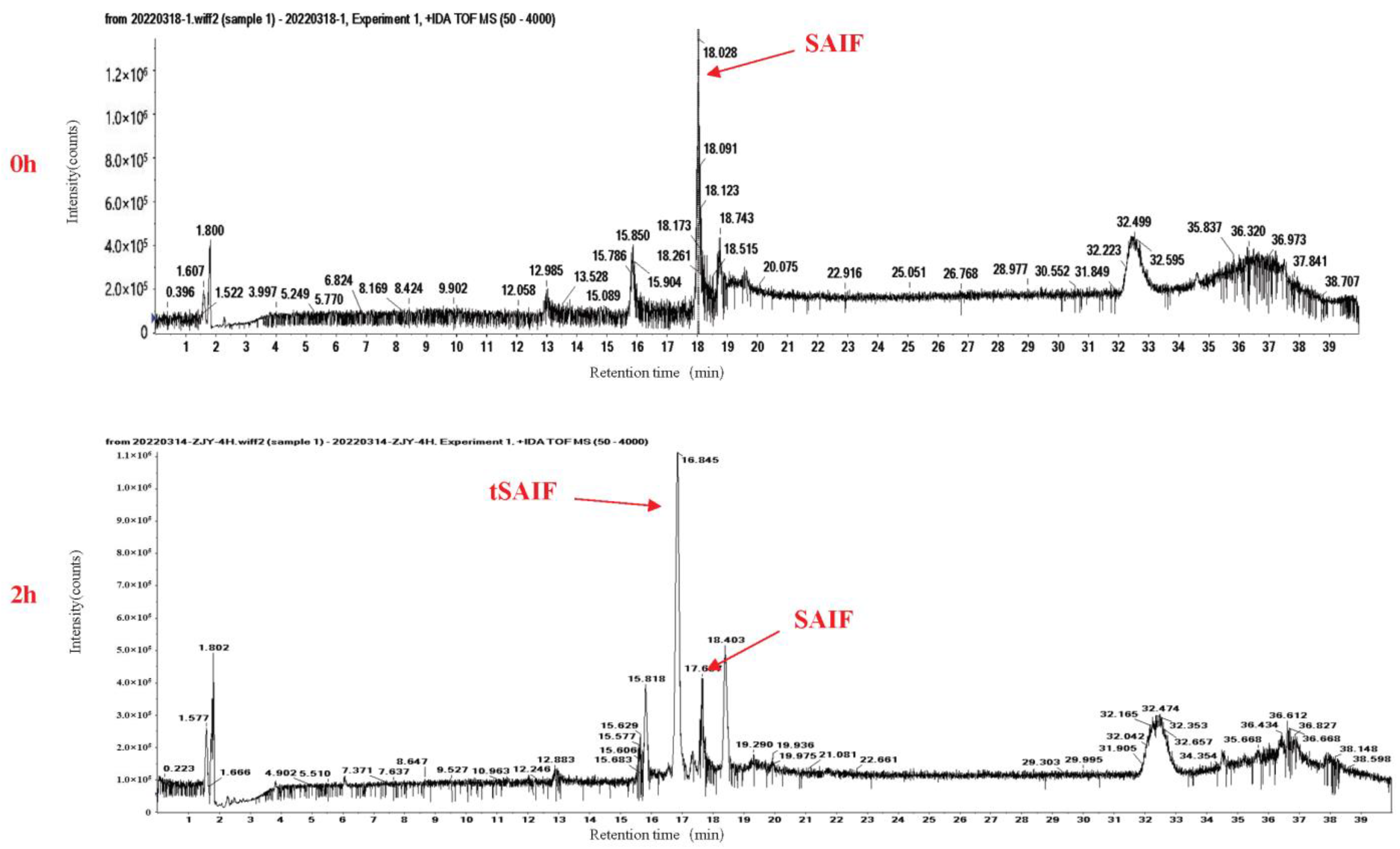
2.1. The Analysis and Obtaining of the ctSAIF Peptide Sequence
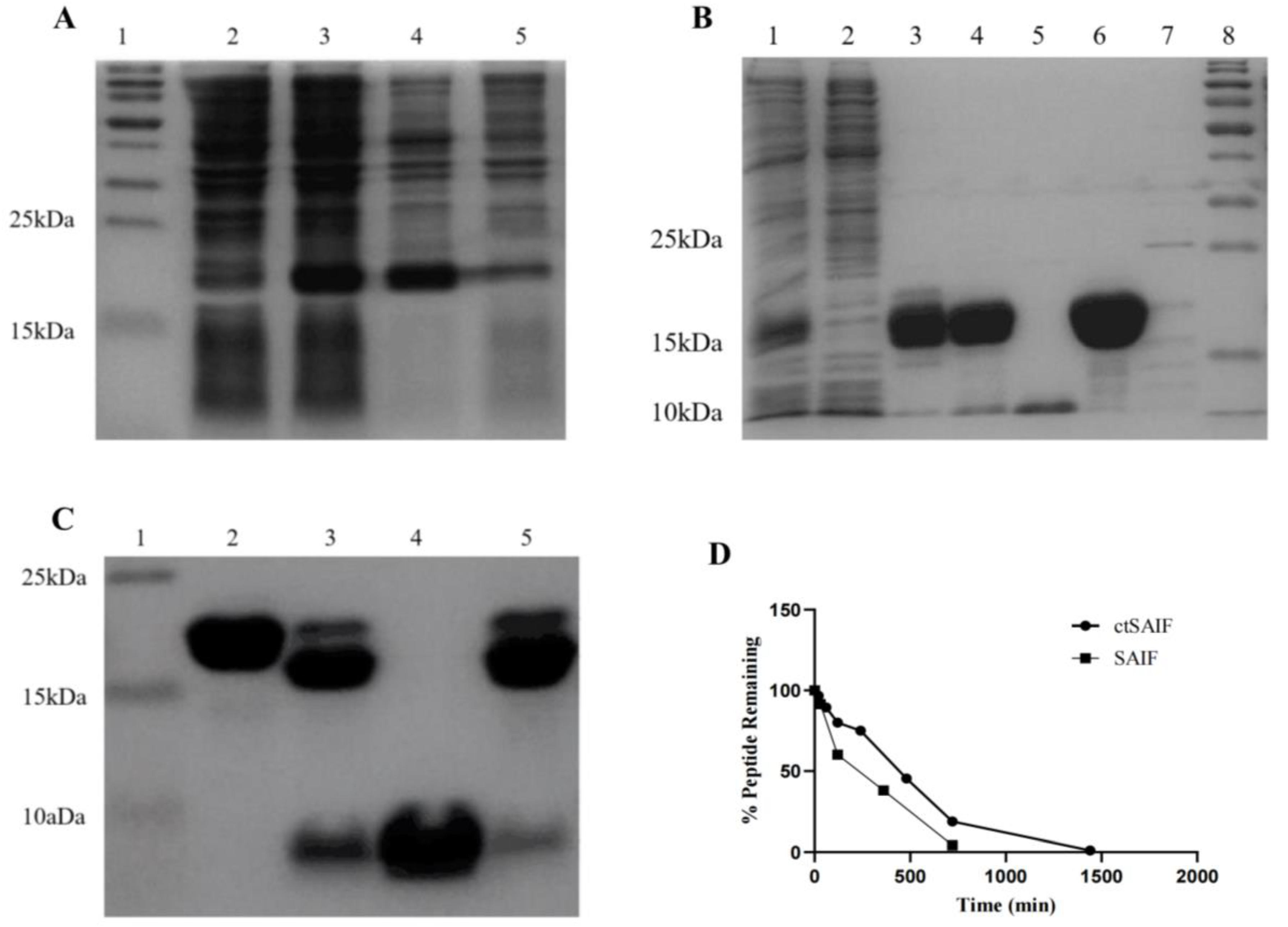
2.2. Expression, Purification and Characterization of LctSAIF in E. coli
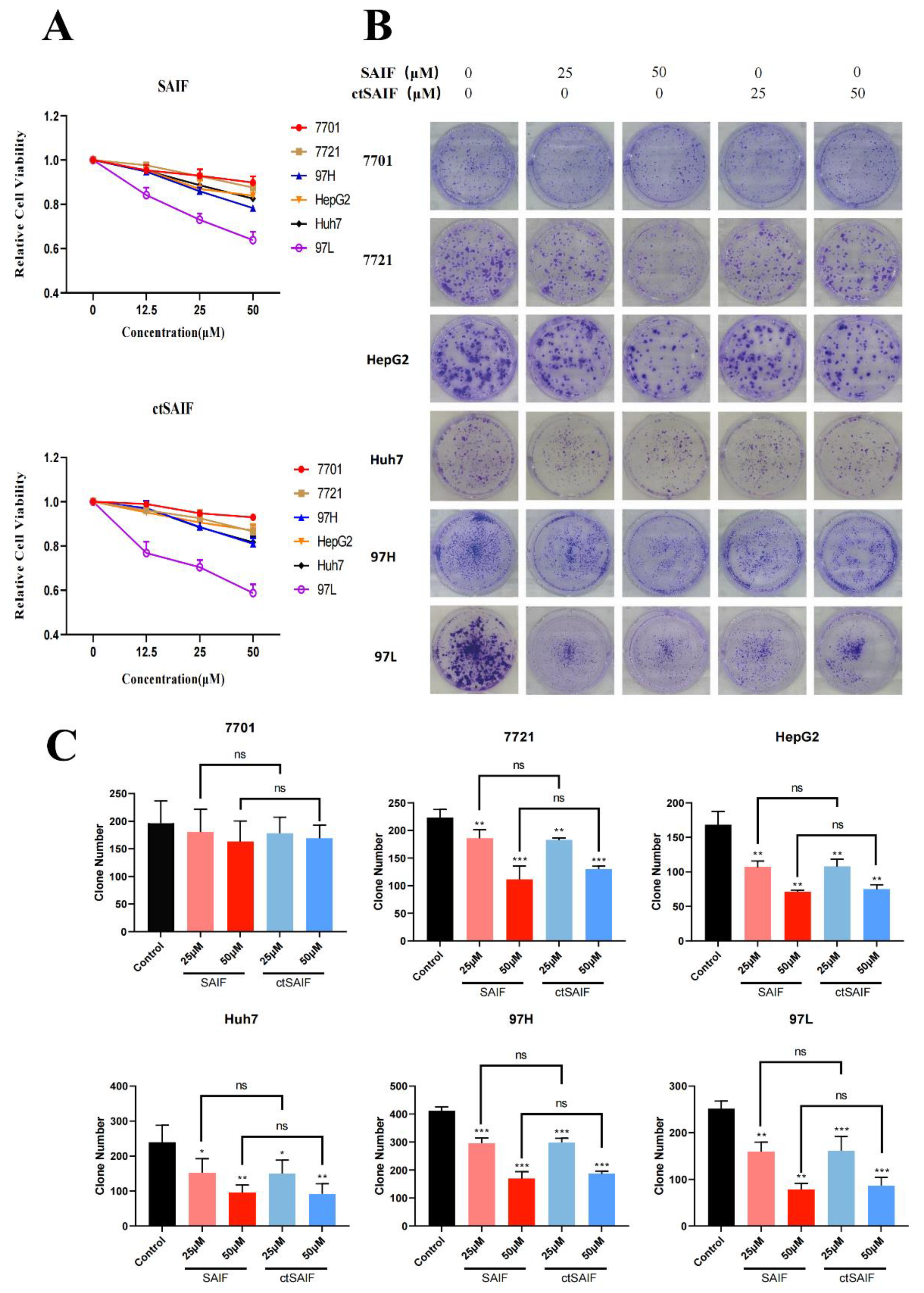
2.3. Inhibition of Hepatocellular Carcinoma Cell Proliferation Using ctSAIF
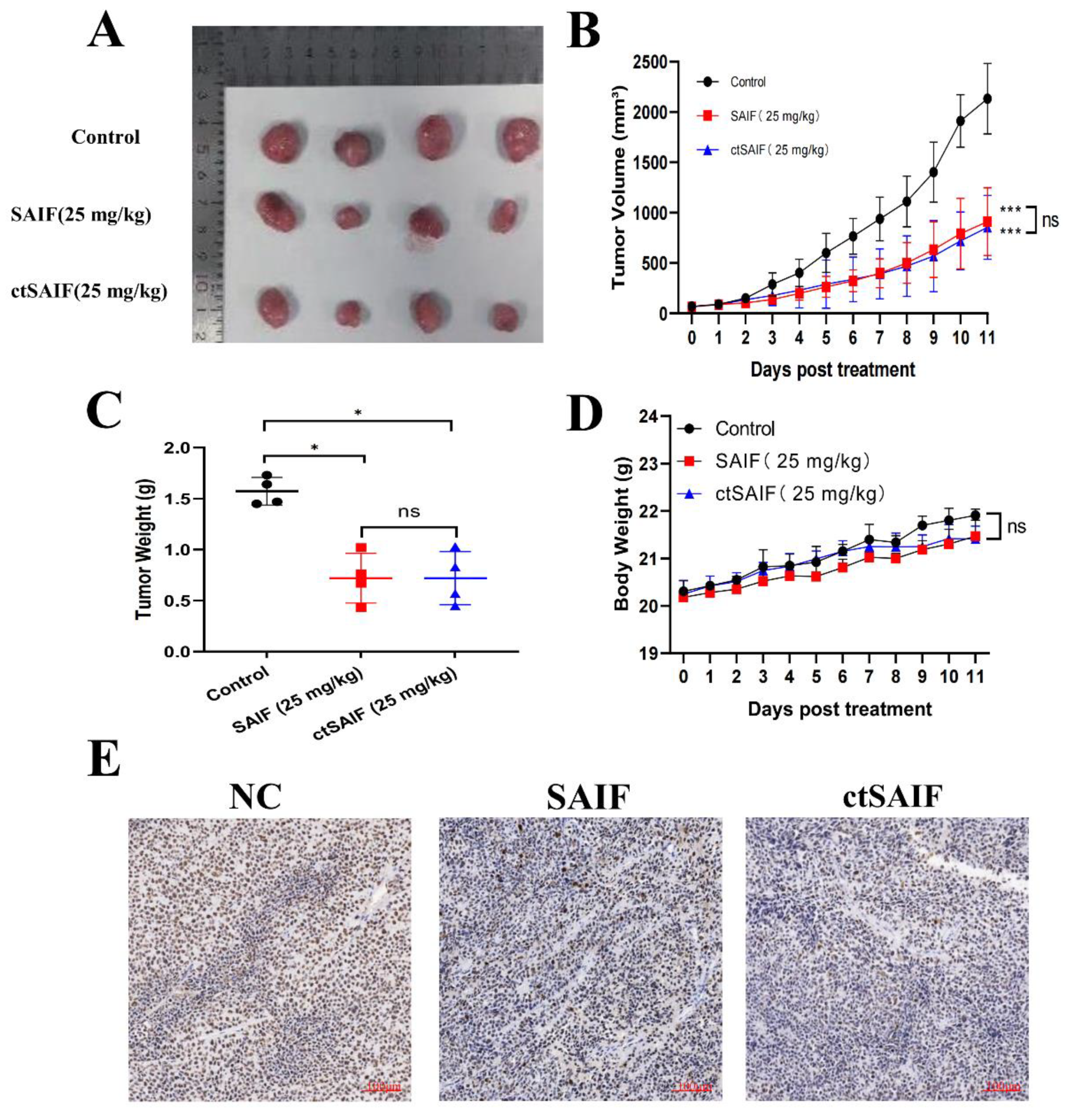
2.4. Inhibition of Liver Cancer Transplant Growth Using ctSAIF
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Materials
4.3. Cell Proliferation Assay (CCK-8 Assay)
4.4. Cell Colony Formation Assay
4.5. In Vitro Serum Stability Assay of Peptides
4.6. Mass Spectrometry Analysis
4.7. Construction of Recombinant Expression Vector
4.8. Expression and Purification of Cyclic Peptide ctSAIF
4.9. Xenograft Model
4.10. Histopathological Examination
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Ferry, D.R.; El-Gazzaz, G.; Mirza, D.F.; James, N.D.; McMaster, P.; Kerr, D.J. Hepatocellular carcinoma. Ann. Oncol. 2001, 12, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Hernandez–Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef]
- Zhu, A.X.; Sahani, D.V.; Duda, D.G.; Di Tomaso, E.; Ancukiewicz, M.; Catalano, O.A.; Sindhwani, V.; Blaszkowsky, L.S.; Yoon, S.S.; Lahdenranta, J. Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: A phase II study. J. Clin. Oncol. 2009, 27, 3027. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, H.; Meng, N.; Lu, W.; Li, G.; Han, Y.; Dai, X.; Xia, Y.; Song, X.; Yang, S. YL529, a novel, orally available multikinase inhibitor, potently inhibits angiogenesis and tumour growth in preclinical models. Br. J. Pharmacol. 2013, 169, 1766–1780. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Marx, V. Watching peptide drugs grow up. Chem. Eng. News 2005, 83, 17–24. [Google Scholar]
- Madsbad, S.; Kielgast, U.; Asmar, M.; Deacon, C.F.; Torekov, S.S.; Holst, J. An overview of once-weekly glucagon-like peptide-1 receptor agonists—Available efficacy and safety data and perspectives for the future. Diabetes Obes. Metab. 2011, 13, 394–407. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, H.; Cui, Z.; Lv, Y.; Zhang, W.; Ma, X.; Zhang, J.; Sun, B.; Zhou, D.; Yuan, L. A peptide derived from apoptin inhibits glioma growth. Oncotarget 2017, 8, 31119. [Google Scholar] [CrossRef] [PubMed]
- Langer, M.; Kratz, F.; Rothen-Rutishauser, B.; Wunderli-Allenspach, H.; Beck-Sickinger, A.G. Novel peptide conjugates for tumor-specific chemotherapy. J. Med. Chem. 2001, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef] [PubMed]
- Periti, P.; Mazzei, T.; Mini, E. Clinical pharmacokinetics of depot leuprorelin. Clin. Pharmacokinet. 2002, 41, 485–504. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anti-Cancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Scaringi, C.; Minniti, G.; Caporello, P.; Enrici, R.M. Integrin inhibitor cilengitide for the treatment of glioblastoma: A brief overview of current clinical results. Anticancer Res. 2012, 32, 4213–4223. [Google Scholar]
- Parrasia, S.; Szabò, I.; Zoratti, M.; Biasutto, L. Peptides as Pharmacological Carriers to the Brain: Promises, Shortcomings and Challenges. Mol. Pharm. 2022, 19, 3700–3729. [Google Scholar] [CrossRef]
- Gullbo, J.; Wickström, M.; Tullberg, M.; Ehrsson, H.; Lewensohn, R.; Nygren, P.; Luthman, K.; Larsson, R. Activity of hydrolytic enzymes in tumour cells is a determinant for anti-tumour efficacy of the melphalan containing prodrug J1. J. Drug Target. 2003, 11, 355–363. [Google Scholar] [CrossRef]
- Hamman, J.H.; Enslin, G.M.; Kotzé, A.F. Oral delivery of peptide drugs: Barriers and developments. BioDrugs 2005, 19, 165–177. [Google Scholar] [CrossRef]
- Ocak, M.; Helbok, A.; Rangger, C.; Peitl, P.K.; Nock, B.A.; Morelli, G.; Eek, A.; Sosabowski, J.K.; Breeman, W.A.; Reubi, J.C. Comparison of biological stability and metabolism of CCK2 receptor targeting peptides, a collaborative project under COST BM0607. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1426–1435. [Google Scholar] [CrossRef]
- Fang, L.; Hou, S.; Xue, L.; Zheng, F.; Zhan, C.-G. Amino-acid mutations to extend the biological half-life of a therapeutically valuable mutant of human butyrylcholinesterase. Chem.-Biol. Interact. 2014, 214, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.M.; Sibanda, B.L. Amino and carboxy-terminal regions in globular proteins. J. Mol. Biol. 1983, 167, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Yang, H.; Fan, Q.; Jia, D.; Tao, Z.; Wan, L.; Lu, X. Enhancing the circulating half-life and the antitumor effects of a tumor-selective cytotoxic peptide by exploiting endogenous serum albumin as a drug carrier. Int. J. Pharm. 2016, 499, 195–204. [Google Scholar] [CrossRef]
- Heinemann, U.; Hahn, M. Circular permutation of polypeptide chains: Implications for protein folding and stability. Prog. Biophys. Mol. Biol. 1995, 64, 121–143. [Google Scholar] [CrossRef] [PubMed]
- Murage, E.N.; Gao, G.; Bisello, A.; Ahn, J.-M. Development of potent glucagon-like peptide-1 agonists with high enzyme stability via introduction of multiple lactam bridges. J. Med. Chem. 2010, 53, 6412–6420. [Google Scholar] [CrossRef] [PubMed]
- Aboye, T.L.; Camarero, J.A. Biological synthesis of circular polypeptides. J. Biol. Chem. 2012, 287, 27026–27032. [Google Scholar] [CrossRef]
- Jagadish, K.; Camarero, J.A. Cyclotides, a promising molecular scaffold for peptide-based therapeutics. Pept. Sci. 2010, 94, 611–616. [Google Scholar] [CrossRef]
- Antos, J.M.; Popp, M.W.-L.; Ernst, R.; Chew, G.-L.; Spooner, E.; Ploegh, H.L. A straight path to circular proteins. J. Biol. Chem. 2009, 284, 16028–16036. [Google Scholar] [CrossRef]
- Lai, X.; Tang, J.; ElSayed, M.E. Recent advances in proteolytic stability for peptide, protein, and antibody drug discovery. Expert Opin. Drug Discov. 2021, 16, 1467–1482. [Google Scholar] [CrossRef]
- Garcia, A.E.; Tai, K.P.; Puttamadappa, S.S.; Shekhtman, A.; Ouellette, A.J.; Camarero, J.A. Biosynthesis and antimicrobial evaluation of backbone-cyclized α-defensins. Biochemistry 2011, 50, 10508–10519. [Google Scholar] [CrossRef]
- Scott, C.P.; Abel-Santos, E.; Wall, M.; Wahnon, D.C.; Benkovic, S.J. Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 13638–13643. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.A.; McGaw, L.J. Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.E.; ACamarero, J. Biological activities of natural and engineered cyclotides, a novel molecular scaffold for peptide-based therapeutics. Curr. Mol. Pharmacol. 2010, 3, 153–163. [Google Scholar] [CrossRef] [PubMed]
- de Veer, S.J.; White, A.M.; Craik, D.J. Sunflower trypsin Inhibitor-1 (SFTI-1): Sowing seeds in the fields of chemistry and biology. Angew. Chem. Int. Ed. 2021, 60, 8050–8071. [Google Scholar] [CrossRef]
- Xie, Q.; Yao, S.; Chen, X.; Xu, L.; Peng, W.; Zhang, L.; Zhang, Q.; Liang, X.-F.; Hong, A. A polypeptide from shark troponin I can inhibit angiogenesis and tumor growth. Mol. Biol. Rep. 2012, 39, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Goeddel, D.V.; Kleid, D.G.; Bolivar, F.; Heyneker, H.L.; Yansura, D.G.; Crea, R.; Hirose, T.; Kraszewski, A.; Itakura, K.; Riggs, A.D. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc. Natl. Acad. Sci. USA 1979, 76, 106–110. [Google Scholar] [CrossRef]
- Brandl, F.; Busslinger, S.; Zangemeister-Wittke, U.; Plückthun, A. Optimizing the anti-tumor efficacy of protein-drug conjugates by engineering the molecular size and half-life. J. Control. Release 2020, 327, 186–197. [Google Scholar] [CrossRef]
- Strohl, W.R. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Butt, T.R.; Edavettal, S.C.; Hall, J.P.; Mattern, M.R. SUMO fusion technology for difficult-to-express proteins. Protein Expr. Purif. 2005, 43, 1–9. [Google Scholar] [CrossRef]
- Kordbacheh, S.; Kasko, A.M. Peptide and protein engineering by modification of backbone and sidechain functional groups. Polym. Int. 2021, 70, 889–896. [Google Scholar] [CrossRef]
- Riener, C.K.; Kada, G.; Gruber, H.J. Quick measurement of protein sulfhydryls with Ellman’s reagent and with 4, 4′-dithiodipyridine. Anal. Bioanal. Chem. 2002, 373, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, Y.; Shen, P.Y.; Dai, X.-Q.; Wang, S.; Smillie, L.B.; Sandford, R.; Chen, X.-Z. Troponin I binds polycystin-L and inhibits its calcium-induced channel activation. Biochemistry 2003, 42, 7618–7625. [Google Scholar] [CrossRef]
- Chen, C.; Liu, J.-B.; Bian, Z.-P.; Xu, J.-D.; Wu, H.-F.; Gu, C.-R.; Shi, Y.; Zhang, J.-N.; Chen, X.-J.; Yang, D. Cardiac troponin I is abnormally expressed in non-small cell lung cancer tissues and human cancer cells. Int. J. Clin. Exp. Pathol. 2014, 7, 1314. [Google Scholar]
- Casas-Tintó, S.; Maraver, A.; Serrano, M.; Ferrús, A. Troponin-I enhances and is required for oncogenic overgrowth. Oncotarget 2016, 7, 52631. [Google Scholar] [CrossRef]
- Sheng, J.-J.; Jin, J.-P. TNNI1, TNNI2 and TNNI3: Evolution, regulation, and protein structure–function relationships. Gene 2016, 576, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Hoffend, J.; Altmann, A.; Kiessling, F.; Strauss, L.; Koczan, D.; Mier, W.; Eisenhut, M.; Kinscherf, R.; Haberkorn, U. Troponin I overexpression inhibits tumor growth, perfusion, and vascularization of morris hepatoma. J. Nucl. Med. 2006, 47, 1506–1514. [Google Scholar]
- Nyberg, P.; Xie, L.; Kalluri, R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005, 65, 3967–3979. [Google Scholar] [CrossRef]
- Liu, G.; Chen, T.; Ding, Z.; Wang, Y.; Wei, Y.; Wei, X. Inhibition of FGF-FGFR and VEGF-VEGFR signalling in cancer treatment. Cell Prolif. 2021, 54, e13009. [Google Scholar] [CrossRef]
- Amini, A.; Masoumi Moghaddam, S.; Morris, D.L.; Pourgholami, M.H. The critical role of vascular endothelial growth factor in tumor angiogenesis. Curr. Cancer Drug Targets 2012, 12, 23–43. [Google Scholar] [CrossRef]
- Peng, K.; Bai, Y.; Zhu, Q.; Hu, B.; Xu, Y. Targeting VEGF–neuropilin interactions: A promising antitumor strategy. Drug Discov. Today 2019, 24, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, L.; Liu, X.; Chen, C.; Wang, B.; Wang, W.; Hu, C.; Yu, K.; Qi, Z.; Liu, Q. Discovery of a highly selective VEGFR2 kinase inhibitor CHMFL-VEGFR2-002 as a novel anti-angiogenesis agent. Acta Pharm. Sin. B 2020, 10, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Ghiselli, G. Heparin binding proteins as therapeutic target: An historical account and current trends. Medicines 2019, 6, 80. [Google Scholar] [CrossRef]
- Ma, S.-N.; Mao, Z.-X.; Wu, Y.; Liang, M.-X.; Wang, D.-D.; Chen, X.; Chang, P.-A.; Zhang, W.; Tang, J.-H. The anti-cancer properties of heparin and its derivatives: A review and prospect. Cell Adhes. Migr. 2020, 14, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Teran, M.; Nugent, M.A. Synergistic binding of vascular endothelial growth factor-A and its receptors to heparin selectively modulates complex affinity. J. Biol. Chem. 2015, 290, 16451–16462. [Google Scholar] [CrossRef]
- Kowalczykowski, S.C.; Dixon, D.A.; Eggleston, A.K.; Lauder, S.D.; Rehrauer, W.M. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 1994, 58, 401–465. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.; Hevener, K.E.; Rice, A.J.; Patel, K.; Johnson, M.E.; Lee, H. High-level expression, purification, and characterization of Staphylococcus aureus dihydroorotase (PyrC) as a cleavable His-SUMO fusion. Protein Expr. Purif. 2013, 88, 98–106. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Solvent A (0.1%TFA in Double-Distilled Water) | Solvent B (0.1%TFA in Acetonitrile) |
|---|---|---|
| 0 | 95% | 5% |
| 27 | 75% | 25% |
| 28 | 5% | 95% |
| 33 | 5% | 95% |
| 40 | 95% | 5% |
| 45 | 95% | 5% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Lei, M.; Xie, J.; Guo, S.; Li, W.; Ren, X.; Wang, T.; Lin, S.; Xie, Q.; Chen, X. Discovery and Characteristics of a Novel Antitumor Cyclopeptide Derived from Shark. Bioengineering 2023, 10, 674. https://doi.org/10.3390/bioengineering10060674
Li F, Lei M, Xie J, Guo S, Li W, Ren X, Wang T, Lin S, Xie Q, Chen X. Discovery and Characteristics of a Novel Antitumor Cyclopeptide Derived from Shark. Bioengineering. 2023; 10(6):674. https://doi.org/10.3390/bioengineering10060674
Chicago/Turabian StyleLi, Fu, Minghua Lei, Junye Xie, Shujun Guo, Weicai Li, Xiujuan Ren, Teng Wang, Songxiong Lin, Qiuling Xie, and Xiaojia Chen. 2023. "Discovery and Characteristics of a Novel Antitumor Cyclopeptide Derived from Shark" Bioengineering 10, no. 6: 674. https://doi.org/10.3390/bioengineering10060674
APA StyleLi, F., Lei, M., Xie, J., Guo, S., Li, W., Ren, X., Wang, T., Lin, S., Xie, Q., & Chen, X. (2023). Discovery and Characteristics of a Novel Antitumor Cyclopeptide Derived from Shark. Bioengineering, 10(6), 674. https://doi.org/10.3390/bioengineering10060674