
Transcranial Electromagnetic Treatment Stops Alzheimer’s Disease Cognitive Decline over a 2½-Year Period: A Pilot Study

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
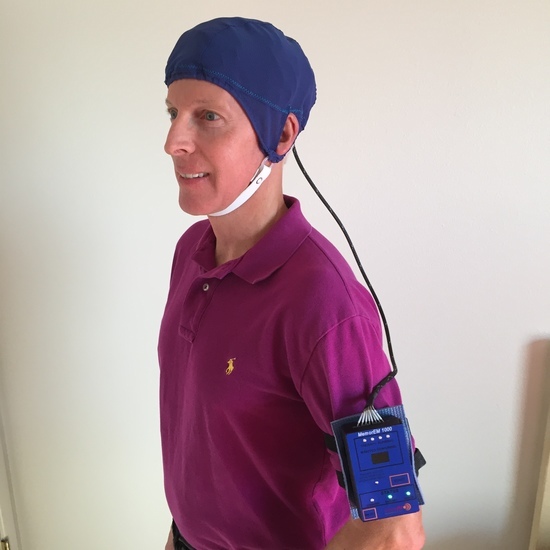
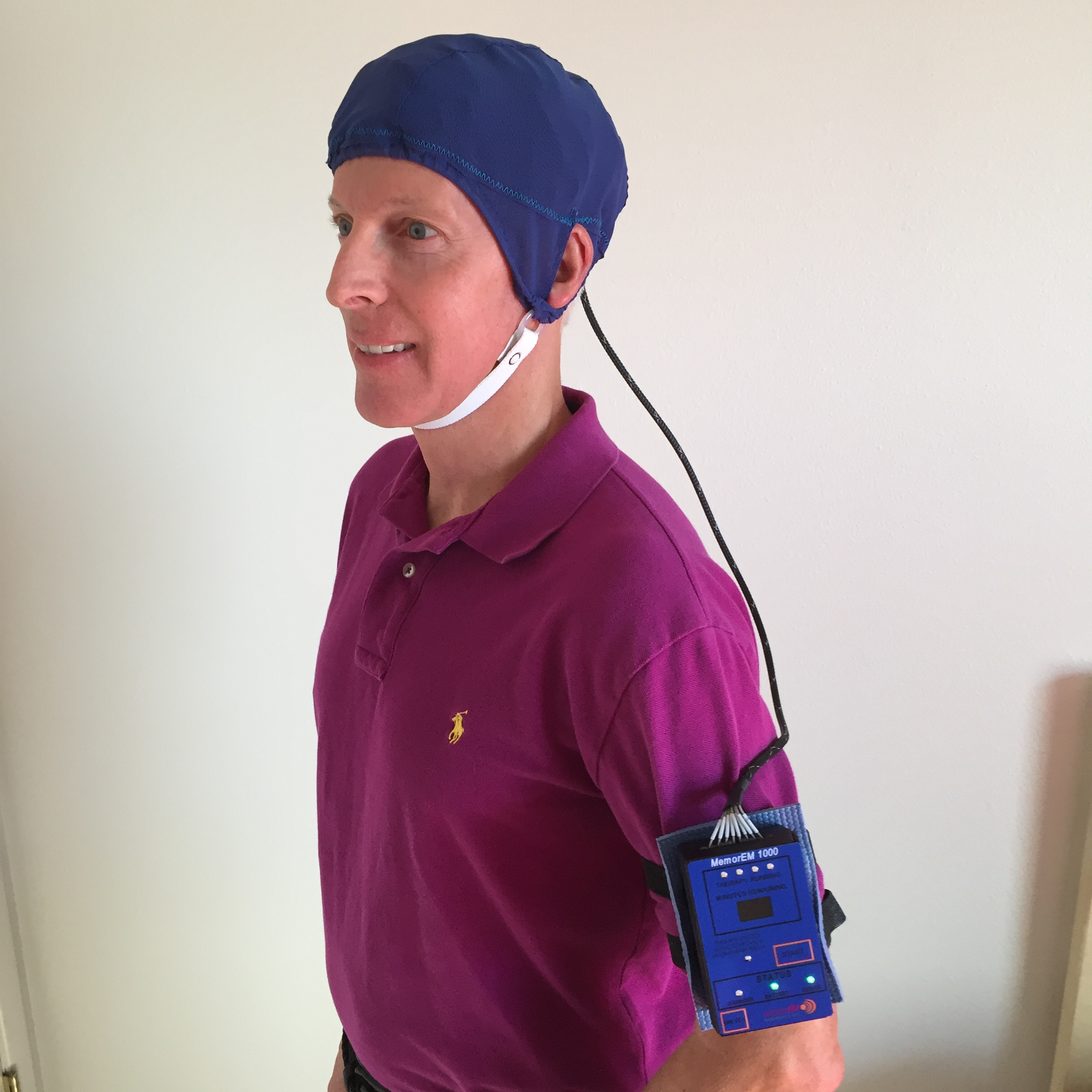
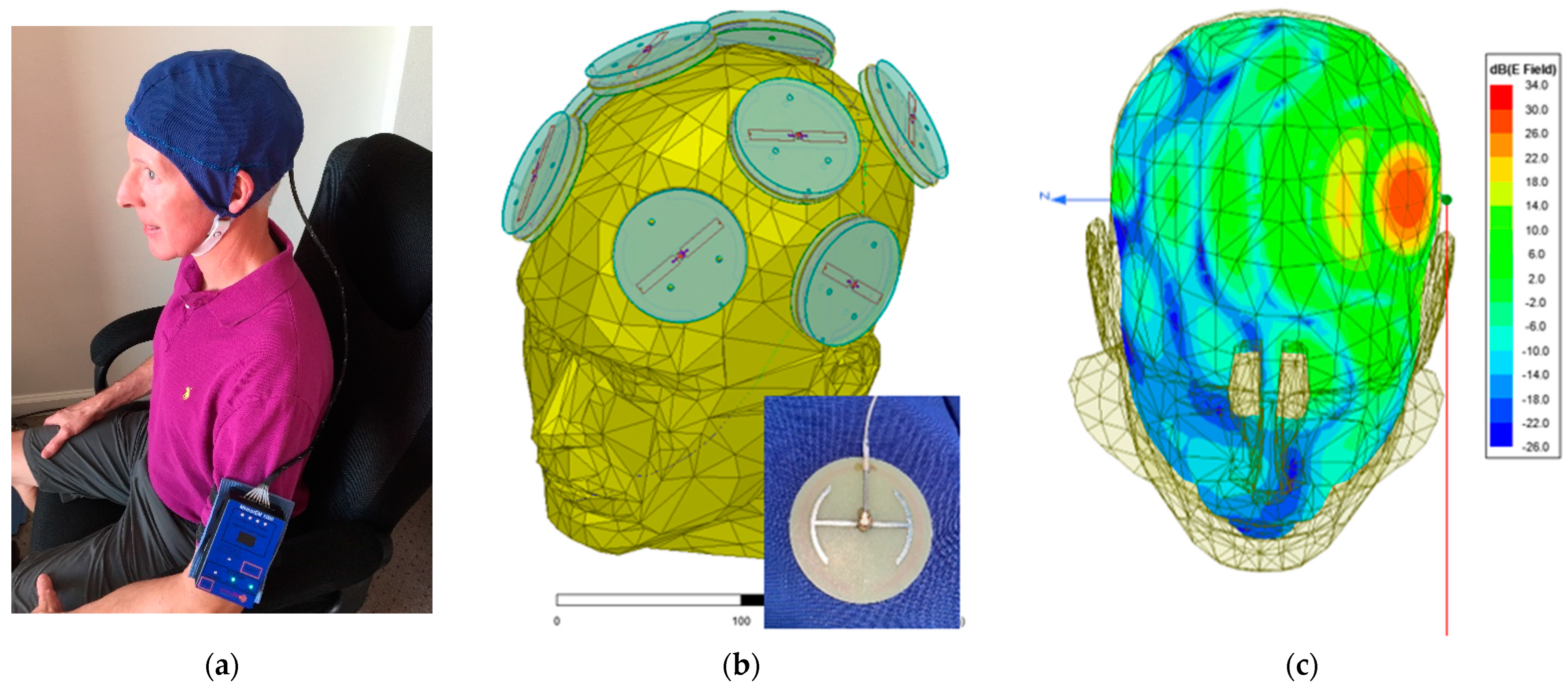
2.2. Investigational Device
2.3. General Protocol
2.4. Subject Screening
2.5. Baseline Evaluations
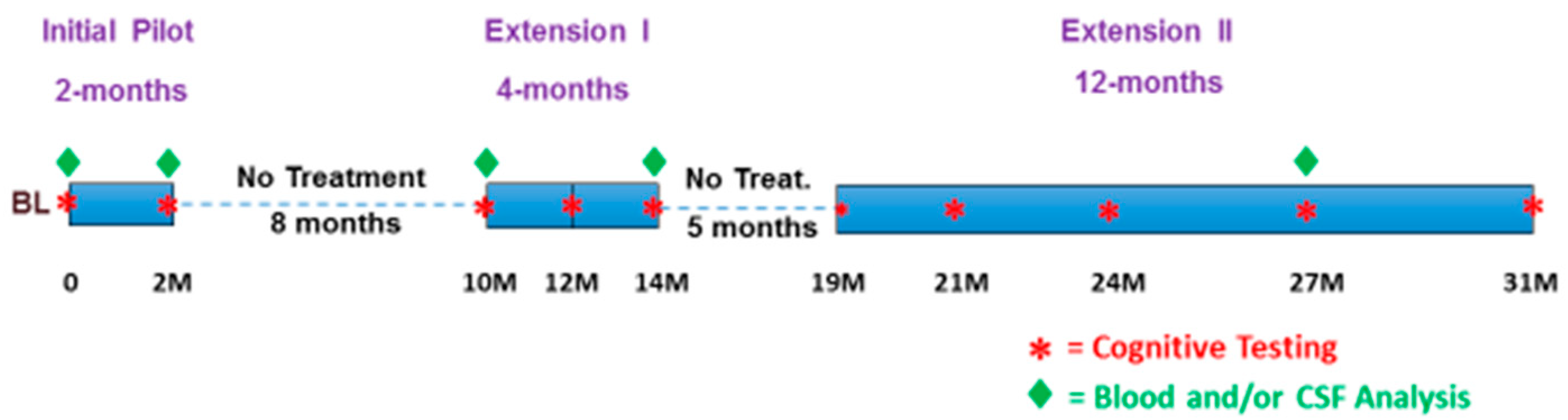
2.6. The 31 Months of Treatment/No-Treatment Periods
2.7. Patient Monitoring and Safety
2.8. Blood and CSF Processing/Analysis
2.9. Human C-Reactive Protein (CRP) Determination
2.10. Human Aβ1-40, Aβ1-42, and Oligomeric Aβ Determination (9A9)
2.11. Human Total Tau (t-tau) and Phospho-Tau (p-tau) Determinations
2.12. Statistical Analyses
3. Results
3.1. Long-Term Safety of TEMT and TEMT’s Effects on Brain Regional Volumes
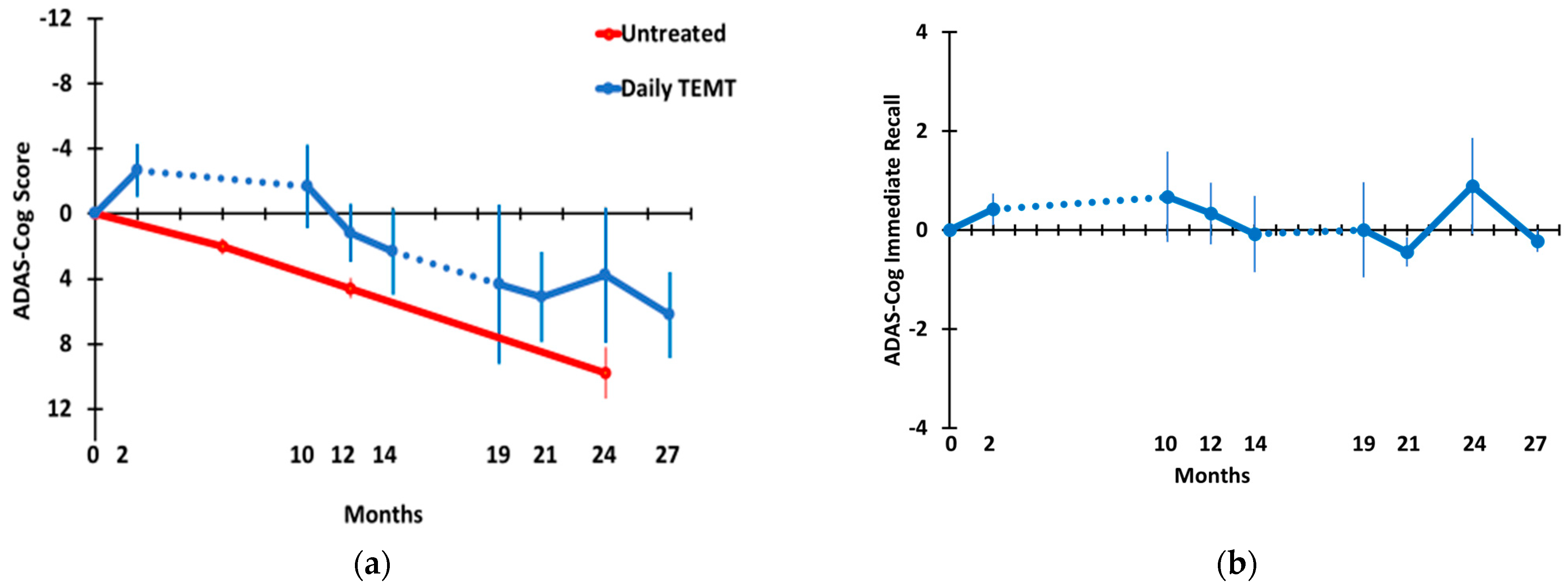
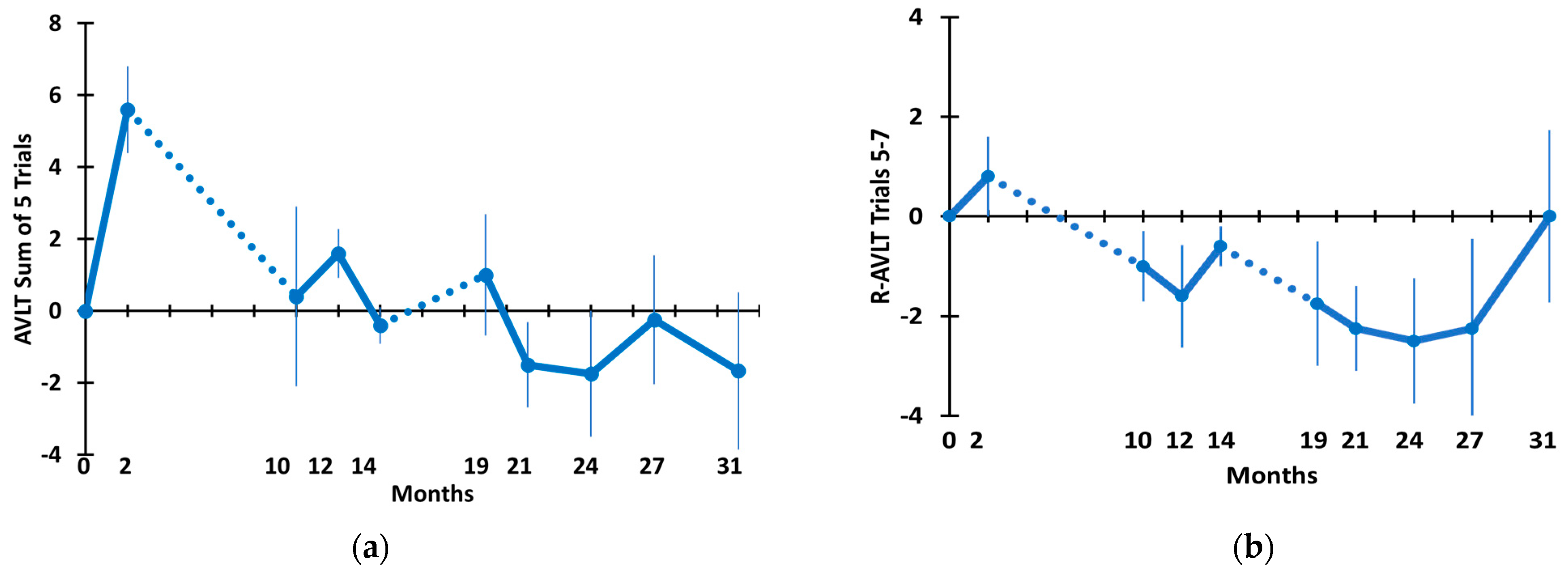
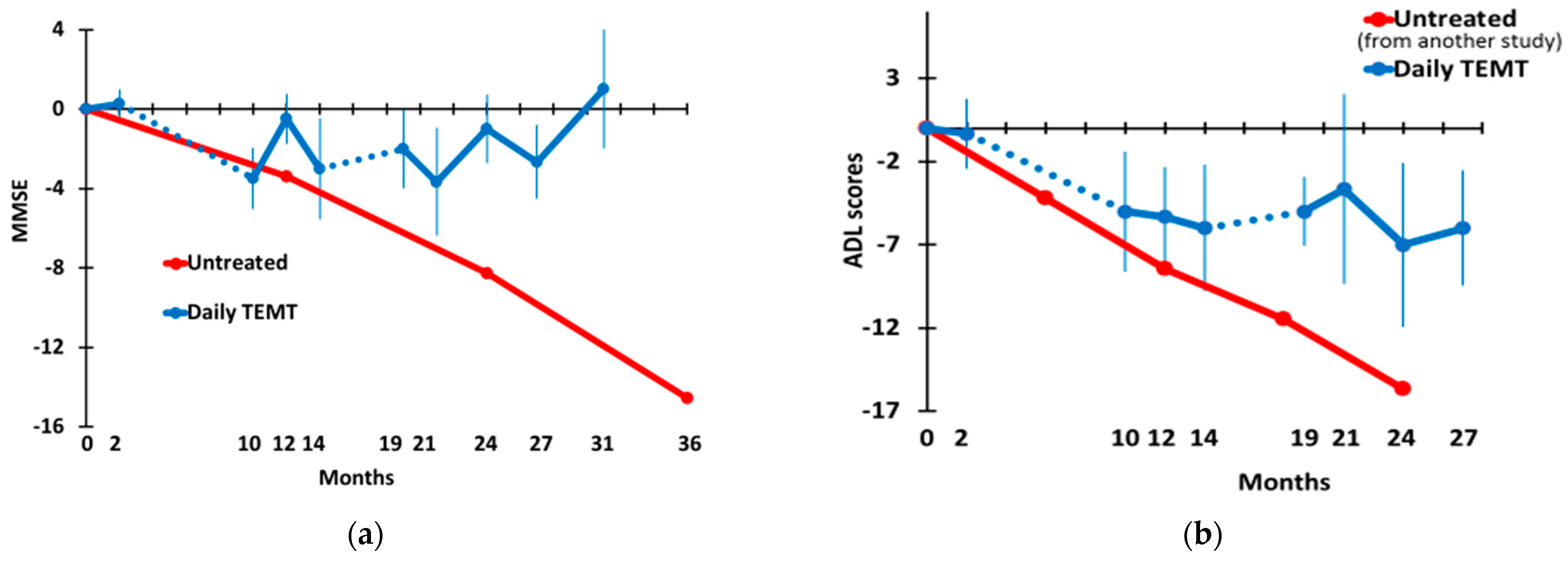
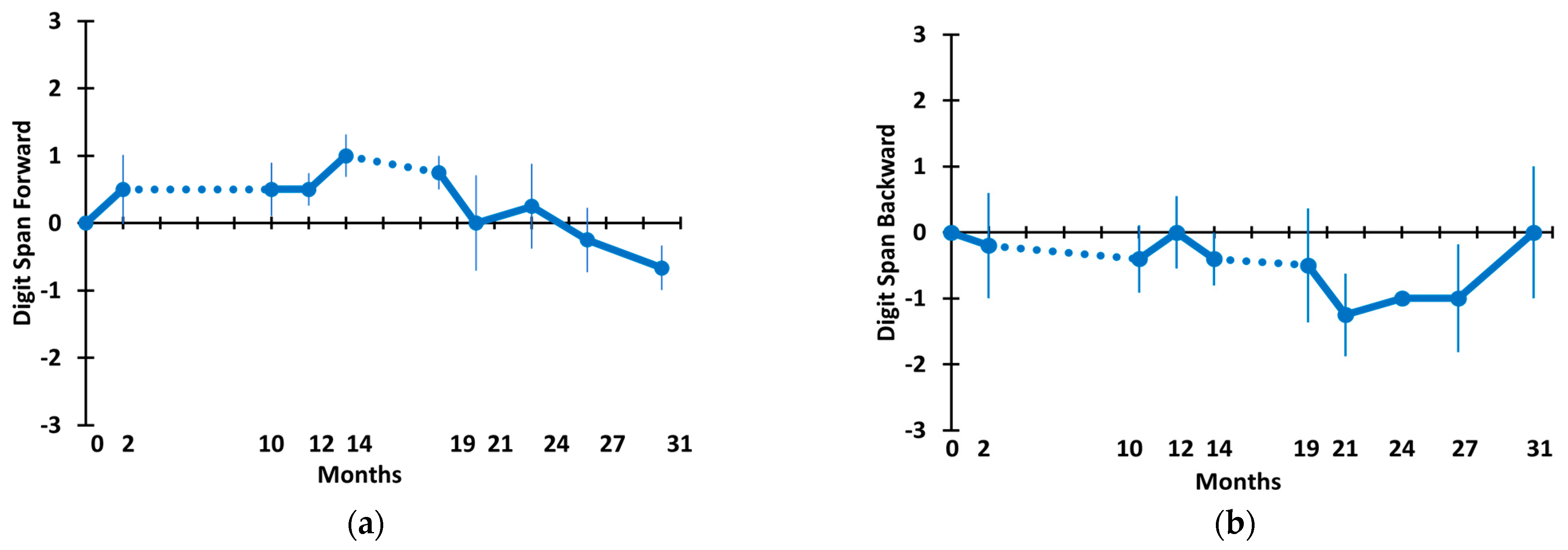
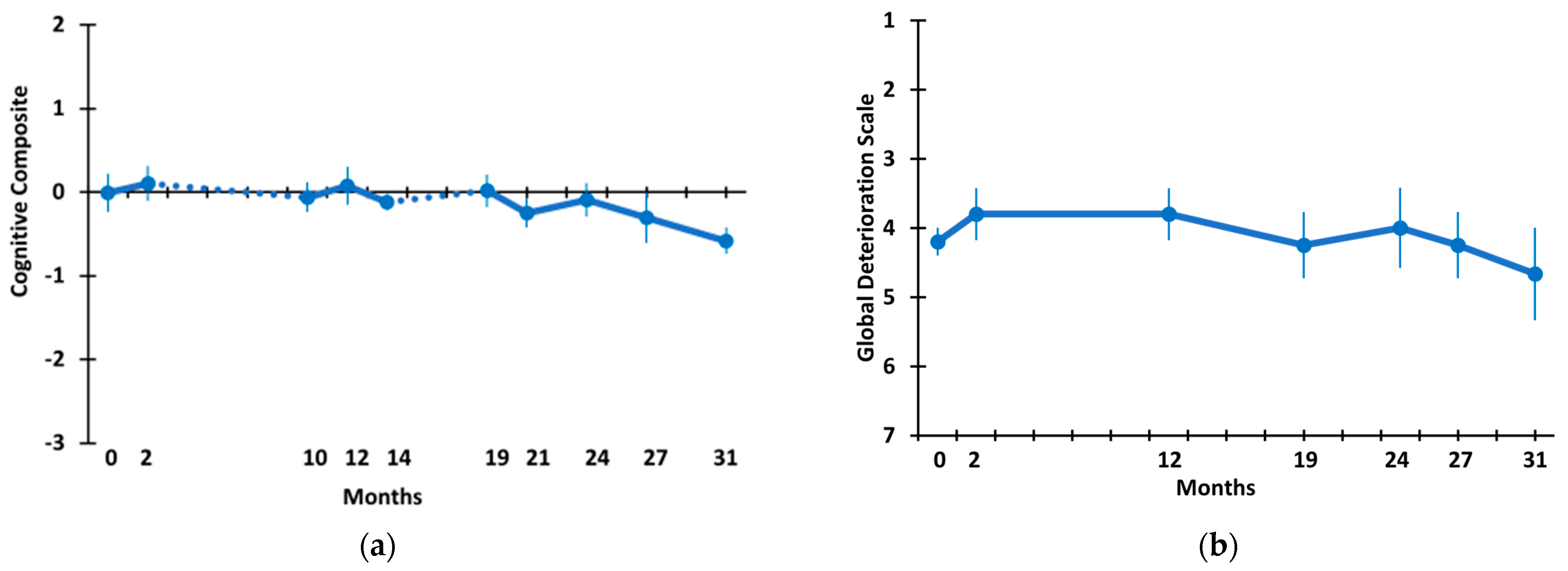
3.2. Long-Term TEMT Effects on Cognition/Functional Measures in AD Subjects over a 2½-Year Period
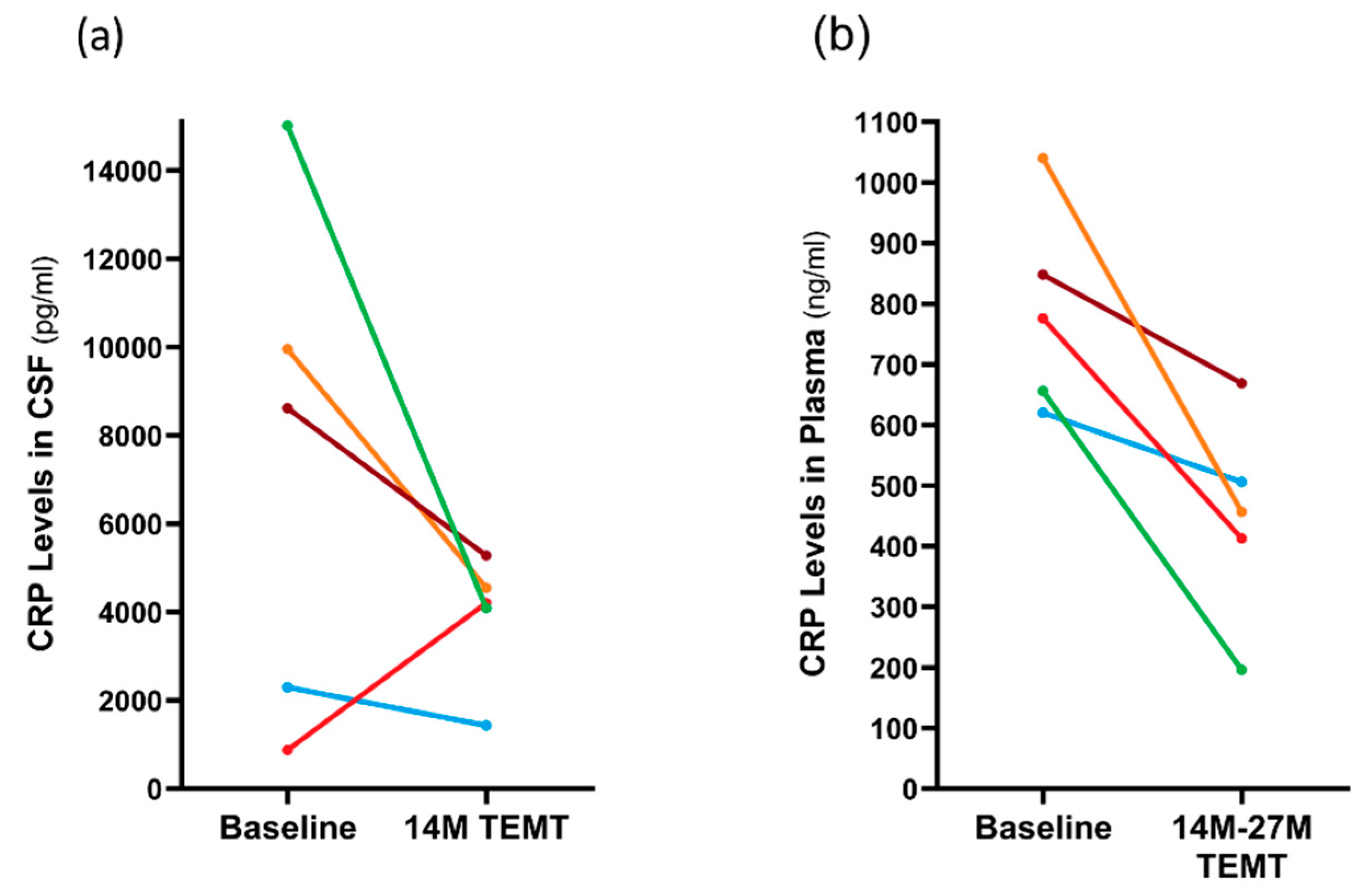
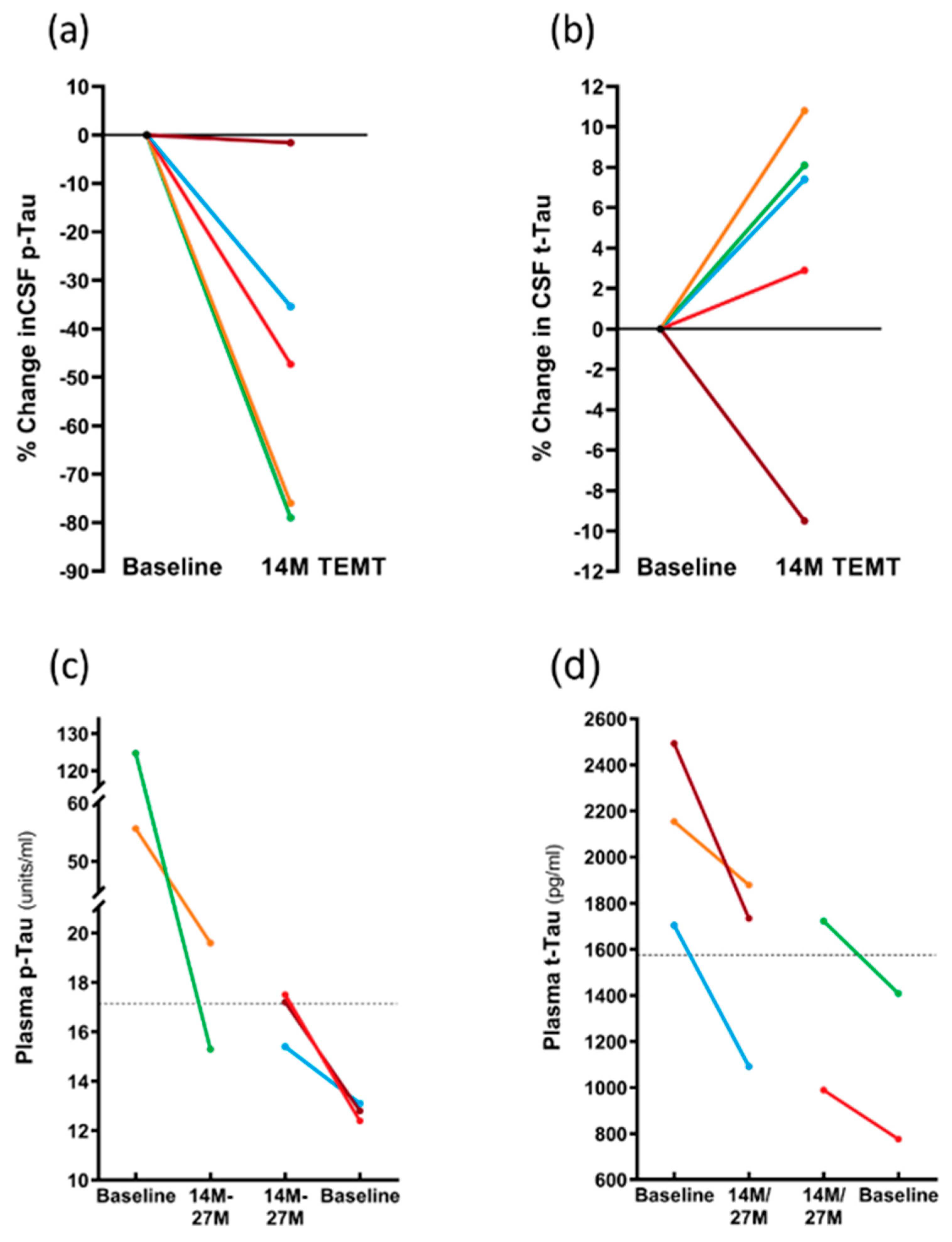
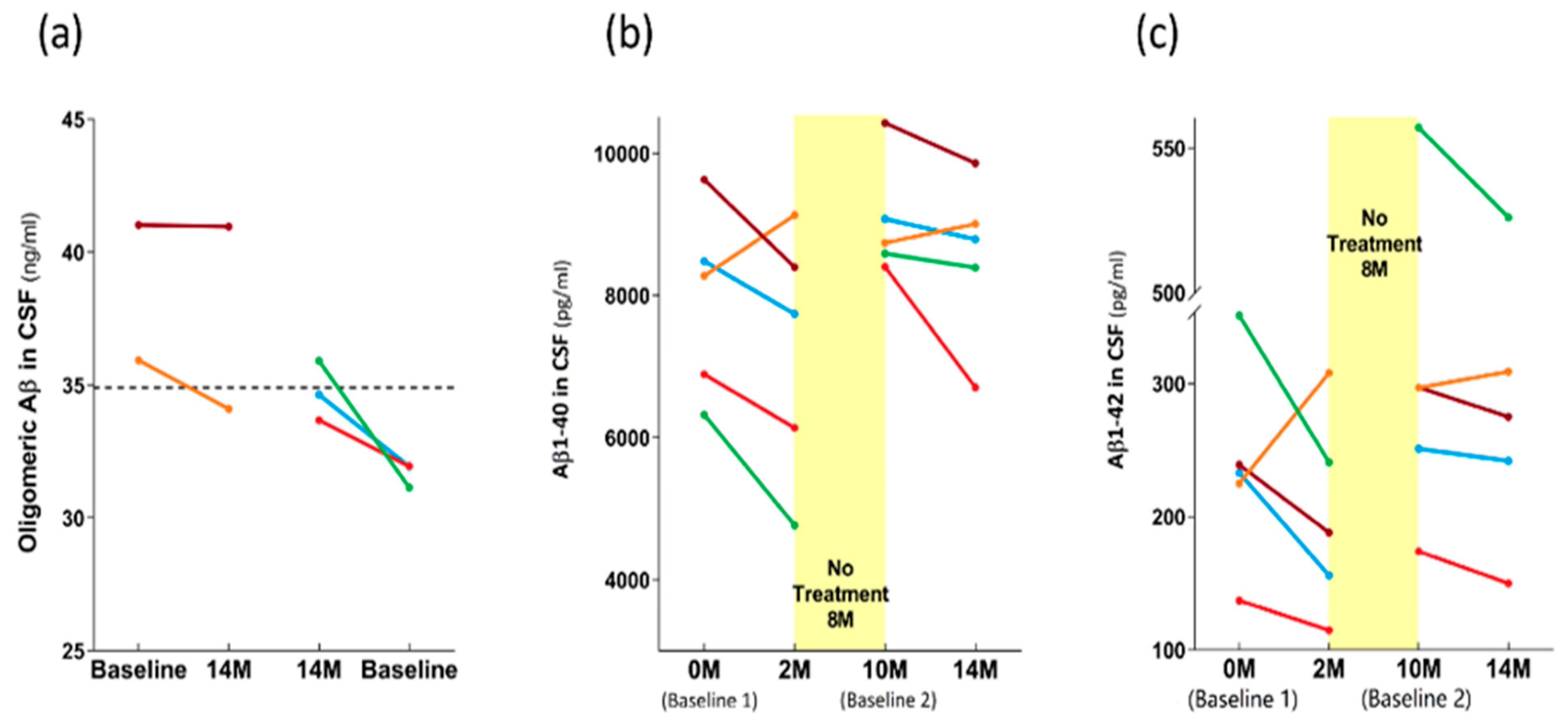
3.3. Long-Term Effects of TEMT on AD Markers in Both CSF and Plasma over a 2+ Year Period
4. Discussion
4.1. Safety of TEMT
4.2. Long-Term Cognitive Benefits of TEMT
4.3. Long-Term Effects of TEMT on AD Markers in Blood and CSF
4.4. Mechanisms of TEMT Action
4.5. Other Neuromodulatory Approaches against AD and Their Mechanisms of Action
4.6. Study Limitations
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Disease International. Available online: https://www.alz.co.uk/research/statistics (accessed on 29 January 2022).
- Alzheimer’s Association (USA). Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 29 January 2022).
- Levin, J.; Voglein, J.; Quiroz, Y.; Bateman, R.; Ghisays, V.; Lopera, F.; McDade, E.; Reiman, E.; Tariot, P.N.; Morris, J.C. Testing the amyloid cascade hypothesis: Prevention trials in autosomal dominant Alzheimer Disease. Alzheimers Dement. 2022. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Alzheimer’s disease drug development and the problem of the blood-brain barrier. Alzheimer’s Dement. 2009, 5, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.T.; Heegaard, N.H. Analysis of protein aggregation in neurodegenerative disease. Anal. Chem. 2013, 85, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.Y.; Teplow, D.B. Amyloid-β proteins and Alzheimer’s Disease. Alzheimer’s Res. Ther. 2013, 5, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Goure, W.F.; Krafft, G.A.; Jerecic, J.; Hefti, F. Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimer’s Res. Ther. 2014, 6, 42–56. [Google Scholar] [CrossRef]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Gerson, J.; Kayed, R. Formation and propagation of tau oligomeric seeds. Front. Neurol. 2013, 4, 93. [Google Scholar] [CrossRef]
- Guerrero-Muñoz, M.J.; Gerson, J.; Castillo-Carranza, D.L. Tau oligomers: The toxic player at synapses in Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 464. [Google Scholar] [CrossRef]
- Fa, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Puma, D.D.L.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular tau oligomers produce an immediate impairment of LTP and emory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef]
- Cline, E.; Bicca, M.; Viola, K.; Kirsten, L. The amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Dragicevic, N.; Bradshaw, P.C.; Mamcartz, M.; Lin, X.; Wang, L.; Cao, C.; Arendash, G. Long-term electromagnetic field treatment enhances brain mitochondrial function of both Alzheimer’s transgenic mice and normal mice: A mechanism for electromagnetic field-induced cognitive benefit? ” Neuroscience 2011, 185, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Zussy, C.; Brureau, A.; Keller, E.; Marchal, S.; Blayo, C.; Delair, B.; Ixart, G.; Maurice, T.; Givalois, L. Alzheimer’s disease related markers, cellular toxicity and behavioral deficits induced six weeks after oligomeric amyloid-beta peptide injection in rats. PLoS ONE 2013, 8, e53117. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choi, H.; Min, J.; Kim, B.; Lee, S.; Yun, J.W.; Choi, M.-S.; Chang, K.-T.; Lee, D.-S. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. Neurochemistry 2015, 132, 687–702. [Google Scholar] [CrossRef]
- Zempel, H.; Thies, E.; Mankelkow, E.; Mandelkow, E.-M. Abeta oligomers cause localized calcium elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D. Soluble amyloid-beta protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Mandelkow, E. Linking amyloid-beta and tau: Amyloid-beta induced synaptic dysfunction via local wreckage of the neuronal cytoskeleton. Neurodegener. Dis. 2012, 10, 64–72. [Google Scholar] [CrossRef]
- Umeda, T.; Ramser, E.; Yamashita, M.; Nakajima, K.; Mori, H.; Silverman, M.; Tomiyama, T. Intracellular amyloid-beta oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol. Comm. 2015, 3, 51. [Google Scholar] [CrossRef]
- Takahashi, R.; Almeida, G.; Kearney, F.; Yu, F.; Lin, M.; Milner, T.; Gouras, G. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 2004, 24, 3592–3599. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.; Castillo-Carranza, D.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegen. 2011, 6, 39. [Google Scholar] [CrossRef]
- Nisbet, R.; Polanco, J.-C.; Ittner, L.M.; Jurgen, G. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef]
- Arendash, G.W.; Sanchez-Ramos, J.; Mori, T.; Mamcarz, M.; Lin, X.; Runfeldt, M.; Wang, l.; Zhang, G.; Sava, V.; Tan, J.; et al. Electromagnetic field treatment protects against and reverses cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimer’s Dis. 2010, 19, 191–210. [Google Scholar] [CrossRef]
- Arendash, G.W.; Mori, T.; Dorsey, M.; Gonzalez, R.; Tajiri, N.; Borlongan, C. Electromagnetic treatment to old Alzheimer’s mice reverses β-amyloid deposition, modifies cerebral blood flow, and provides selected cognitive benefit. PLoS ONE 2012, 7, e35751. [Google Scholar]
- Mori, T.; Arendash, G.W. Electromagnetic field treatment enhances neuronal activity: Linkage to cognitive benefit and therapeutic implications for Alzheimer’s Disease. J. Alzheimer’s Dis. Parkinsonism 2011, 1, 2. [Google Scholar] [CrossRef]
- Arendash, G.W.; Cao, C.; Zhao, H.; Baranowski, R.; Shen, N.; Yang, H.; Wang, J. Transcranial electromagnetic treatment disaggregates Aβ, tau, and α-synuclein oligomers in Alzheimer’s brain homogenates. Being Submitted. 2022.
- Arendash, G.; Cao, C.; Abulaban, H.; Baranowski, R.; Wisniewski, G.; Becerra, L.; Andel, R.; Lin, X.; Zhang, X.; Wittwer, D.; et al. A Clinical Trial of Transcranial Electromagnetic Treatment in Alzheimer’s Disease: Cognitive Enhancement and Associated Changes in Cerebrospinal Fluid, Blood, and Brain Imaging. J. Alzheimer’s Dis. 2019, 71, 57–82. [Google Scholar] [CrossRef]
- Knopman, D.; Jack, C.; Wiste, H.; Lundt, E.; Weigand, S.; Vemuri, P.; Lowe, V.J.; Kantarci, K.; Gunter, J.L.; Senjem, M.L.; et al. 18Ffluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol. Aging 2014, 35, 2096–2106. [Google Scholar] [CrossRef]
- Jack, C.; Knopman, D.; Weigand, S.; Wiste, H.; Vemuri, P.; Lowe, V.; Kantarci, K.; Gunter, J.; Senjem, M.; Ivnik, R.; et al. An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann. Neurol. 2012, 71, 765–775. [Google Scholar] [CrossRef]
- Jack, C.; Wiste, H.; Weigand, S.; Rocca, W.; Knopman, D.; Mielke, M.; Lowe, V.; Vemuri, P.; Mielke, M.; Roberts, R.; et al. Age-specific population frequencies of cerebral beta-amyloidosis and neurodegeneration among people with normal cognitive function aged 50–89 years: A cross-section study. Lancet Neurol. 2014, 13, 997–1005. [Google Scholar] [CrossRef]
- Niemantsverdriet, E.; Ottoy, J.; Somers, C.; De Roeck, E.; Struyfs, H.; Soetewey, F.; Verhaeghe, J.; Van den Bossche, R.; Van Mossevelde, S.; Goeman, J.; et al. The cerebrospinal fluid Aβ1-42/Aβ1-40 ratio improves concordance with amyloid-PET for diagnosing Alzheimer’s disease in a clinical setting. J. Alzheimer’s Dis. 2017, 60, 561–576. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; He, J.-S.; Bao, F.-X.; Sun, W.-M.; Dai, X.-X.; Wang, X.-B.; Li, Y.-Q.; Zhen, X.-X.; Hu, H.-G.; et al. Preparation and characterization of a monoclonal antibody with high affinity for soluble Aβ oligomers. Hybridoma 2009, 28, 349–354. [Google Scholar]
- Gueorguieva, R.; Krystal, J.H. Move over ANOVA. Arch. Gen. Psychiatry 2004, 61, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Podhorna, J.; Krahnke, T.; Shear, M.; Harrison, J. Alzheimer’s Disease Neuroimaging Initiative: Alzheimer’s Disease assessment scale-Cognitive subscale variants in mild cognitive impairment and mild Alzheimer’s disease; change over time and the effect of enrichment strategies. Alzheimer’s Res. Ther. 2004, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.-S.; Libon, D.; Rascovsky, K.; Grossman, M.; Xie, S.W. Differential longitudinal decline on the mini-mental state examination in Frontotemporal Lobar Degeneration and Alzheimer’s Disease. Alzheimer Dis. Assoc. Disord. 2013, 27, 310–315. [Google Scholar] [CrossRef]
- Callahan, C.; Boustani, M.; Schmid, A.; LaMantia, M.; Austrom, M.; Miller, D.; Gao, S.; Ferguson, D.; Lane, K.; Hendrie, H.; et al. Targeted functional decline in Alzheimer’s Disease: A randomized trial. Ann. Intern. Med. 2017, 166, 164–171. [Google Scholar] [CrossRef]
- Arendash, G.W. Transcranial electromagnetic treatment against Alzheimer’s disease: Why it has the potential to trump Alzheimer’s disease drug development. J. Alzheimer’s Dis. 2012, 32, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W. Review of the evidence that Transcranial Electromagnetic Treatment will be a safe and effective therapeutic against Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 53, 753–771. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kazui, H.; Matsumoto, K.; Nakano, Y.; Yasuda, M.; Mori, E. Does donepezil treatment slow the progression of hippocampal atrophy in patients with Alzheimer’s disease? Am. J. Psychiatry 2005, 162, 676–682. [Google Scholar] [CrossRef]
- Dubois, B.; Chupin, M.; Hampel, H.; Lista, S.; Cavedo, E.; Croisele, B.; Tisserand, G.L.; Touchon, J.; Bonafe, A.; Ousset, P.J.; et al. Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1041–1049. [Google Scholar] [CrossRef]
- Bentwich, J.; Dobronevsky, E.; Aichenbaum, S.; Shorer, R.; Peretz, R.; Khaigrekht, M.; Marton, R.G.; Rabey, J.M. Beneficial effect of repetitive transcranial magnetic stimulation combined with cognitive training for the treatment of Alzheimer’s disease: A proof of concept study. J. Neural. Transm. 2011, 118, 463–471. [Google Scholar] [CrossRef]
- Gerriero, F.; Botarelli, E.; Mele, G.; Polo, L.; Zoncu, D.; Renati, P.; Sgarlata, C.; Rollone, M.; Ricevuti, G.; Maurizi, N.; et al. An innovative intervention for the treatment of cognitive impairment—Emisymmetric bilateral stimulation improves cognitive functions in Alzheimer’s disease and mild cognitive impairment: An open-label study. Neuropsychiatr. Dis. Treat. 2015, 11, 2391. [Google Scholar] [CrossRef]
- Mathews, M.; Abner, E.; Kryscio, R.; Jicha, G.; Cooper, G.; Smith, C.; Caban-Holt, A.; Schmitt, F. Diagnostic accuracy and practice effects in the National Alzheimer’s Coordinating Center Uniform Data Set neuropsychological battery. Alzheimers Dement. 2014, 11, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, N.; Samtani, M.; Farnum, M.; Yang, E.; Novak, G.; Grudman, M.; Narayan, V.; DiBernardo, A.; Alzheimer’s Disease Neuroimaging Initiative. The ADAS-cog revised: Novel composite scales based on ADAS-cog to improve efficiency in MCI and early AD trials. Alzheimer’s Dement. 2013, 9, S21–S31. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, D.; Acevedo, A.; Luis, C.; Crum, T.; Barker, W.; Duara, R. Semantic interference deficits and the detection of mild Alzheimer’s disease and mild cognitive impairment without dementia. J. Int. Neuropsychol. Soc. 2004, 10, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Ayutyanont, N.; Langbaum, J.; Hendrix, S.; Chen, K.; Fliesher, S.; Fresenhahn, M.; Ward, M.; Aguirre, C.; Acosta-Baena, N.; Madrigal, L.; et al. The Alzheimer’s prevention initiative composite cognitive test score: Sample size estimates for the evaluation of preclinical Alzheimer’s disease treatments in presenilin 1E280A mutation carriers. J. Clin. Psychiatry 2014, 75, 652–660. [Google Scholar] [CrossRef][Green Version]
- Malek-Ahmadi, M.; Chen, K.; Perez, S.; He, A.; Mufson, E. Cognitive composite score association with Alzheimer’s disease plaque and tangle pathology. Alzheimer’s Res. Ther. 2018, 10, 90. [Google Scholar] [CrossRef]
- Langbaum, J.; Ellison, N.; Caputo, A.; Thomas, R.; Langlois, C.; Riviere, M.-E.; Graf, A.; Lopez, C.L.; Reiman, E.M.; Tariot, P.N.; et al. The Alzheimer’s prevention initiative composite cognitive test: A practical measure for tracking cognitive decline in preclinical Alzheimer’s disease. Alzheimers Res. Ther. 2012, 12, 66. [Google Scholar] [CrossRef]
- Wessels, A.; Andersen, S.; Dowsett, S.; Siemers, E.R. The integrated Alzheimer’s Disease rating scale (iADRS) Findings from the EXPEDITION3 trial. J. Prev. Alzheimer’s Dis. 2018, 5, 134–136. [Google Scholar] [CrossRef]
- Mintun, M.; Lo, A.; Duggan Evens, C.; Wessels, A.; Ardayfio, P.; Andersen, S.; Shcherbinin, S.; Sparks, J.; Sims, J.; Brys, M.; et al. Donanemab in early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Shen, X.-N.; Niu, L.-D.; Wang, Y.-J.; Cao, X.-P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.-T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598. [Google Scholar] [CrossRef]
- Natale, G.; Clouston, S.; Smith, D. Elevated C-reactive protein in Alzheimer’s Disease without depression in older adults: Finds from the health and retirement study. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 673–682. [Google Scholar] [CrossRef]
- Al-Baradie, R.; Pu, S.; Liu, D.; Zeinolabediny, Y.; Ferris, G.; Sanfel, C.; Corpas, R.; Garcia-Lara, E.; Alsagaby, A.; Alshehri, B.; et al. Monomeric C-reactive protein localized in the cerebral tissue of damaged vascular brain regions is associated with neuro-inflammation and neurodegeneration—An immunohistochemical study. Front. Immunol. 2021, 12, 644213. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Howell, J.; Ozturk, T.; Gangishetti, U.; Kollhof, A.; Hatcher-Martin, J.; Hatcher-Martin, J.M.; Anderson, A.M.; Tyor, W.R. CSF cytokines in aging, multiple sclerosis, and dementia. Front. Immunol. 2019, 10, 480. [Google Scholar] [CrossRef] [PubMed]
- Fila, M. Re-balancing of inflammation and abeta immunity as a therapeutic for Alzheimer’s disease-view from the bedside. CNS Neurol. Disord. Drug Targets 2010. [CrossRef]
- Sopova, K.; Gatsiou, K.; Stellos, K.; Laske, C. Dysregulation of neurotrophic and haematopoietic growth factors in Alzheimer’s disease: From pathophysiology to novel treatment strategies. Curr. Alzheimer Res. 2014, 11, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Webers, A.; Heneka, M.; Gleeson, P. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Candore, G.; Caruso, C.; Jirillo, E.; Magrone, T.; Vasto, S. Low grade inflammation as a common pathogenetic denominator in age-related diseases: Novel drug targets for anti-ageing strategies and successful ageing achievement. Curr. Pharm. Des. 2010, 16, 584–596. [Google Scholar] [CrossRef]
- Cao, C.; Abulaban, H.; Baranowski, R.; Wang, Y.; Bai, I.; Lin, X.; Shen, N.; Zhang, X.; Arendash, G.W. Transcranial Electromagnetic Treatment “rebalances” blood and brain cytokine levels in Alzheimer’s patients: A new mechanism for reversal of their cognitive impairment. Front. Aging Neurosci. 2022, 14, 829049. [Google Scholar] [CrossRef]
- Iqbal, K.; Gong, C.-X.; Liu, F. Hyperphosphorylation-induced tau oligomers. Front. Neurol. 2013, 4, 112. [Google Scholar] [CrossRef]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020, 11, 1683. [Google Scholar] [CrossRef]
- Wallin, A.; Blennow, K.; Andreasen, N.; Minthon, L. CSF biomarkers for Alzheimer’s disease: Levels of beta-amyloid, tau, phosphorylated tau related to clinical symptoms and survival. Dement. Geriatr. Cogn. Disord. 2006, 21, 131–138. [Google Scholar] [CrossRef]
- Niemantsverdriet, E.; Valckx, S.; Bjerke, M.; Engelborghs, S. Alzheimer’s Disease CSF Biomarkers: Clinical indications and rational use. Acta Neurol. Belg. 2017, 117, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Dumurgier, J.; Ayrignac, X.; Marelli, C.; Alcolea, D.; Ormaechea, J.F.; Thouvenot, E.; Delaby, C.; Hirtz, C.; Vialaret, J.; et al. Cerebrospinal fluid A-beta 1-40 peptides increase in Alzheimer’s disease and are highly correlated with phosphor-tau in control individuals. Alzheimer’s Res. Ther. 2020, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Kang, G.; Kwon, J.; Choi, H.; Pack, J.; Kim, N.; Lee, Y.-S.; Lee, H.-J. 1950 MHz electromagnetic fields ameliorate A-beta pathology in Alzheimer’s disease mice. Curr. Alzheimer Res. 2015, 12, 481–492. [Google Scholar] [CrossRef]
- Son, Y.; Kim, J.; Jeong, Y.; Jeong, Y.; Kwon, J.; Choi, H.-D. Long-term RF exposure on behavior and cerebral glucose metabolism in 5xFAD mice. Neurosci. Lett. 2018, 666, 64–69. [Google Scholar] [CrossRef] [PubMed]
- English, N.; Solomentsev, G.; O’Brien, P. Nonequilibrium molecular dynamics study of electric and low-frequency microwave fields on hen egg white lysozyme. J. Chem. Phys. 2009, 131, 035106. [Google Scholar] [CrossRef] [PubMed]
- Gerner, C.; Haudek, V.; Schandl, U.; Bayer, B.; Gundacker, N.; Hutter, H.; Mosgoeller, W. Increased protein synthesis by cells exposed to a 1800 MHz RF mobile phone electromagnetic field. Int. Arch. Occup. Environm. Health 2010, 83, 691–702. [Google Scholar] [CrossRef]
- Todorova, N.; Bentveizen, A.; English, N.; Yarovsky, I. Electromagnetic-field effects on structure and dynamics of amyloidogenic peptides. J. Chem. Physics. 2016, 144, 085101. [Google Scholar] [CrossRef]
- Blank, M.; Goodman, R. Initial interactions in electromagnetic field-induced biosynthesis. J Cell Physiol 2004, 199, 359–363. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: Oxford, England, 1997. [Google Scholar]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef]
- Tansey, M.; Goldberg, M. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef]
- Kustrimovic, N.; Marino, F.; Cosentino, M. Peripheral immunity, immunoaging, and Neuroinflammation in Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 3719–3753. [Google Scholar] [CrossRef] [PubMed]
- King, E.; Thomas, A. Systemic inflammation in Lewy Body Diseases: A systematic review. Alzheimer Dis. Assoc. Disord. 2017, 31, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Usenko, T.; Nikolaev, M.; Miliukhina, I.; Bezrukova, A.; Senkevich, K.; Gomzyakova, N.; Beltceva, Y.A.; Zalutskaya, N.M.; Gracheva, E.V.; Timofeeva, A.A.; et al. Plasma cytokine profile in synucleinophaties with dementia. J. Clin. Neurosci. 2020, 78, 323–326. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.; Baloh, R. Inflammation in ALC/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef]
- Bright, F.; Chan, G.; van Hummel, A.; Ittner, L.; Yazi, D. TDP-43 and inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Int. J. Mol. Sci. 2021, 22, 7781. [Google Scholar] [CrossRef]
- Assenza, G.; Capone, F.; di Biase, L.; Ferreri, F.; Florio, L.; Guerra, A.; Marano, M.; Paolucci, M.; Ranieri, F.; Salomone, G.; et al. Oscillatory activities in neurological disorders of elderly: Biomarkers to target for neuromodulation. Front. Aging Neurosci. 2017, 9, 189. [Google Scholar] [CrossRef]
- Sanches, C.; Stengel, C.; Godard, J.; Mertz, J.; Teichmann, M.; Migliaccio, R.; Valero-Cabre, A. Past, present, and future of non-invasive brain stimulation approaches to treat cognitive impairment in neurodegenerative diseases: Time for a comprehensive critical review. Front. Aging Neurosci. 2021, 12, 578339. [Google Scholar] [CrossRef]
- Cantone, M.; Di Pino, G.; Capone, F.; Piombo, M.; Chiarello, D.; Cheeran, B.; Pennisi, G.; Di Lazzaro, V. The contribution of transcranial magnetic stimulation in the diagnosis and in the management of dementia. Clin. Neurophysiol. 2014, 125, 1509–1532. [Google Scholar] [CrossRef]
- Sabbagh, M.; Sadowsky, C.; Tousi, B.; Agronin, M.; Alva, G.; Armon, C.; Bernick, C.; Keegan, A.P.; Karantzoulis, S.; Baror, E.; et al. Effects of a combined transcranial magnetic stimulation (TMS) and cognitive training intervention in patients with Alzheimer’s disease. Alzheimers Dement. 2020, 16, 641–650. [Google Scholar] [CrossRef]
- Minoura, I.; Muto, E. Dielectric measurement of individual microtubules using the electro-orientation method. Biophys. J. 2006, 90, 3739–3748. [Google Scholar] [CrossRef]
- Jagust, L.; Landau, S.; Shar, L.; Trjanowski, J.; Koeppe, R.; Reiman, E.; Foster, N.; Petersen, R.; Weiner, M.; Price, J.; et al. Relationships between biomarkers in aging and dementia. Neurology 2009, 73, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Landau, S.; Harvey, D.; Madison, C.; Koeppe, R.; Reiman, E.; Foster, N.; Weiner, M.; Jagust, W.; Alzheimer’s Disease Neuroimaging Initiative. Associations between cognitive, functional and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 2011, 32, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Laxton, A.; Tang-Wai, D.; McAndrews, M.; Zumsteg, D.; Wennberg, R.; Keren, R.; Wherrett, J.; Naglie, G.; Hamani, C.; Smith, G.S.; et al. A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann. Neurol. 2010, 68, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Saltmarche, A.; Naeser, M.; Ho, K.; Hamblin, M.; Lim, L. Significant improvement in cognition in mild to moderately severe dementia cases treated with transcranial plus intranasal photobiomodulation: Case series report. Photomed. Laser Surg. 2017, 35, 432–441. [Google Scholar] [CrossRef]
- He, Z.; Colon-Motas, K.; Pybus, A.; Piendel, L.; Seppa, J.; Walker, M.L.; Manzanares, C.M.; Qiu, D.; Miocinovic, S.; Wood, L.B.; et al. A feasibility trial of gamma sensory flicker for patients with prodromal Alzheimer’s disease. Alzheimers Dement. 2021, 7, e12178. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | 1 | 2 | 3 | 4 | 5 | Mean |
|---|---|---|---|---|---|---|
| Age | 77 | 76 | 66 | 63 | 65 | 69.4 |
| Gender | F | F | M | F | F | ----- |
| ApoE Genotype | 2/3 | 3/3 | 3/3 | 3/3 | 2/3 | ----- |
| MMSE Score | 26 | 19 | 21 | 17 | 19 | 20.4 |
| GDS Rating | 4 | 4 | 3 | 5 | 4 | 4.0 |
| Education | 19 | 16 | 15 | 13 | 14 | 15.4 |
| Anat. MRI Analysis | a,b | a,b | a | b | a | ----- |
| PET AD Sign. ROI | 1.10 | 1.06 | 1.10 | 1.32 | 1.40 | 1.20 |
| Aβ1-42/t-tau Ratio | 1.48 | 1.02 | 0.93 | 0.61 | ----- | 1.01 |
| ADAS-cog13 Score | 24.0 | 26.7 | 30.3 | 30.7 | 37.3 | 29.8 |
| COGNITIVE DOMAIN | Est. | SEM | p-Value | Baseline vs. 27–31 M |
|---|---|---|---|---|
| ADAS-Cog Overall | p= 0.144 (t = −2.34) | |||
| Intercept | −0.7 | 0.29 | 0.091 | |
| Change over time | 0.02 | 0.02 | 0.457 | |
| ADAS Immediate Recall | p= 0.423 (t = 1.00) | |||
| Intercept | 0.11 | 1.21 | 0.931 | |
| Change over time | −0.01 | 0.03 | 0.848 | |
| Rey AVLT Sum of 5 Trials | p= 0.525 (t = 0.762) | |||
| Intercept | 0.44 | 0.43 | 0.365 | |
| Change over time | −0.03 | 0.03 | 0.239 | |
| Rey AVLT Retroactive Interference | p= 1.000 (t = 0.000) | |||
| Intercept | 0.66 | 0.48 | 0.246 | |
| Change over time | −0.11 | 0.04 | 0.051 | |
| MMSE | ||||
| Intercept | 0.66 | 0.23 | 0.067 | p= 0.287 (t = 1.44) |
| Change over time | −0.05 | 0.03 | 0.266 | |
| ADL | p= 0.225 (t = 1.73) | |||
| Intercept | 0.67 | 0.38 | 0.177 | |
| Change over time | −0.06 | 0.03 | 0.137 | |
| Digits Forward | p= 0.184 (t = 2.00) | |||
| Intercept | −0.13 | 0.38 | 0.754 | |
| Change over time | 0.05 | 0.03 | 0.153 | |
| Digits Backward | p= 0.667 (t = 0.50) | |||
| Intercept | 0.23 | 0.41 | 0.606 | |
| Change over time | −0.03 | 0.03 | 0.342 | |
| Cognitive composite | p= 0.328 (t = 1.28) | |||
| Intercept | 0.17 | 0.2 | 0.456 | |
| Change over time | −0.01 | 0.01 | 0.399 | |
| Global Deterioration Scale (GDS) | p= 0.423 (t = −1.00) | |||
| (Qualitative, by caregiver) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arendash, G.; Abulaban, H.; Steen, S.; Andel, R.; Wang, Y.; Bai, Y.; Baranowski, R.; McGarity, J.; Scritsmier, L.; Lin, X.; et al. Transcranial Electromagnetic Treatment Stops Alzheimer’s Disease Cognitive Decline over a 2½-Year Period: A Pilot Study. Medicines 2022, 9, 42. https://doi.org/10.3390/medicines9080042
Arendash G, Abulaban H, Steen S, Andel R, Wang Y, Bai Y, Baranowski R, McGarity J, Scritsmier L, Lin X, et al. Transcranial Electromagnetic Treatment Stops Alzheimer’s Disease Cognitive Decline over a 2½-Year Period: A Pilot Study. Medicines. 2022; 9(8):42. https://doi.org/10.3390/medicines9080042
Chicago/Turabian StyleArendash, Gary, Haitham Abulaban, Susan Steen, Ross Andel, Yanhong Wang, Yun Bai, Rob Baranowski, Jon McGarity, Lyle Scritsmier, Xiaoyang Lin, and et al. 2022. "Transcranial Electromagnetic Treatment Stops Alzheimer’s Disease Cognitive Decline over a 2½-Year Period: A Pilot Study" Medicines 9, no. 8: 42. https://doi.org/10.3390/medicines9080042
APA StyleArendash, G., Abulaban, H., Steen, S., Andel, R., Wang, Y., Bai, Y., Baranowski, R., McGarity, J., Scritsmier, L., Lin, X., Shen, N., Aljassabi, A., Li, Y., & Cao, C. (2022). Transcranial Electromagnetic Treatment Stops Alzheimer’s Disease Cognitive Decline over a 2½-Year Period: A Pilot Study. Medicines, 9(8), 42. https://doi.org/10.3390/medicines9080042