

Significance of Pulmonary Endothelial Injury and the Role of Cyclooxygenase-2 and Prostanoid Signaling


Abstract
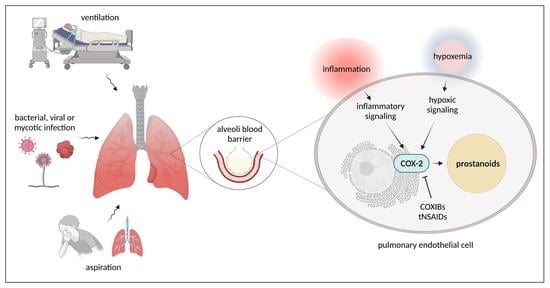
1. Introduction
2. Pulmonary Endothelial Dysfunction as a Result of Infections
2.1. Bacterial Infections
2.2. Viral Infections
2.3. Mycotic Infections
3. Pulmonary Endothelial Dysfunction as a Result of Mechanical Stress and Aspiration
4. Cyclooxygenase Signaling Pathways
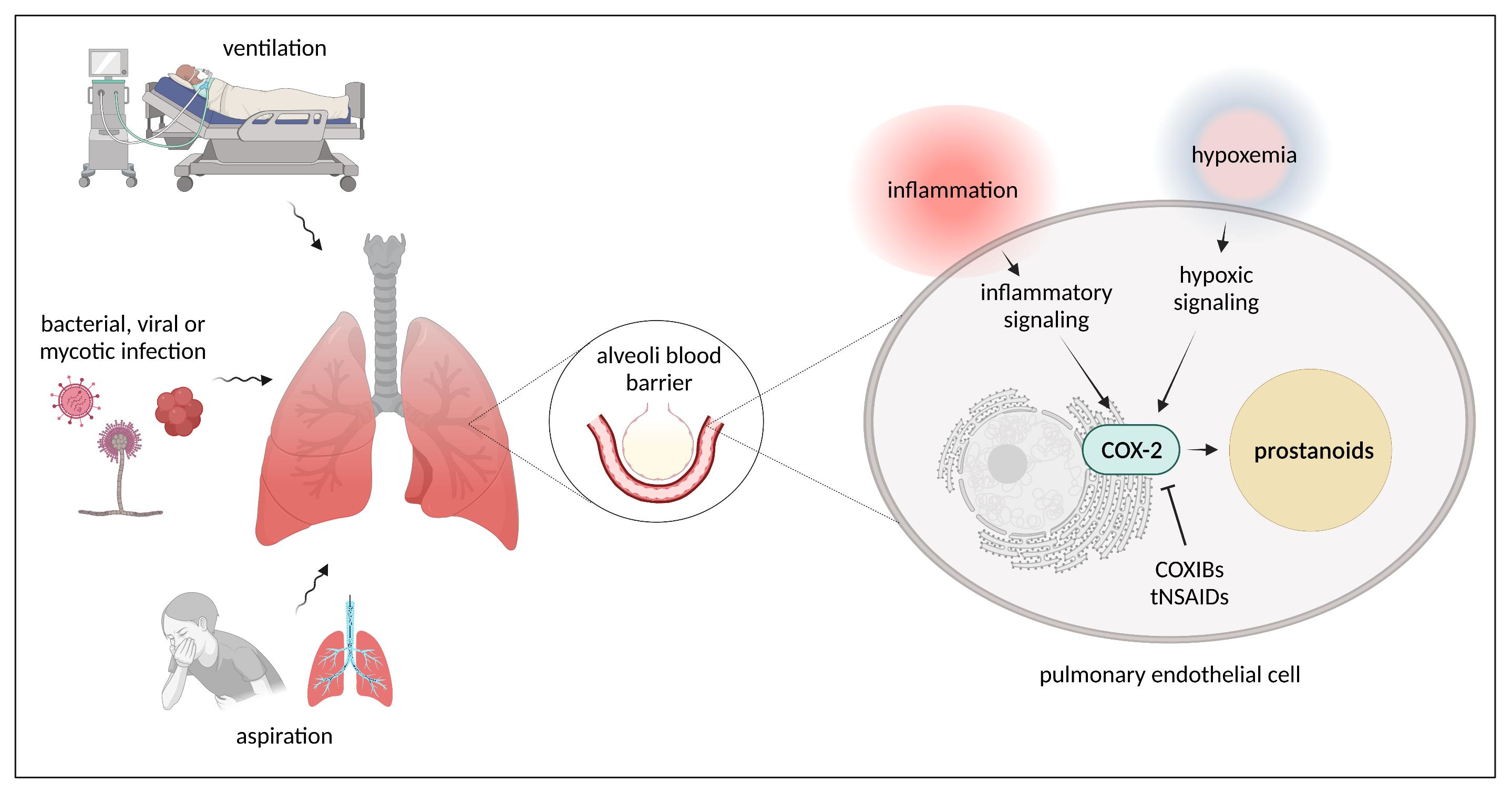
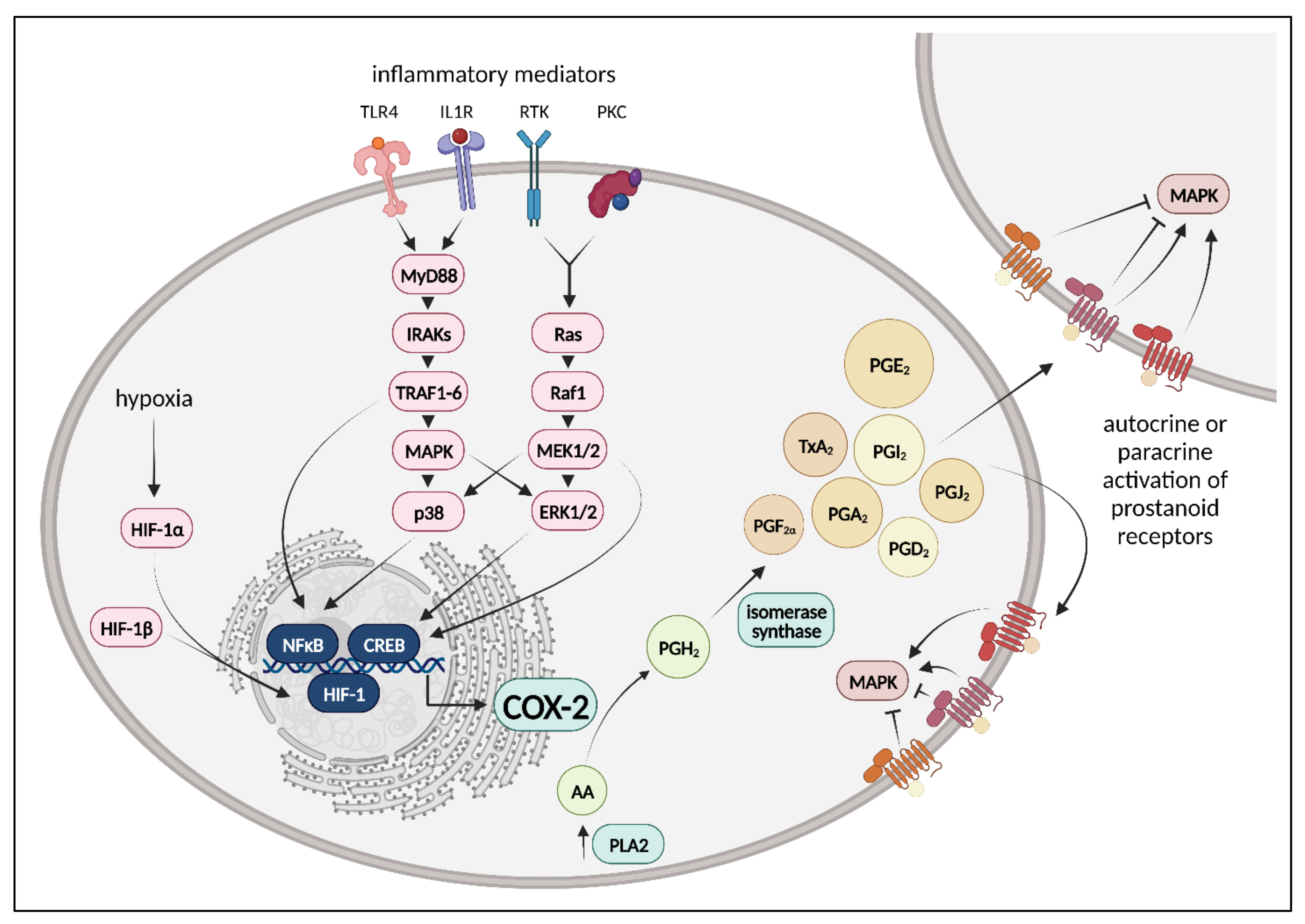
4.1. Cyclooxygenases, Prostanoids, Prostanoid Receptors, and Downstream Signaling
4.2. COX Pathways in Healthy and Injured Lung Endothelium
4.2.1. COX Pathways in Healthy Lung Endothelium
4.2.2. COX Pathways in Injured Lung Endothelium
Influence of Infections (Bacterial, Viral, Mycotic) on COX Pathways
COX-Pathways and ARDS
COX-Pathways and Hypoxia
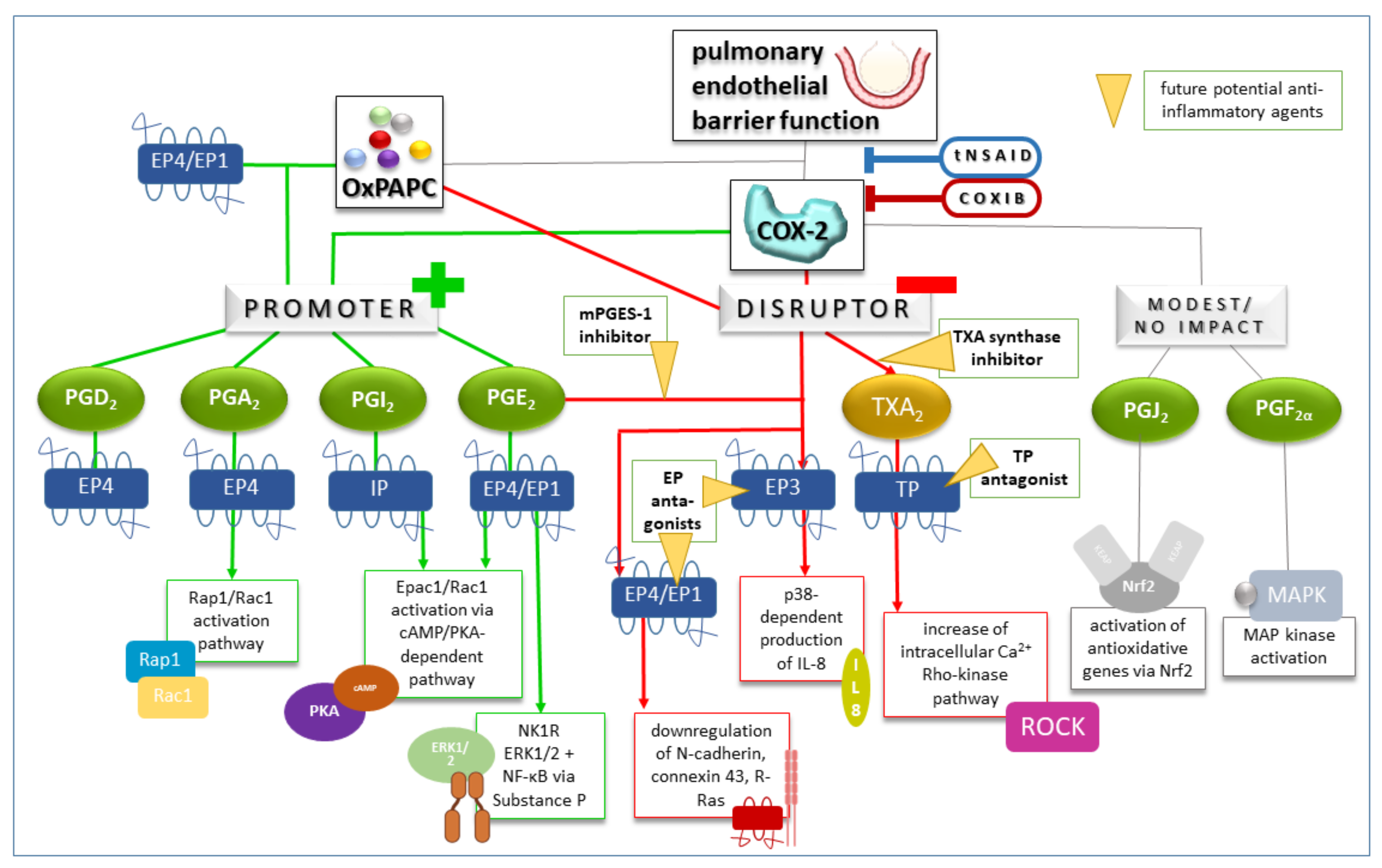
4.3. Therapeutic Approaches to Inhibit Cyclooxygenases in Lung Injury and Treatment Response
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| ACE2 | angiotensin-converting enzyme 2 |
| ADAM | A disintegrin and metalloproteinase domain-containing protein |
| AIM2 | absence in melanoma protein 2 |
| ALI | acute lung injury |
| ARDS | Acute Respiratory Distress Syndrome |
| BMP-2 | bone morphogenetic protein-2 |
| CotH | coat protein homolog |
| COX | cyclooxygenase |
| COXIB | selective COX-2 inhibitor |
| CREB | cAMP response element-binding protein |
| CV-2 | crossveinless-2 |
| EC | endothelial cells |
| EGFR | epidermal growth factor receptor |
| ENaC | epithelial sodium channel |
| ERK-1/2 | extracellular signal-regulated kinase 1/2 |
| HDAC | histone deacetylase |
| HIF | hypoxia inducible factor |
| HMGB | high mobility group box |
| HMVEC | human microvascular endothelial cell |
| HPAEC | human pulmonary artery endothelial cells |
| HPMVEC | human pulmonary microvascular endothelial cell |
| HPS | hepatopulmonary syndrome |
| HPV | hypoxic pulmonary vasoconstriction |
| Hsp | heat shock protein |
| HUVEC | human umbilical vein endothelial cell |
| ICAM | intercellular adhesion molecule |
| IL | interleukin |
| IPA | invasive pulmonary aspergillosis |
| JAK/STAT3 | Janus kinase signal transducer and activator of transcription |
| LasB | elastase B |
| LPS | lipopolysaccharide |
| LTA | lipoteichoic acid |
| MyD88 | myeloid differentiation primary response 88 |
| NFAT | nuclear factor of activated T cells |
| NF-κB | nuclear factor kappa B |
| NLRP3 | nucleotide-binding domain and leucine-rich repeat family pyrin domain-containing 3 |
| NK1R | neurokinin-1 receptor |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PAMP | pathogen-associated membrane pattern |
| PGD2 | prostaglandin D2 |
| PGE2 | prostaglandin E2 |
| PGF2α | prostaglandin F2α |
| PGG2 | prostaglandin G2 |
| PGH2 | prostaglandin H2 |
| PGI2 | prostaglandin I2 |
| PKA | protein kinase A |
| PKC | protein kinase C |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| Rap1 | Ras-related protein 1 |
| S1P | sphingosine-1-phosphate |
| TLR | toll-like receptor |
| TNF | tumor necrosis factor |
| TSLP | thymic stromal lymphopoietin |
| TXA2 | thromboxane A2 |
| VASP | vasodilator-stimulated phosphoprotein |
| VCAM | vascular cell adhesion molecule |
| VE cadherin | vascular endothelial cadherin |
| VEGF | vascular endothelial growth factor |
| VILI | ventilator induced lung injury |
| ZO-1 | zona occludens- 1 –protein |
References
- Green, C.E.; Turner, A.M. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Respir. Res. 2017, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, N.A.; Kotanidou, A.; Catravas, J.D.; Orfanos, S.E. Endothelial pathomechanisms in acute lung injury. Vasc. Pharmacol. 2008, 49, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E. Endothelial cell-cell junctions: Happy together. Nat. Rev. Mol. Cell Biol. 2004, 5, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Regan, E.R.; Aird, W.C. Dynamical systems approach to endothelial heterogeneity. Circ. Res. 2012, 111, 110–130. [Google Scholar] [CrossRef]
- Harb, R.; Whiteus, C.; Freitas, C.; Grutzendler, J. In vivo imaging of cerebral microvascular plasticity from birth to death. J. Cereb. Blood Flow Metab. 2013, 33, 146–156. [Google Scholar] [CrossRef]
- Gillich, A.; Zhang, F.; Farmer, C.G.; Travaglini, K.J.; Tan, S.Y.; Gu, M.; Zhou, B.; Feinstein, J.A.; Krasnow, M.A.; Metzger, R.J. Capillary cell-type specialization in the alveolus. Nature 2020, 586, 785–789. [Google Scholar] [CrossRef]
- Troeger, C.; Blacker, B.; Khalil, I.A.; Rao, P.C.; Cao, J.; Zimsen, S.R.M.; Albertson, S.B.; Deshpande, A.; Farag, T.; Abebe, Z.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1191–1210. [Google Scholar] [CrossRef]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef]
- Catravas, J.D.; Snead, C.; Dimitropoulou, C.; Chang, A.S.Y.; Lucas, R.; Verin, A.D.; Black, S.M. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vasc. Pharmacol. 2010, 52, 175–181. [Google Scholar] [CrossRef]
- Gong, P.; Angelini, D.J.; Yang, S.; Xia, G.; Cross, A.S.; Mann, D.; Bannerman, D.D.; Vogel, S.N.; Goldblum, S.E. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J. Biol. Chem. 2008, 283, 13437–13449. [Google Scholar] [CrossRef]
- Chatterjee, A.; Snead, C.; Yetik-Anacak, G.; Antonova, G.; Zeng, J.; Catravas, J.D. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L755–L763. [Google Scholar] [CrossRef]
- Jean-Baptiste, E. Cellular mechanisms in sepsis. J. Intensive Care Med. 2007, 22, 63–72. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Baumgarten, G.; Knuefermann, P.; Wrigge, H.; Putensen, C.; Stapel, H.; Fink, K.; Meyer, R.; Hoeft, A.; Grohé, C. Role of Toll-like receptor 4 for the pathogenesis of acute lung injury in Gram-negative sepsis. Eur. J. Anaesthesiol. 2006, 23, 1041–1048. [Google Scholar] [CrossRef]
- Park, I.; Kim, M.; Choe, K.; Song, E.; Seo, H.; Hwang, Y.; Ahn, J.; Lee, S.-H.; Lee, J.H.; Jo, Y.H.; et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur. Respir. J. 2019, 53, 1800786. [Google Scholar] [CrossRef]
- Wiener-Kronish, J.P.; Albertine, K.H.; Matthay, M.A. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J. Clin. Investig. 1991, 88, 864–875. [Google Scholar] [CrossRef]
- Okajima, K.; Harada, N. Regulation of inflammatory responses by sensory neurons: Molecular mechanism(s) and possible therapeutic applications. Curr. Med. Chem. 2006, 13, 2241–2251. [Google Scholar] [CrossRef]
- Inagawa, R.; Okada, H.; Takemura, G.; Suzuki, K.; Takada, C.; Yano, H.; Ando, Y.; Usui, T.; Hotta, Y.; Miyazaki, N.; et al. Ultrastructural Alteration of Pulmonary Capillary Endothelial Glycocalyx During Endotoxemia. Chest 2018, 154, 317–325. [Google Scholar] [CrossRef]
- Christaki, E.; Opal, S.M. Is the mortality rate for septic shock really decreasing? Curr. Opin. Crit. Care 2008, 14, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Bannerman, D.D.; Goldblum, S.E. Endotoxin induces endothelial barrier dysfunction through protein tyrosine phosphorylation. Am. J. Physiol. 1997, 273, L217–L226. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N.; Handa, V.; Dimitropoulou, C.; Rafikov, R.; Snead, C.; Kumar, S.; Joshi, A.; Thangjam, G.; Fulton, D.; Black, S.M.; et al. LPS induces pp60c-src-mediated tyrosine phosphorylation of Hsp90 in lung vascular endothelial cells and mouse lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L883–L893. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Dimitropoulou, C.; Thangjam, G.; Snead, C.; Feldman, S.; Barabutis, N.; Fulton, D.; Hou, Y.; Kumar, S.; Patel, V.; et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am. J. Respir. Cell Mol. Biol. 2014, 50, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Kása, A.; Csortos, C.; Verin, A.D. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 2015, 3, e974448. [Google Scholar] [CrossRef]
- Kovacs-Kasa, A.; Gorshkov, B.A.; Kim, K.-M.; Kumar, S.; Black, S.M.; Fulton, D.J.; Dimitropoulou, C.; Catravas, J.D.; Verin, A.D. The protective role of MLCP-mediated ERM dephosphorylation in endotoxin-induced lung injury in vitro and in vivo. Sci. Rep. 2016, 6, 39018. [Google Scholar] [CrossRef]
- Kovacs-Kasa, A.; Kim, K.M.; Cherian-Shaw, M.; Black, S.M.; Fulton, D.J.; Verin, A.D. Extracellular adenosine-induced Rac1 activation in pulmonary endothelium: Molecular mechanisms and barrier-protective role. J. Cell. Physiol. 2018, 233, 5736–5746. [Google Scholar] [CrossRef]
- Kuhlmann, N.; Wroblowski, S.; Knyphausen, P.; Boor, S.d.; Brenig, J.; Zienert, A.Y.; Meyer-Teschendorf, K.; Praefcke, G.J.K.; Nolte, H.; Krüger, M.; et al. Structural and Mechanistic Insights into the Regulation of the Fundamental Rho Regulator RhoGDIα by Lysine Acetylation. J. Biol. Chem. 2016, 291, 5484–5499. [Google Scholar] [CrossRef]
- Fontaine, S.N.; Sabbagh, J.J.; Baker, J.; Martinez-Licha, C.R.; Darling, A.; Dickey, C.A. Cellular factors modulating the mechanism of tau protein aggregation. Cell. Mol. Life Sci. 2015, 72, 1863–1879. [Google Scholar] [CrossRef]
- Kovacs-Kasa, A.; Kovacs, L.; Cherian-Shaw, M.; Patel, V.; Meadows, M.L.; Fulton, D.J.; Su, Y.; Verin, A.D. Inhibition of Class IIa HDACs improves endothelial barrier function in endotoxin-induced acute lung injury. J. Cell. Physiol. 2021, 236, 2893–2905. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef]
- Pietrocola, G.; Arciola, C.R.; Rindi, S.; Di Poto, A.; Missineo, A.; Montanaro, L.; Speziale, P. Toll-like receptors (TLRs) in innate immune defense against Staphylococcus aureus. Int. J. Artif. Organs 2011, 34, 799–810. [Google Scholar] [CrossRef]
- Pai, A.B.; Patel, H.; Prokopienko, A.J.; Alsaffar, H.; Gertzberg, N.; Neumann, P.; Punjabi, A.; Johnson, A. Lipoteichoic acid from Staphylococcus aureus induces lung endothelial cell barrier dysfunction: Role of reactive oxygen and nitrogen species. PLoS ONE 2012, 7, e49209. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Sheng, S.; Shao, Z. Staphylococcal alpha-hemolysin can form hexamers in phospholipid bilayers. J. Mol. Biol. 1998, 276, 325–330. [Google Scholar] [CrossRef]
- Becker, K.A.; Fahsel, B.; Kemper, H.; Mayeres, J.; Li, C.; Wilker, B.; Keitsch, S.; Soddemann, M.; Sehl, C.; Kohnen, M.; et al. Staphylococcus aureus Alpha-Toxin Disrupts Endothelial-Cell Tight Junctions via Acid Sphingomyelinase and Ceramide. Infect. Immun. 2018, 86, e00606-17. [Google Scholar] [CrossRef]
- Lucas, R.; Yang, G.; Gorshkov, B.A.; Zemskov, E.A.; Sridhar, S.; Umapathy, N.S.; Jezierska-Drutel, A.; Alieva, I.B.; Leustik, M.; Hossain, H.; et al. Protein kinase C-α and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am. J. Respir. Cell Mol. Biol. 2012, 47, 445–453. [Google Scholar] [CrossRef]
- Zhou, A.; Wang, H.; Lan, K.; Zhang, X.; Xu, W.; Yin, Y.; Li, D.; Yuan, J.; He, Y. Apoptosis induced by pneumolysin in human endothelial cells involves mitogen-activated protein kinase phosphorylation. Int. J. Mol. Med. 2012, 29, 1025–1030. [Google Scholar] [CrossRef]
- N’Guessan, P.D.; Schmeck, B.; Ayim, A.; Hocke, A.C.; Brell, B.; Hammerschmidt, S.; Rosseau, S.; Suttorp, N.; Hippenstiel, S. Streptococcus pneumoniae R6x induced p38 MAPK and JNK-mediated caspase-dependent apoptosis in human endothelial cells. Thromb. Haemost. 2005, 94, 295–303. [Google Scholar] [CrossRef]
- Czikora, I.; Alli, A.A.; Sridhar, S.; Matthay, M.A.; Pillich, H.; Hudel, M.; Berisha, B.; Gorshkov, B.; Romero, M.J.; Gonzales, J.; et al. Epithelial Sodium Channel-α Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front. Immunol. 2017, 8, 842. [Google Scholar] [CrossRef]
- Epelman, S.; Stack, D.; Bell, C.; Wong, E.; Neely, G.G.; Krutzik, S.; Miyake, K.; Kubes, P.; Zbytnuik, L.D.; Ma, L.L.; et al. Different domains of Pseudomonas aeruginosa exoenzyme S activate distinct TLRs. J. Immunol. 2004, 173, 2031–2040. [Google Scholar] [CrossRef]
- Huber, P.; Bouillot, S.; Elsen, S.; Attrée, I. Sequential inactivation of Rho GTPases and Lim kinase by Pseudomonas aeruginosa toxins ExoS and ExoT leads to endothelial monolayer breakdown. Cell. Mol. Life Sci. 2014, 71, 1927–1941. [Google Scholar] [CrossRef] [PubMed]
- Sayner, S.L.; Frank, D.W.; King, J.; Chen, H.; VandeWaa, J.; Stevens, T. Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ. Res. 2004, 95, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, C.D.; Alexeyev, M.; Pastukh, V.; Balczon, R.; Stevens, T. Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability. J. Biol. Chem. 2012, 287, 25407–25418. [Google Scholar] [CrossRef] [PubMed]
- Golovkine, G.; Faudry, E.; Bouillot, S.; Voulhoux, R.; Attrée, I.; Huber, P. VE-cadherin cleavage by LasB protease from Pseudomonas aeruginosa facilitates type III secretion system toxicity in endothelial cells. PLoS Pathog. 2014, 10, e1003939. [Google Scholar] [CrossRef]
- Tsan, M.F.; Cao, X.; White, J.E.; Sacco, J.; Lee, C.Y. Pertussis toxin-induced lung edema. Role of manganese superoxide dismutase and protein kinase C. Am. J. Respir. Cell Mol. Biol. 1999, 20, 465–473. [Google Scholar] [CrossRef]
- Wang, H.; Rogers, T.J.; Paton, J.C.; Paton, A.W. Differential effects of Escherichia coli subtilase cytotoxin and Shiga toxin 2 on chemokine and proinflammatory cytokine expression in human macrophage, colonic epithelial, and brain microvascular endothelial cell lines. Infect. Immun. 2014, 82, 3567–3579. [Google Scholar] [CrossRef]
- Morinaga, N.; Yahiro, K.; Matsuura, G.; Watanabe, M.; Nomura, F.; Moss, J.; Noda, M. Two distinct cytotoxic activities of subtilase cytotoxin produced by shiga-toxigenic Escherichia coli. Infect. Immun. 2007, 75, 488–496. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, M.H.; Tesh, V.L. Shiga toxins expressed by human pathogenic bacteria induce immune responses in host cells. J. Microbiol. 2013, 51, 724–730. [Google Scholar] [CrossRef]
- Wong, K.T.; Shieh, W.-J.; Kumar, S.; Norain, K.; Abdullah, W.; Guarner, J.; Goldsmith, C.S.; Chua, K.B.; Lam, S.K.; Tan, C.T.; et al. Nipah Virus Infection. Am. J. Pathol. 2002, 161, 2153–2167. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Hickey, A.C.; Smith, M.A.; Chan, Y.-P.; Wang, L.-F.; Mattapallil, J.J.; Geisbert, J.B.; Bossart, K.N.; Broder, C.C. Development of an acute and highly pathogenic nonhuman primate model of Nipah virus infection. PLoS ONE 2010, 5, e10690. [Google Scholar] [CrossRef]
- Jessie, K.; Fong, M.Y.; Devi, S.; Lam, S.K.; Wong, K.T. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J. Infect. Dis. 2004, 189, 1411–1418. [Google Scholar] [CrossRef]
- Zaki, S.R.; Greer, P.W.; Coffield, L.M.; Goldsmith, C.S.; Nolte, K.B.; Foucar, K.; Feddersen, R.M.; Zumwalt, R.E.; Miller, G.L.; Khan, A.S. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am. J. Pathol. 1995, 146, 552–579. [Google Scholar]
- Olofsson, S.; Bergström, T. Glycoconjugate glycans as viral receptors. Ann. Med. 2005, 37, 154–172. [Google Scholar] [CrossRef]
- Seternes, T.; Sørensen, K.; Smedsrød, B. Scavenger endothelial cells of vertebrates: A nonperipheral leukocyte system for high-capacity elimination of waste macromolecules. Proc. Natl. Acad. Sci. USA 2002, 99, 7594–7597. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.A.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Cheung, C.Y.; Leung, C.Y.H.; Nicholls, J.M. Innate immune responses to influenza A H5N1: Friend or foe? Trends Immunol. 2009, 30, 574–584. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Puerta-Guardo, H.; Glasner, D.R.; Harris, E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12, e1005738. [Google Scholar] [CrossRef]
- Gorbunova, E.E.; Simons, M.J.; Gavrilovskaya, I.N.; Mackow, E.R. The Andes Virus Nucleocapsid Protein Directs Basal Endothelial Cell Permeability by Activating RhoA. mBio 2016, 7, e01747-16. [Google Scholar] [CrossRef]
- Kolli, D.; Gupta, M.R.; Sbrana, E.; Velayutham, T.S.; Chao, H.; Casola, A.; Garofalo, R.P. Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am. J. Respir. Cell Mol. Biol. 2014, 51, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Haeberle, H.A.; Takizawa, R.; Casola, A.; Brasier, A.R.; Dieterich, H.J.; Van Rooijen, N.; Gatalica, Z.; Garofalo, R.P. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J. Infect. Dis. 2002, 186, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.E.; Cleary, D.W.; Clarke, S.C. Secondary Bacterial Infections Associated with Influenza Pandemics. Front. Microbiol. 2017, 8, 1041. [Google Scholar] [CrossRef] [PubMed]
- Shafran, N.; Shafran, I.; Ben-Zvi, H.; Sofer, S.; Sheena, L.; Krause, I.; Shlomai, A.; Goldberg, E.; Sklan, E.H. Secondary bacterial infection in COVID-19 patients is a stronger predictor for death compared to influenza patients. Sci. Rep. 2021, 11, 12703. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.M.; Diavatopoulos, D.A.; Ferwerda, G.; Pickkers, P.; Jonge, M.I.d.; Kox, M. The endotoxin-induced pulmonary inflammatory response is enhanced during the acute phase of influenza infection. Intensive Care Med. Exp. 2018, 6, 15. [Google Scholar] [CrossRef]
- Ru, Y.X.; Li, Y.C.; Zhao, Y.; Zhao, S.X.; Yang, J.P.; Zhang, H.M.; Pang, T.X. Multiple organ invasion by viruses: Pathological characteristics in three fatal cases of the 2009 pandemic influenza A/H1N1. Ultrastruct. Pathol. 2011, 35, 155–161. [Google Scholar] [CrossRef]
- Nakajima, N.; Van Tin, N.; Sato, Y.; Thach, H.N.; Katano, H.; Diep, P.H.; Kumasaka, T.; Thuy, N.T.; Hasegawa, H.; San, L.T.; et al. Pathological study of archival lung tissues from five fatal cases of avian H5N1 influenza in Vietnam. Mod. Pathol. 2013, 26, 357–369. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mavunda, K.; Krilov, L.R. Current State of Respiratory Syncytial Virus Disease and Management. Infect. Dis. Ther. 2021, 10, 5–16. [Google Scholar] [CrossRef]
- Arnold, R.; Konig, W. Respiratory syncytial virus infection of human lung endothelial cells enhances selectively intercellular adhesion molecule-1 expression. J. Immunol. 2005, 174, 7359–7367. [Google Scholar] [CrossRef]
- Lay, M.K.; Cespedes, P.F.; Palavecino, C.E.; Leon, M.A.; Diaz, R.A.; Salazar, F.J.; Mendez, G.P.; Bueno, S.M.; Kalergis, A.M. Human metapneumovirus infection activates the TSLP pathway that drives excessive pulmonary inflammation and viral replication in mice. Eur. J. Immunol. 2015, 45, 1680–1695. [Google Scholar] [CrossRef]
- Bugatti, A.; Marsico, S.; Fogli, M.; Roversi, S.; Messali, S.; Bosisio, D.; Giagulli, C.; Caruso, A.; Sozzani, S.; Fiorentini, S.; et al. Human Metapneumovirus Establishes Persistent Infection in Lung Microvascular Endothelial Cells and Primes a Th2-Skewed Immune Response. Microorganisms 2020, 8, 824. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, P1043–P1057.E15. [Google Scholar] [CrossRef]
- He, L.; Mäe, M.A.; Muhl, L.; Sun, Y.; Pietilä, R.; Nahar, K.; Liébanas, E.V.; Fagerlund, M.J.; Oldner, A.; Liu, J.; et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2—Implications for microvascular inflammation and hypercoagulopathy in COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F.; Gallagher, T. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Widagdo, W.; Sooksawasdi Na Ayudhya, S.; Hundie, G.B.; Haagmans, B.L. Host Determinants of MERS-CoV Transmission and Pathogenesis. Viruses 2019, 11, 280. [Google Scholar] [CrossRef]
- Geimonen, E.; Neff, S.; Raymond, T.; Kocer, S.S.; Gavrilovskaya, I.N.; Mackow, E.R. Pathogenic and nonpathogenic hantaviruses differentially regulate endothelial cell responses. Proc. Natl. Acad. Sci. USA 2002, 99, 13837–13842. [Google Scholar] [CrossRef]
- Mori, M.; Rothman, A.L.; Kurane, I.; Montoya, J.M.; Nolte, K.B.; Norman, J.E.; Waite, D.C.; Koster, F.T.; Ennis, F.A. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J. Infect. Dis. 1999, 179, 295–302. [Google Scholar] [CrossRef]
- Patterson, T.F.; Kirkpatrick, W.R.; White, M.; Hiemenz, J.W.; Wingard, J.R.; Dupont, B.; Rinaldi, M.G.; Stevens, D.A.; Graybill, J.R. Invasive aspergillosis. Disease spectrum, treatment practices, and outcomes. I3 Aspergillus Study Group. Medicine 2000, 79, 250–260. [Google Scholar] [CrossRef]
- Upton, A.; Kirby, K.A.; Carpenter, P.; Boeckh, M.; Marr, K.A. Invasive aspergillosis following hematopoietic cell transplantation: Outcomes and prognostic factors associated with mortality. Clin. Infect. Dis. 2007, 44, 531–540. [Google Scholar] [CrossRef]
- Al-Bader, N.; Vanier, G.; Liu, H.; Gravelat, F.N.; Urb, M.; Hoareau, C.M.-Q.; Campoli, P.; Chabot, J.; Filler, S.G.; Sheppard, D.C. Role of trehalose biosynthesis in Aspergillus fumigatus development, stress response, and virulence. Infect. Immun. 2010, 78, 3007–3018. [Google Scholar] [CrossRef]
- Ejzykowicz, D.E.; Cunha, M.M.; Rozental, S.; Solis, N.V.; Gravelat, F.N.; Sheppard, D.C.; Filler, S.G. The Aspergillus fumigatus transcription factor Ace2 governs pigment production, conidiation and virulence. Mol. Microbiol. 2009, 72, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Ejzykowicz, D.E.; Solis, N.V.; Gravelat, F.N.; Chabot, J.; Li, X.; Sheppard, D.C.; Filler, S.G. Role of Aspergillus fumigatus DvrA in host cell interactions and virulence. Eukaryot. Cell 2010, 9, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Joosten, L.A.B.; Devesa, I.; Mora-Montes, H.M.; Kanneganti, T.-D.; Dinarello, C.A.; van der Meer, J.W.M.; Gow, N.A.R.; Kullberg, B.J.; Netea, M.G. Bypassing pathogen-induced inflammasome activation for the regulation of interleukin-1beta production by the fungal pathogen Candida albicans. J. Infect. Dis. 2009, 199, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Spellberg, B.; Avanessian, V.; Fu, Y.; Edwards, J.E. Rhizopus oryzae adheres to, is phagocytosed by, and damages endothelial cells in vitro. Infect. Immun. 2005, 73, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Chiang, L.Y.; Sheppard, D.C.; Gravelat, F.N.; Patterson, T.F.; Filler, S.G. Aspergillus fumigatus stimulates leukocyte adhesion molecules and cytokine production by endothelial cells in vitro and during invasive pulmonary disease. Infect. Immun. 2008, 76, 3429–3438. [Google Scholar] [CrossRef]
- Phan, Q.T.; Belanger, P.H.; Filler, S.G. Role of hyphal formation in interactions of Candida albicans with endothelial cells. Infect. Immun. 2000, 68, 3485–3490. [Google Scholar] [CrossRef]
- Lopes Bezerra, L.M.; Filler, S.G. Interactions of Aspergillus fumigatus with endothelial cells: Internalization, injury, and stimulation of tissue factor activity. Blood 2004, 103, 2143–2149. [Google Scholar] [CrossRef]
- Frater, J.L.; Hall, G.S.; Procop, G.W. Histologic features of zygomycosis: Emphasis on perineural invasion and fungal morphology. Arch. Pathol. Lab. Med. 2001, 125, 375–378. [Google Scholar] [CrossRef]
- Shaukat, A.; Bakri, F.; Young, P.; Hahn, T.; Ball, D.; Baer, M.R.; Wetzler, M.; Slack, J.L.; Loud, P.; Czuczman, M.; et al. Invasive filamentous fungal infections in allogeneic hematopoietic stem cell transplant recipients after recovery from neutropenia: Clinical, radiologic, and pathologic characteristics. Mycopathologia 2005, 159, 181–188. [Google Scholar] [CrossRef]
- Kamai, Y.; Lossinsky, A.S.; Liu, H.; Sheppard, D.C.; Filler, S.G. Polarized response of endothelial cells to invasion by Aspergillus fumigatus. Cell. Microbiol. 2009, 11, 170–182. [Google Scholar] [CrossRef]
- Banoth, B.; Tuladhar, S.; Karki, R.; Sharma, B.R.; Briard, B.; Kesavardhana, S.; Burton, A.; Kanneganti, T.-D. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J. Biol. Chem. 2020, 295, 18276–18283. [Google Scholar] [CrossRef]
- Stergiopoulou, T.; Meletiadis, J.; Roilides, E.; Kleiner, D.E.; Schaufele, R.; Roden, M.; Harrington, S.; Dad, L.; Segal, B.; Walsh, T.J. Host-dependent patterns of tissue injury in invasive pulmonary aspergillosis. Am. J. Clin. Pathol. 2007, 127, 349–355. [Google Scholar] [CrossRef]
- Coelho, C.; Bocca, A.L.; Casadevall, A. The tools for virulence of Cryptococcus neoformans. Adv. Appl. Microbiol. 2014, 87, 1–41. [Google Scholar] [CrossRef]
- Gilbert, A.S.; Wheeler, R.T.; May, R.C. Fungal Pathogens: Survival and Replication within Macrophages. Cold Spring Harb. Perspect. Med. 2014, 5, a019661. [Google Scholar] [CrossRef]
- Opitz, B.; Hippenstiel, S.; Eitel, J.; Suttorp, N. Extra- and intracellular innate immune recognition in endothelial cells. Thromb. Haemost. 2007, 98, 319–326. [Google Scholar]
- Netea, M.G.; van der Graaf, C.A.A.; Vonk, A.G.; Verschueren, I.; van der Meer, J.W.M.; Kullberg, B.J. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J. Infect. Dis. 2002, 185, 1483–1489. [Google Scholar] [CrossRef]
- Pietrella, D.; Pandey, N.; Gabrielli, E.; Pericolini, E.; Perito, S.; Kasper, L.; Bistoni, F.; Cassone, A.; Hube, B.; Vecchiarelli, A. Secreted aspartic proteases of Candida albicans activate the NLRP3 inflammasome. Eur. J. Immunol. 2013, 43, 679–692. [Google Scholar] [CrossRef]
- Pietrella, D.; Rachini, A.; Pandey, N.; Schild, L.; Netea, M.; Bistoni, F.; Hube, B.; Vecchiarelli, A. The Inflammatory response induced by aspartic proteases of Candida albicans is independent of proteolytic activity. Infect. Immun. 2010, 78, 4754–4762. [Google Scholar] [CrossRef]
- Kasper, L.; König, A.; Koenig, P.-A.; Gresnigt, M.S.; Westman, J.; Drummond, R.A.; Lionakis, M.S.; Groß, O.; Ruland, J.; Naglik, J.R.; et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 2018, 9, 4260. [Google Scholar] [CrossRef]
- Naglik, J.R.; Gaffen, S.L.; Hube, B. Candidalysin: Discovery and function in Candida albicans infections. Curr. Opin. Microbiol. 2019, 52, 100–109. [Google Scholar] [CrossRef]
- Phan, Q.T.; Myers, C.L.; Fu, Y.; Sheppard, D.C.; Yeaman, M.R.; Welch, W.H.; Ibrahim, A.S.; Edwards, J.E.; Filler, S.G. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 2007, 5, e64. [Google Scholar] [CrossRef] [PubMed]
- Briard, B.; Fontaine, T.; Samir, P.; Place, D.E.; Muszkieta, L.; Malireddi, R.K.S.; Karki, R.; Christgen, S.; Bomme, P.; Vogel, P.; et al. Galactosaminogalactan activates the inflammasome to provide host protection. Nature 2020, 588, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Briard, B.; Malireddi, R.K.S.; Kanneganti, T.-D. Role of inflammasomes/pyroptosis and PANoptosis during fungal infection. PLoS Pathog. 2021, 17, e1009358. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lee, M.J.; Solis, N.V.; Phan, Q.T.; Swidergall, M.; Ralph, B.; Ibrahim, A.S.; Sheppard, D.C.; Filler, S.G. Aspergillus fumigatus CalA binds to integrin α5β1 and mediates host cell invasion. Nat. Microbiol. 2016, 2, 16211. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, J.H.; Hwang, B.Y.; Kim, H.S.; Lee, J.J. Anti-angiogenic activities of gliotoxin and its methylthioderivative, fungal metabolites. Arch. Pharm. Res. 2001, 24, 397–401. [Google Scholar] [CrossRef]
- Liu, M.; Spellberg, B.; Phan, Q.T.; Fu, Y.; Fu, Y.; Lee, A.S.; Edwards, J.E.; Filler, S.G.; Ibrahim, A.S. The endothelial cell receptor GRP78 is required for mucormycosis pathogenesis in diabetic mice. J. Clin. Investig. 2010, 120, 1914–1924. [Google Scholar] [CrossRef]
- Gebremariam, T.; Liu, M.; Luo, G.; Bruno, V.; Phan, Q.T.; Waring, A.J.; Edwards, J.E.; Filler, S.G.; Yeaman, M.R.; Ibrahim, A.S. CotH3 mediates fungal invasion of host cells during mucormycosis. J. Clin. Investig. 2014, 124, 237–250. [Google Scholar] [CrossRef]
- Tremblay, L.N.; Slutsky, A.S. Ventilator-induced lung injury: From the bench to the bedside. Intensive Care Med. 2006, 32, 24–33. [Google Scholar] [CrossRef]
- Birukov, K.G.; Jacobson, J.R.; Flores, A.A.; Ye, S.Q.; Birukova, A.A.; Verin, A.D.; Garcia, J.G.N. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L785–L797. [Google Scholar] [CrossRef]
- Birukova, A.A.; Rios, A.; Birukov, K.G. Long-term cyclic stretch controls pulmonary endothelial permeability at translational and post-translational levels. Exp. Cell Res. 2008, 314, 3466–3477. [Google Scholar] [CrossRef]
- Wolfson, R.K.; Mapes, B.; Garcia, J.G.N. Excessive mechanical stress increases HMGB1 expression in human lung microvascular endothelial cells via STAT3. Microvasc. Res. 2014, 92, 50–55. [Google Scholar] [CrossRef]
- Wolfson, R.K.; Chiang, E.T.; Garcia, J.G.N. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc. Res. 2011, 81, 189–197. [Google Scholar] [CrossRef]
- Vestweber, D.; Winderlich, M.; Cagna, G.; Nottebaum, A.F. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009, 19, 8–15. [Google Scholar] [CrossRef]
- Dejana, E.; Lampugnani, M.G.; Martinez-Estrada, O.; Bazzoni, G. The molecular organization of endothelial junctions and their functional role in vascular morphogenesis and permeability. Int. J. Dev. Biol. 2000, 44, 743–748. [Google Scholar]
- Schulte, D.; Küppers, V.; Dartsch, N.; Broermann, A.; Li, H.; Zarbock, A.; Kamenyeva, O.; Kiefer, F.; Khandoga, A.; Massberg, S.; et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 2011, 30, 4157–4170. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zimmerman, G.A. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am. J. Respir. Cell Mol. Biol. 2005, 33, 319–327. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Chiang, C.-H.; Chuang, C.-H.; Liu, S.-L.; Jheng, Y.-H.; Ryu, J.H. Spillover of cytokines and reactive oxygen species in ventilator-induced lung injury associated with inflammation and apoptosis in distal organs. Respir. Care 2014, 59, 1422–1432. [Google Scholar] [CrossRef]
- Ma, H.; Feng, X.; Ding, S. Hesperetin attenuates ventilator-induced acute lung injury through inhibition of NF-κB-mediated inflammation. Eur. J. Pharmacol. 2015, 769, 333–341. [Google Scholar] [CrossRef]
- Iwaki, M.; Ito, S.; Morioka, M.; Iwata, S.; Numaguchi, Y.; Ishii, M.; Kondo, M.; Kume, H.; Naruse, K.; Sokabe, M.; et al. Mechanical stretch enhances IL-8 production in pulmonary microvascular endothelial cells. Biochem. Biophys. Res. Commun. 2009, 389, 531–536. [Google Scholar] [CrossRef]
- Miyao, N.; Suzuki, Y.; Takeshita, K.; Kudo, H.; Ishii, M.; Hiraoka, R.; Nishio, K.; Tamatani, T.; Sakamoto, S.; Suematsu, M.; et al. Various adhesion molecules impair microvascular leukocyte kinetics in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1059–L1068. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Zemskov, E.A.; Sun, X.; Wang, H.; Yegambaram, M.; Wu, X.; Garcia-Flores, A.; Song, S.; Tang, H.; Kangath, A.; et al. Activation of the mechanosensitive Ca2+ channel TRPV4 induces endothelial barrier permeability via the disruption of mitochondrial bioenergetics. Redox Biol. 2021, 38, 101785. [Google Scholar] [CrossRef] [PubMed]
- Su, K.; Bo, L.; Jiang, C.; Deng, X.; Zhao, Y.-Y.; Minshall, R.D.; Hu, G. TLR4 is required for macrophage efferocytosis during resolution of ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L787–L801. [Google Scholar] [CrossRef] [PubMed]
- Determann, R.M.; Royakkers, A.; Wolthuis, E.K.; Vlaar, A.P.; Choi, G.; Paulus, F.; Hofstra, J.J.; de Graaff, M.J.; Korevaar, J.C.; Schultz, M.J. Ventilation with lower tidal volumes as compared with conventional tidal volumes for patients without acute lung injury: A preventive randomized controlled trial. Crit. Care 2010, 14, R1. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate in coagulation and inflammation. Semin. Immunopathol. 2012, 34, 73–91. [Google Scholar] [CrossRef]
- Shea, B.S.; Brooks, S.F.; Fontaine, B.A.; Chun, J.; Luster, A.D.; Tager, A.M. Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am. J. Respir. Cell Mol. Biol. 2010, 43, 662–673. [Google Scholar] [CrossRef]
- London, N.R.; Zhu, W.; Bozza, F.A.; Smith, M.C.P.; Greif, D.M.; Sorensen, L.K.; Chen, L.; Kaminoh, Y.; Chan, A.C.; Passi, S.F.; et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci. Transl. Med. 2010, 2, 23ra19. [Google Scholar] [CrossRef]
- Jones, C.A.; Nishiya, N.; London, N.R.; Zhu, W.; Sorensen, L.K.; Chan, A.C.; Lim, C.J.; Chen, H.; Zhang, Q.; Schultz, P.G.; et al. Slit2-Robo4 signalling promotes vascular stability by blocking Arf6 activity. Nat. Cell Biol. 2009, 11, 1325–1331. [Google Scholar] [CrossRef]
- Wolfson, R.K.; Lang, G.; Jacobson, J.; Garcia, J.G.N. Therapeutic Strategies to Limit Lung Endothelial Cell Permeability. In The Pulmonary Endothelium; Voelkel, N.F., Rounds, S., Eds.; Wiley-Blackwell: Oxford, UK, 2009; pp. 337–354. [Google Scholar] [CrossRef]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef]
- Knight, P.R.; Druskovich, G.; Tait, A.R.; Johnson, K.J. The role of neutrophils, oxidants, and proteases in the pathogenesis of acid pulmonary injury. Anesthesiology 1992, 77, 772–778. [Google Scholar] [CrossRef]
- Davidson, B.A.; Knight, P.R.; Helinski, J.D.; Nader, N.D.; Shanley, T.P.; Johnson, K.J. The role of tumor necrosis factor-alpha in the pathogenesis of aspiration pneumonitis in rats. Anesthesiology 1999, 91, 486–499. [Google Scholar] [CrossRef]
- Folkesson, H.G.; Matthay, M.A.; Hébert, C.A.; Broaddus, V.C. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J. Clin. Investig. 1995, 96, 107–116. [Google Scholar] [CrossRef]
- Knight, P.R.; Rutter, T.; Tait, A.R.; Coleman, E.; Johnson, K. Pathogenesis of gastric particulate lung injury: A comparison and interaction with acidic pneumonitis. Anesth. Analg. 1993, 77, 754–760. [Google Scholar] [CrossRef]
- Raghavendran, K.; Davidson, B.A.; Mullan, B.A.; Hutson, A.D.; Russo, T.A.; Manderscheid, P.A.; Woytash, J.A.; Holm, B.A.; Notter, R.H.; Knight, P.R. Acid and particulate-induced aspiration lung injury in mice: Importance of MCP-1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L134–L143. [Google Scholar] [CrossRef]
- Nawa, S.; Shimizu, A.; Kino, K.; Soga, H.; Shimizu, N. Development of an experimental model of an acute respiratory failure by intratracheal sea water infusion: A comparison with a conventional oleic acid induction. Res. Exp. Med. 1994, 194, 25–33. [Google Scholar] [CrossRef]
- Xinmin, D.; Yunyou, D.; Chaosheng, P.; Huasong, F.; Pingkun, Z.; Jiguang, M.; Zhiqian, X.; Qinzhi, X. Dexamethasone treatment attenuates early seawater instillation-induced acute lung injury in rabbits. Pharmacol. Res. 2006, 53, 372–379. [Google Scholar] [CrossRef]
- Li, P.-C.; Wang, B.-R.; Li, C.-C.; Lu, X.; Qian, W.-S.; Li, Y.-J.; Jin, F.-G.; Mu, D.-G. Seawater inhalation induces acute lung injury via ROS generation and the endoplasmic reticulum stress pathway. Int. J. Mol. Med. 2018, 41, 2505–2516. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, L.; Dong, M.; Li, Z.; Jin, F. Endothelial Semaphorin 7A promotes inflammation in seawater aspiration-induced acute lung injury. Int. J. Mol. Sci. 2014, 15, 19650–19661. [Google Scholar] [CrossRef]
- Li, J.; Xu, M.; Fan, Q.; Xie, X.; Zhang, Y.; Mu, D.; Zhao, P.; Zhang, B.; Cao, F.; Wang, Y.; et al. Tanshinone IIA ameliorates seawater exposure-induced lung injury by inhibiting aquaporins (AQP) 1 and AQP5 expression in lung. Respir. Physiol. Neurobiol. 2011, 176, 39–49. [Google Scholar] [CrossRef]
- Williams, C.S.; Mann, M.; DuBois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [PubMed]
- Rajakariar, R.; Yaqoob, M.M.; Gilroy, D.W. COX-2 in inflammation and resolution. Mol. Interv. 2006, 6, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.R.; Zeldin, D.C. The lung HETEs (and EETs) up. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1–H10. [Google Scholar] [CrossRef] [PubMed]
- Oni-Orisan, A.; Deng, Y.; Schuck, R.N.; Theken, K.N.; Edin, M.L.; Lih, F.B.; Molnar, K.; DeGraff, L.; Tomer, K.B.; Zeldin, D.C.; et al. Dual modulation of cyclooxygenase and CYP epoxygenase metabolism and acute vascular inflammation in mice. Prostaglandins Other Lipid Mediat. 2013, 104–105, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kis, B.; Snipes, J.A.; Busija, D.W. Acetaminophen and the cyclooxygenase-3 puzzle: Sorting out facts, fictions, and uncertainties. J. Pharmacol. Exp. Ther. 2005, 315, 1–7. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Kirkby, N.S.; Ahmetaj-Shala, B.; Armstrong, P.C.; Crescente, M.; Ferreira, P.; Pires, M.E.L.; Vaja, R.; Warner, T.D. Cyclooxygenases and the cardiovascular system. Pharmacol. Ther. 2021, 217, 107624. [Google Scholar] [CrossRef]
- Straus, D.S.; Glass, C.K. Cyclopentenone prostaglandins: New insights on biological activities and cellular targets. Med. Res. Rev. 2001, 21, 185–210. [Google Scholar] [CrossRef]
- Mbonye, U.R.; Song, I. Posttranscriptional and posttranslational determinants of cyclooxygenase expression. BMB Rep. 2009, 42, 552–560. [Google Scholar] [CrossRef]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, H.; Luo, L.; Lin, J.; Li, D.; Zheng, S.; Huang, H.; Yan, S.; Yang, J.; Hao, Y.; et al. Resolvin D1 Improves the Resolution of Inflammation via Activating NF-kappaB p50/p50-Mediated Cyclooxygenase-2 Expression in Acute Respiratory Distress Syndrome. J. Immunol. 2017, 199, 2043–2054. [Google Scholar] [CrossRef]
- Hirata, T.; Narumiya, S. Prostanoid receptors. Chem. Rev. 2011, 111, 6209–6230. [Google Scholar] [CrossRef]
- Breyer, R.M.; Bagdassarian, C.K.; Myers, S.A.; Breyer, M.D. Prostanoid Receptors: Subtypes and Signaling. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 661–690. [Google Scholar] [CrossRef]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid Receptors: Structures, Properties, and Functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar] [CrossRef]
- Bos, C.L.; Richel, D.J.; Ritsema, T.; Peppelenbosch, M.P.; Versteeg, H.H. Prostanoids and prostanoid receptors in signal transduction. Int. J. Biochem. Cell Biol. 2004, 36, 1187–1205. [Google Scholar] [CrossRef]
- Alfranca, A.; Iñiguez, M.A.; Fresno, M.; Redondo, J.M. Prostanoid signal transduction and gene expression in the endothelium: Role in cardiovascular diseases. Cardiovasc. Res. 2006, 70, 446–456. [Google Scholar] [CrossRef]
- Ashton, A.W.; Cheng, Y.; Helisch, A.; Ware, J.A. Thromboxane A2 receptor agonists antagonize the proangiogenic effects of fibroblast growth factor-2: Role of receptor internalization, thrombospondin-1, and αvβ3. Circ. Res. 2004, 94, 735–742. [Google Scholar] [CrossRef]
- Dormond, O.; Bezzi, M.; Mariotti, A.; Ruëgg, C. Prostaglandin E2 promotes integrin αVβ3-dependent endothelial cell adhesion, Rac-activation, and spreading through cAMP/PKA-dependent signaling. J. Biol. Chem. 2002, 277, 45838–45846. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Peri, K.; Ribeiro-da-Silva, A.; Almazan, G.; Shichi, H.; Hou, X.; Varma, D.R.; Chemtob, S. Localization of functional prostaglandin E2 receptors EP3 and EP4 in the nuclear envelope. J. Biol. Chem. 1999, 274, 15719–15724. [Google Scholar] [CrossRef]
- Kömhoff, M.; Lesener, B.; Nakao, K.; Seyberth, H.W.; Nüsing, R.M. Localization of the prostacyclin receptor in human kidney. Kidney Int. 1998, 54, 1899–1908. [Google Scholar] [CrossRef]
- Murata, T.; Lin, M.I.; Aritake, K.; Matsumoto, S.; Narumiya, S.; Ozaki, H.; Urade, Y.; Hori, M.; Sessa, W.C. Role of prostaglandin D2 receptor DP as a suppressor of tumor hyperpermeability and angiogenesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 20009–20014. [Google Scholar] [CrossRef]
- Chen, J.; Champa-Rodriguez, M.; Woodward, D. Identification of a prostanoid FP receptor population producing endothelium-dependent vasorelaxation in the rabbit jugular vein. Br. J. Pharmacol. 1995, 116, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Oskolkova, O.; Gawlak, G.; Tian, Y.F.; Ke, Y.B.; Sarich, N.; Son, S.; Andreasson, K.; Bochkov, V.N.; Birukova, A.A.; Birukov, K.G. Prostaglandin E receptor-4 receptor mediates endothelial barrier-enhancing and anti-inflammatory effects of oxidized phospholipids. FASEB J. 2017, 31, 4187–4202. [Google Scholar] [CrossRef] [PubMed]
- Starosta, V.; Wu, T.; Zimman, A.; Pham, D.; Tian, X.; Oskolkova, O.; Bochkov, V.; Berliner, J.A.; Birukova, A.A.; Birukov, K.G. Differential regulation of endothelial cell permeability by high and low doses of oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine. Am. J. Respir. Cell Mol. Biol. 2012, 46, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Göggel, R.; Hoffman, S.; Nüsing, R.; Narumiya, S.; Uhlig, S. Platelet-Activating Factor–induced Pulmonary Edema Is Partly Mediated by Prostaglandin E2, E-Prostanoid 3-Receptors, and Potassium Channels. Am. J. Respir. Crit. Care Med. 2002, 166, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Rittchen, S.; Rohrer, K.; Platzer, W.; Knuplez, E.; Bärnthaler, T.; Marsh, L.M.; Atallah, R.; Sinn, K.; Klepetko, W.; Sharma, N.; et al. Prostaglandin D2 strengthens human endothelial barrier by activation of E-type receptor 4. Biochem. Pharmacol. 2020, 182, 114277. [Google Scholar] [CrossRef]
- Ke, Y.; Oskolkova, O.V.; Sarich, N.; Tian, Y.; Sitikov, A.; Tulapurkar, M.E.; Son, S.; Birukova, A.A.; Birukov, K.G. Effects of prostaglandin lipid mediators on agonist-induced lung endothelial permeability and inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L710–L721. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Shala, F.; Elghazouli, Y.; Warner, T.D.; Gaston-Massuet, C.; Crescente, M.; Armstrong, P.C.; Herschman, H.R.; Kirkby, N.S. Cell-Specific Gene Deletion Reveals the Antithrombotic Function of COX1 and Explains the Vascular COX1/Prostacyclin Paradox. Circ. Res. 2019, 125, 847–854. [Google Scholar] [CrossRef]
- Kirkby, N.S.; Lundberg, M.H.; Harrington, L.S.; Leadbeater, P.D.; Milne, G.L.; Potter, C.M.; Al-Yamani, M.; Adeyemi, O.; Warner, T.D.; Mitchell, J.A. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc. Natl. Acad. Sci. USA 2012, 109, 17597–17602. [Google Scholar] [CrossRef]
- Boegehold, M.A. Endothelium-dependent control of vascular tone during early postnatal and juvenile growth. Microcirculation 2010, 17, 394–406. [Google Scholar] [CrossRef]
- Fukunaga, K.; Kohli, P.; Bonnans, C.; Fredenburgh, L.E.; Levy, B.D. Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J. Immunol. 2005, 174, 5033–5039. [Google Scholar] [CrossRef]
- Huang, T.-H.; Fang, P.-H.; Li, J.-M.; Ling, H.-Y.; Lin, C.-M.; Shi, C.-S. Cyclooxygenase-2 Activity Regulates Recruitment of VEGF-Secreting Ly6Chigh Monocytes in Ventilator-Induced Lung Injury. Int. J. Mol. Sci. 2019, 20, 1771. [Google Scholar] [CrossRef]
- Lee, I.T.; Yang, C.M. Inflammatory signalings involved in airway and pulmonary diseases. Mediat. Inflamm. 2013, 2013, 791231. [Google Scholar] [CrossRef]
- Peters, T.; Henry, P.J. Protease-activated receptors and prostaglandins in inflammatory lung disease. Br. J. Pharmacol. 2009, 158, 1017–1033. [Google Scholar] [CrossRef]
- Park, G.Y.; Christman, J.W. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L797–L805. [Google Scholar] [CrossRef]
- Carey, M.A.; Germolec, D.R.; Langenbach, R.; Zeldin, D.C. Cyclooxygenase enzymes in allergic inflammation and asthma. Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 157–162. [Google Scholar] [CrossRef]
- Kytölä, J.; Kääpä, P.; Uotila, P. Meconium Aspiration Stimulates Cyclooxygenase-2 and Nitric Oxide Synthase-2 Expression in Rat Lungs. Pediatr. Res. 2003, 53, 731–736. [Google Scholar] [CrossRef]
- Chen, J.X.; Berry, L.C.; Christman, B.W.; Meyrick, B. Glutathione mediates LPS-stimulated COX-2 expression via early transient p42/44 MAPK activation. J. Cell. Physiol. 2003, 197, 86–93. [Google Scholar] [CrossRef]
- Lee, W.; Kim, J.; Park, E.K.; Bae, J.S. Maslinic Acid Ameliorates Inflammation via the Downregulation of NF-κB and STAT-1. Antioxidants 2020, 9, 106. [Google Scholar] [CrossRef]
- Szymanski, K.V.; Toennies, M.; Becher, A.; Fatykhova, D.; N’Guessan, P.D.; Gutbier, B.; Klauschen, F.; Neuschaefer-Rube, F.; Schneider, P.; Rueckert, J.; et al. Streptococcus pneumoniae-induced regulation of cyclooxygenase-2 in human lung tissue. Eur. Respir. J. 2012, 40, 1458–1467. [Google Scholar] [CrossRef]
- Bormann, T.; Maus, R.; Stolper, J.; Jonigk, D.; Welte, T.; Gauldie, J.; Kolb, M.; Maus, U.A. Role of the COX2-PGE(2) axis in S. pneumoniae-induced exacerbation of experimental fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L377–L392. [Google Scholar] [CrossRef]
- Chen, J.S.; Alfajaro, M.M.; Wei, J.; Chow, R.D.; Filler, R.B.; Eisenbarth, S.C.; Wilen, C.B. Cyclooxgenase-2 is induced by SARS-CoV-2 infection but does not affect viral entry or replication. bioRxiv 2020. [Google Scholar] [CrossRef]
- Loo, J.; Spittle, D.A.; Newnham, M. COVID-19, immunothrombosis and venous thromboembolism: Biological mechanisms. Thorax 2021, 76, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Radi, Z.A.; Meyerholz, D.K.; Ackermann, M.R. Pulmonary cyclooxygenase-1 (COX-1) and COX-2 cellular expression and distribution after respiratory syncytial virus and parainfluenza virus infection. Viral Immunol. 2010, 23, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Cheung, C.Y.; Peiris, J.S. Role of cyclooxygenase-2 in H5N1 viral pathogenesis and the potential use of its inhibitors. Hong Kong Med. J. 2013, 19, 29–35. [Google Scholar]
- Shiraishi, Y.; Asano, K.; Niimi, K.; Fukunaga, K.; Wakaki, M.; Kagyo, J.; Takihara, T.; Ueda, S.; Nakajima, T.; Oguma, T.; et al. Cyclooxygenase-2/Prostaglandin D2/CRTH2 Pathway Mediates Double-Stranded RNA-Induced Enhancement of Allergic Airway Inflammation. J. Immunol. 2008, 180, 541–549. [Google Scholar] [CrossRef]
- Shirey, K.A.; Pletneva, L.M.; Puche, A.C.; Keegan, A.D.; Prince, G.A.; Blanco, J.C.; Vogel, S.N. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. 2010, 3, 291–300. [Google Scholar] [CrossRef]
- Gopalakrishnan, A.; Joseph, J.; Shirey, K.A.; Keegan, A.D.; Boukhvalova, M.S.; Vogel, S.N.; Blanco, J.C.G. Protection against influenza-induced Acute Lung Injury (ALI) by enhanced induction of M2a macrophages: Possible role of PPARγ/RXR ligands in IL-4-induced M2a macrophage differentiation. Front. Immunol. 2022, 13, 968336. [Google Scholar] [CrossRef]
- Kirkby, N.S.; Zaiss, A.K.; Wright, W.R.; Jiao, J.; Chan, M.V.; Warner, T.D.; Herschman, H.R.; Mitchell, J.A. Differential COX-2 induction by viral and bacterial PAMPs: Consequences for cytokine and interferon responses and implications for anti-viral COX-2 directed therapies. Biochem. Biophys. Res. Commun. 2013, 438, 249–256. [Google Scholar] [CrossRef]
- Sanches, J.M.; Rossato, L.; Lice, I.; Alves de Piloto Fernandes, A.M.; Bueno Duarte, G.H.; Rosini Silva, A.A.; de Melo Porcari, A.; de Oliveira Carvalho, P.; Gil, C.D. The role of annexin A1 in Candida albicans and Candida auris infections in murine neutrophils. Microb. Pathog. 2021, 150, 104689. [Google Scholar] [CrossRef]
- Garth, J.M.; Reeder, K.M.; Godwin, M.S.; Mackel, J.J.; Dunaway, C.W.; Blackburn, J.P.; Steele, C. IL-33 Signaling Regulates Innate IL-17A and IL-22 Production via Suppression of Prostaglandin E(2) during Lung Fungal Infection. J. Immunol. 2017, 199, 2140–2148. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, J.; Toki, S.; Goleniewska, K.; Norlander, A.E.; Newcomb, D.C.; Wu, P.; Boyd, K.L.; Kita, H.; Peebles, R.S., Jr. COX Inhibition Increases Alternaria-Induced Pulmonary Group 2 Innate Lymphoid Cell Responses and IL-33 Release in Mice. J. Immunol. 2020, 205, 1157–1166. [Google Scholar] [CrossRef]
- Pereira, P.A.; Trindade, B.C.; Secatto, A.; Nicolete, R.; Peres-Buzalaf, C.; Ramos, S.G.; Sadikot, R.; Bitencourt Cda, S.; Faccioli, L.H. Celecoxib improves host defense through prostaglandin inhibition during Histoplasma capsulatum infection. Mediat. Inflamm. 2013, 2013, 950981. [Google Scholar] [CrossRef]
- Luh, S.-P.; Chiang, C.-H. Acute lung injury/acute respiratory distress syndrome (ALI/ARDS): The mechanism, present strategies and future perspectives of therapies. J. Zhejiang Univ. Sci. B 2007, 8, 60–69. [Google Scholar] [CrossRef]
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef]
- Sio, S.W.; Ang, S.F.; Lu, J.; Moochhala, S.; Bhatia, M. Substance P upregulates cyclooxygenase-2 and prostaglandin E metabolite by activating ERK1/2 and NF-kappaB in a mouse model of burn-induced remote acute lung injury. J. Immunol. 2010, 185, 6265–6276. [Google Scholar] [CrossRef]
- Ohmura, T.; Tian, Y.; Sarich, N.; Ke, Y.; Meliton, A.; Shah, A.S.; Andreasson, K.; Birukov, K.G.; Birukova, A.A. Regulation of lung endothelial permeability and inflammatory responses by prostaglandin A2: Role of EP4 receptor. Mol. Biol. Cell 2017, 28, 1622–1635. [Google Scholar] [CrossRef]
- Perrot, C.Y.; Herrera, J.L.; Fournier-Goss, A.E.; Komatsu, M. Prostaglandin E2 breaks down pericyte–endothelial cell interaction via EP1 and EP4-dependent downregulation of pericyte N-cadherin, connexin-43, and R-Ras. Sci. Rep. 2020, 10, 11186. [Google Scholar] [CrossRef]
- Kobayashi, K.; Horikami, D.; Omori, K.; Nakamura, T.; Yamazaki, A.; Maeda, S.; Murata, T. Thromboxane A2 exacerbates acute lung injury via promoting edema formation. Sci. Rep. 2016, 6, 32109. [Google Scholar] [CrossRef]
- Mochizuki, M.; Ishii, Y.; Itoh, K.; Iizuka, T.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Matsuno, Y.; Hegab, A.E.; Nomura, A.; et al. Role of 15-DeoxyΔ12,14 Prostaglandin J2 and Nrf2 Pathways in Protection against Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2005, 171, 1260–1266. [Google Scholar] [CrossRef]
- Kylhammar, D.; Rådegran, G. The principal pathways involved in the in vivo modulation of hypoxic pulmonary vasoconstriction, pulmonary arterial remodelling and pulmonary hypertension. Acta Physiol. 2017, 219, 728–756. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gao, J.; Chen, B.; Chen, L.; Belguise, K.; Yu, W.; Lu, K.; Wang, X.; Yi, B. Cyclooxygenase-2 promotes pulmonary intravascular macrophage accumulation by exacerbating BMP signaling in rat experimental hepatopulmonary syndrome. Biochem. Pharmacol. 2017, 138, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Liu, C.; Chen, L.; Yang, Z.; Belguise, K.; Wang, X.; Lu, K.; Yan, H.; Yi, B. Cyclooxygenase-2 regulates HPS patient serum induced-directional collective HPMVEC migration via PKC/Rac signaling pathway. Gene 2019, 692, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef]
- Marnett, L.J. The COXIB experience: A look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 265–290. [Google Scholar] [CrossRef]
- Marnett, L.J. Mechanisms of cyclooxygenase-2 inhibition and cardiovascular side effects—The plot thickens. Cancer Prev. Res. 2009, 2, 288–290. [Google Scholar] [CrossRef]
- Atukorala, I.; Hunter, D.J. Valdecoxib: The rise and fall of a COX-2 inhibitor. Expert Opin. Pharmacother. 2013, 14, 1077–1086. [Google Scholar] [CrossRef]
- Lebedeva, E.S.; Kuzubova, N.N.; Titova, O.N.; Surkova, E.A. Effect of cyclooxygenase-2 inhibition on lung inflammation and hypoxia-inducible factor-1 signalling in COPD model. Eur. Respir. J. 2017, 50, PA3926. [Google Scholar] [CrossRef]
- Chong, S.J.; Wong, Y.C.; Wu, J.; Tan, M.H.; Lu, J.; Moochhala, S.M. Parecoxib reduces systemic inflammation and acute lung injury in burned animals with delayed fluid resuscitation. Int. J. Inflamm. 2014, 2014, 972645. [Google Scholar] [CrossRef]
- Li, A.M.; Zhang, L.N.; Li, W.Z. Amelioration of meconium-induced acute lung injury by parecoxib in a rabbit model. Int. J. Clin. Exp. Med. 2015, 8, 6804–6812. [Google Scholar]
- Robertson, J.A.; Sauer, D.; Gold, J.A.; Nonas, S.A. The Role of Cyclooxygenase-2 in Mechanical Ventilation–Induced Lung Injury. Am. J. Respir. Cell. Mol. Biol. 2012, 47, 387–394. [Google Scholar] [CrossRef]
- Meng, F.Y.; Gao, W.; Ju, Y.N. Parecoxib reduced ventilation induced lung injury in acute respiratory distress syndrome. BMC Pharmacol. Toxicol. 2017, 18, 25. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, S.; Zosky, G.R.; Wei, X.; Shu, S.; Wang, D.; Chai, X. Paracoxib Alleviates Ventilator-Induced Lung Injury Through Functional Modulation of Lung-Recruited CD11bloLy6Chi Monocytes. SHOCK 2021, 55, 236–243. [Google Scholar] [CrossRef]
- Qin, J.; Su, X.; Jin, X.; Zhao, J. Parecoxib mitigates lung ischemia-reperfusion injury in rats by reducing oxidative stress and inflammation and up-regulating HO-1 expression. Acta Cir. Bras. 2021, 36, e360901. [Google Scholar] [CrossRef]
- Robb, C.T.; Goepp, M.; Rossi, A.G.; Yao, C. Non-steroidal anti-inflammatory drugs, prostaglandins, and COVID-19. Br. J. Pharmacol. 2020, 177, 4899–4920. [Google Scholar] [CrossRef]
- Verrall, G.M. Scientific Rationale for a Bottom-Up Approach to Target the Host Response in Order to Try and Reduce the Numbers Presenting With Adult Respiratory Distress Syndrome Associated With COVID-19. Is There a Role for Statins and COX-2 Inhibitors in the Prevention and Early Treatment of the Disease? Front. Immunol. 2020, 11, 2167. [Google Scholar]
- Hoxha, M. What about COVID-19 and arachidonic acid pathway? Eur. J. Clin. Pharmacol. 2020, 76, 1501–1504. [Google Scholar] [CrossRef]
- Baghaki, S.; Yalcin, C.E.; Baghaki, H.S.; Aydin, S.Y.; Daghan, B.; Yavuz, E. COX2 inhibition in the treatment of COVID-19: Review of literature to propose repositioning of celecoxib for randomized controlled studies. Int. J. Infect. Dis. 2020, 101, 29–32. [Google Scholar] [CrossRef]
- Hong, W.; Chen, Y.; You, K.; Tan, S.; Wu, F.; Tao, J.; Chen, X.; Zhang, J.; Xiong, Y.; Yuan, F.; et al. Celebrex Adjuvant Therapy on Coronavirus Disease 2019: An Experimental Study. Front. Pharmacol. 2020, 11, 561674. [Google Scholar] [CrossRef]
- Shaban, N.Z.; Sleem, A.A.; Abu-Serie, M.M.; Maher, A.M.; Habashy, N.H. Regulation of the NF-κB signaling pathway and IL-13 in asthmatic rats by aerosol inhalation of the combined active constituents of Punica granatum juice and peel. Biomed. Pharmacother. 2022, 155, 113721. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Toxin | Impact on EC | Sources | |
|---|---|---|---|
| Gram positive | |||
| Staphylococcus aureus | α-toxin | disruption of endothelial-cell tight junctions through (activating acid sphingomyelinase/release of ceramide) loss of barrier function through ADAM10 activation | [34,35] |
| Streptococcus pneumoniae | pneumolysin | activation of Ca2+-dependent enzymes, including PKC-α activation of the NF-κB and p38 MAP kinase pathways | [36,37,38] |
| Listeria monocytogenes | listeriolysin O | dysfunction in the ENaC channel | [39] |
| Gram negative | |||
| Pseudomonas aeruginosa | exoenzyme S and T | activation of TLR-2 and -4 disruption of the actin cytoskeleton and interference of phagocytosis | [40,41] |
| exoenzyme Y and U | microtubule breakdown and tau phosphorylation | [42,43] | |
| LasB | cleavage of VE cadherin | [44] | |
| Bordetella pertussis | pertussis toxin | increase in PKC-mediated endothelial permeability | [45] |
| Shiga toxin such as Escherichia coli | subtilase cytotoxin AB | inhibition of protein synthesis and induction of vacuole formation | [46,47] |
| shigatoxin 2 | increase in cytokine and chemokine expression, e.g., TNF-α, IL-6, IL-8 inhibition of protein synthesis and induction of ribotoxic and ER stress responses | [46,48] |
| Primary Site of Lung Cell Damage | Specific Impact on Pulmonary EC | Sources | |
|---|---|---|---|
| Orthomyxoviridae | |||
| Influenza A | EC and epithelial cells | increase in cytoplasmatic translocation of High-Mobility Group Box 1 (HMGB1); release of HMGB1 via IL-6-receptor and activation of Janus kinase signal transducer and activator of transcription 3 (JAK/STAT3) signaling pathway activation of p38 MAPK and c-Jun N -terminal kinase pathways leading to cytoskeletal rearrangement and hyperpermeability via e.g., ERM (ezrin, radixin and moesin) phosphorylation | [66,67,68] |
| Paramyxoviridae | |||
| RSV | EC and epithelial cells | upregulation of intercellular adhesion molecule 1 (ICAM-1)/vascular cell adhesion molecule 1 (VCAM-1) and E-selectin upregulation (dependent on protein kinase C (PKC), protein kinase A (PKA), p38 MAPK) promotes PMN transmigration | [69] |
| Human Metapneumovirus | epithelial cells, alveolar macrophages and dendritic cells | indirect impact on EC via triggering thymic stromal lymphopoietin (TSLP), IL-8 and IL-33 expression in epithelial cells, cytokines IL-4, IL-5, Interferon γ (IFN-γ), IL-10, and TNF-α | [61,70,71] |
| Coronaviridae | |||
| SARS-CoV2 | ciliated bronchial cells, alveolar cells and EC | dysfunction of bradykinin–kallikrein pathway and RAAS complex by angiotensin-converting enzyme 2 (ACE2) downregulation via ADAM17 mediated ACE2 shedding decrease in platelet-derived growth factor receptor β (PDGFR-β) and Angiopoietin I through pericyte loss | [72,73,74] |
| MERS-CoV | ciliated bronchial cells, alveolar cells and EC | upregulation of proinflammatory cytokines e.g., TNF-α, IL-6, CSF-1 and CSF-3, IL-32 endoplasmic reticulum stress and oxysterols enhance apoptosis | [75] |
| Bunyaviridae | |||
| Hantavirus species | epithelial and EC | induction of transcriptional activation of VEGF and expression of B cell lymphoma 2 (Bcl2) gene activation of NF-κB induction of the expression of the chemokines RANTES (regulated upon activation, normal T cell expressed and presumably secreted) and enhancement of IP-10 infiltration of CD4+ and CD8+ T cells | [76,77] |
| Fungal Species | Pathogens | Specific Impact on Pulmonary EC | Sources |
|---|---|---|---|
| Candida albicans | mannan, chitins, β-1,3-glucans, β-1,6-glucans | recognition by pattern recognition receptors (PRR), e.g., mannose receptors and TLR-2 and -4, presumably enablement of adhesion to and transmigration across EC | [95,96] |
| secreted aspartic proteases (Sap2, Sap6) | potent induction of IL-1β, TNF-α, and IL-6 production, e.g., through activation of NLRP3 inflammasome | [97,98] | |
| candidalysin | formation of pores in host cell membrane, induction of potassium efflux to cause NLRP3 inflammasome activation | [99,100] | |
| Als3 (agglutinin-like sequence 3) | Induces tyrosine phosphorylation of EC proteins, causing microfilament rearrangement resulting in pseudopod production and endocytosis | [101] | |
| Aspergillus fumigatus | galactosamino-galactan | inhibition of translation by ribosome immobilization, induction of endoplasmic reticulum stress and triggers NLRP3 inflammasome activation | [102] |
| dsDNA | induction of AIM2 inflammasome | [103] | |
| thaumatin-like protein CalA | interaction with integrin α5β1 on EC, inducing endocytosis | [104] | |
| gliotoxin | inhibition of NF-κB pathway, anti-angiogenetic activity | [105] | |
| Rhizopus oryzae | coat protein homolog (CotH2 and CotH3) | binding via glucose-regulated protein 78 to EC and induction of endocytosis | [106,107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nickl, R.; Hauser, S.; Pietzsch, J.; Richter, T. Significance of Pulmonary Endothelial Injury and the Role of Cyclooxygenase-2 and Prostanoid Signaling. Bioengineering 2023, 10, 117. https://doi.org/10.3390/bioengineering10010117
Nickl R, Hauser S, Pietzsch J, Richter T. Significance of Pulmonary Endothelial Injury and the Role of Cyclooxygenase-2 and Prostanoid Signaling. Bioengineering. 2023; 10(1):117. https://doi.org/10.3390/bioengineering10010117
Chicago/Turabian StyleNickl, Rosa, Sandra Hauser, Jens Pietzsch, and Torsten Richter. 2023. "Significance of Pulmonary Endothelial Injury and the Role of Cyclooxygenase-2 and Prostanoid Signaling" Bioengineering 10, no. 1: 117. https://doi.org/10.3390/bioengineering10010117
APA StyleNickl, R., Hauser, S., Pietzsch, J., & Richter, T. (2023). Significance of Pulmonary Endothelial Injury and the Role of Cyclooxygenase-2 and Prostanoid Signaling. Bioengineering, 10(1), 117. https://doi.org/10.3390/bioengineering10010117