PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block?


 ,
, 
Abstract
1. Introduction
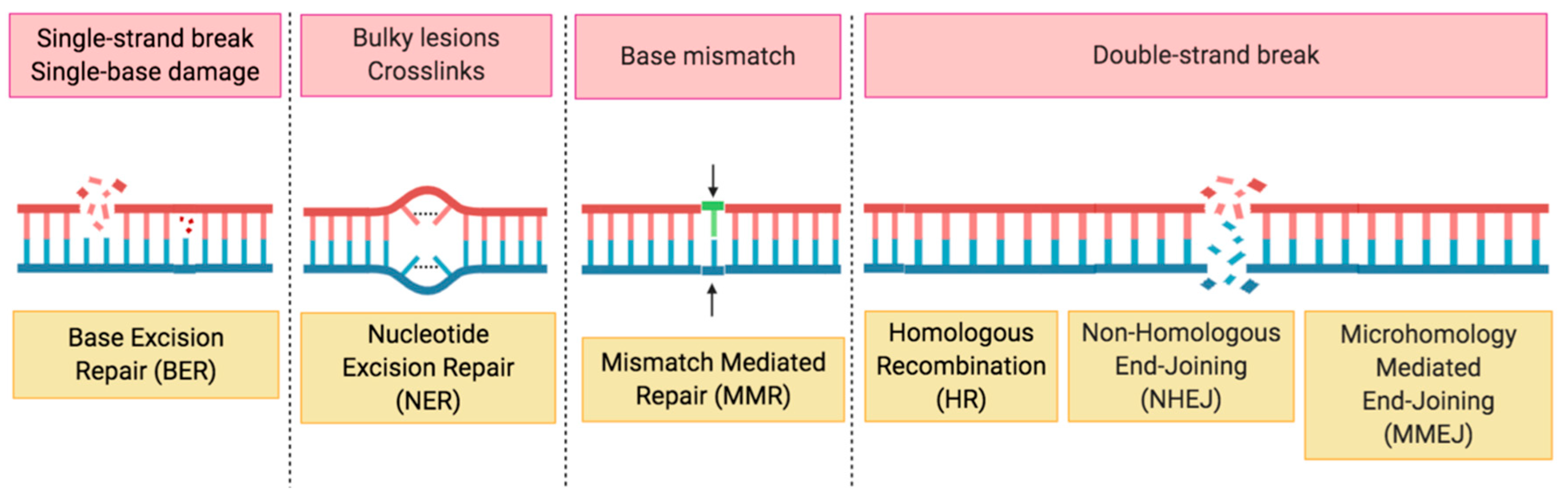
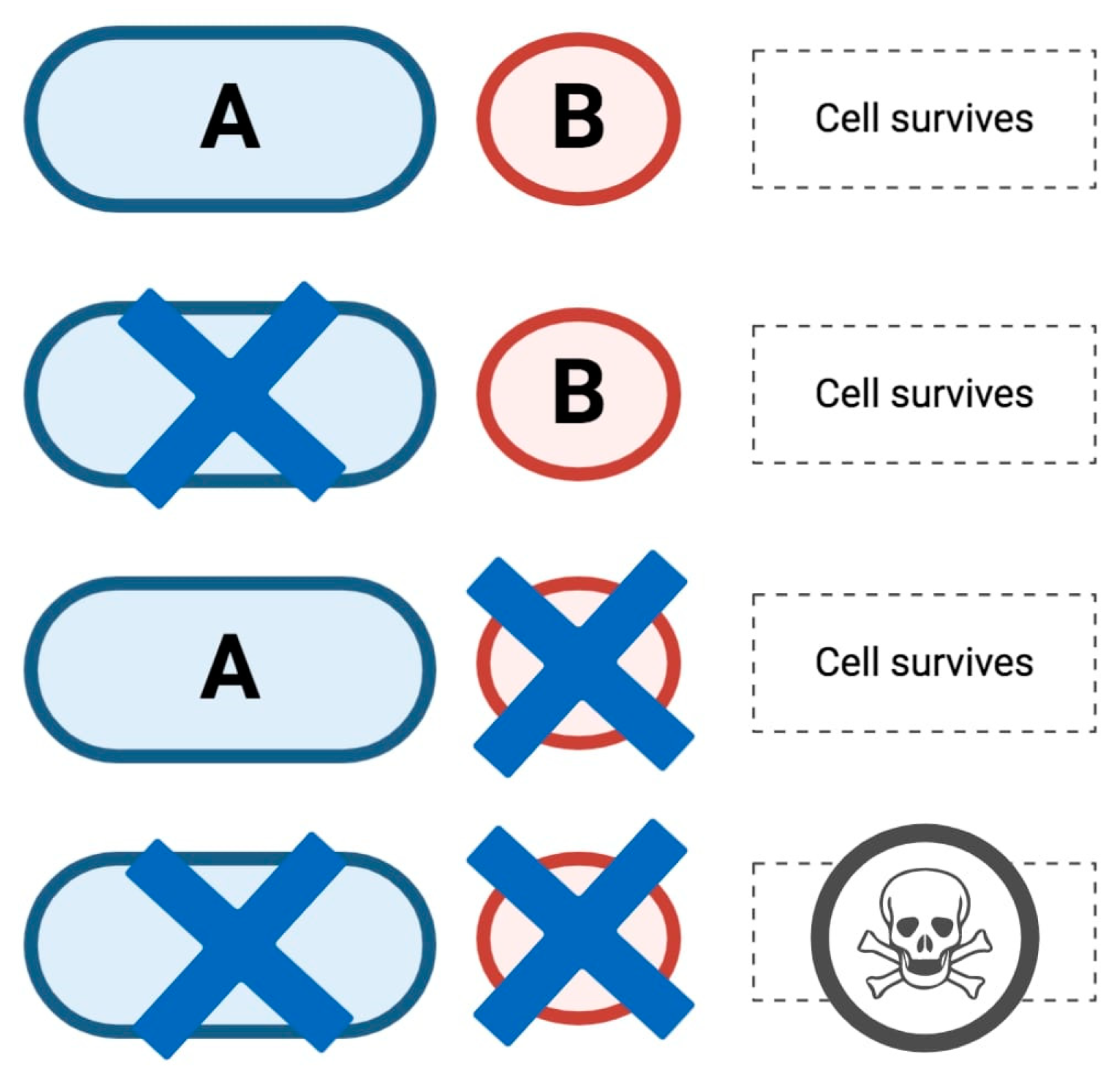
2. HRD, the Role of PARP in DDR and Synthetic Lethality
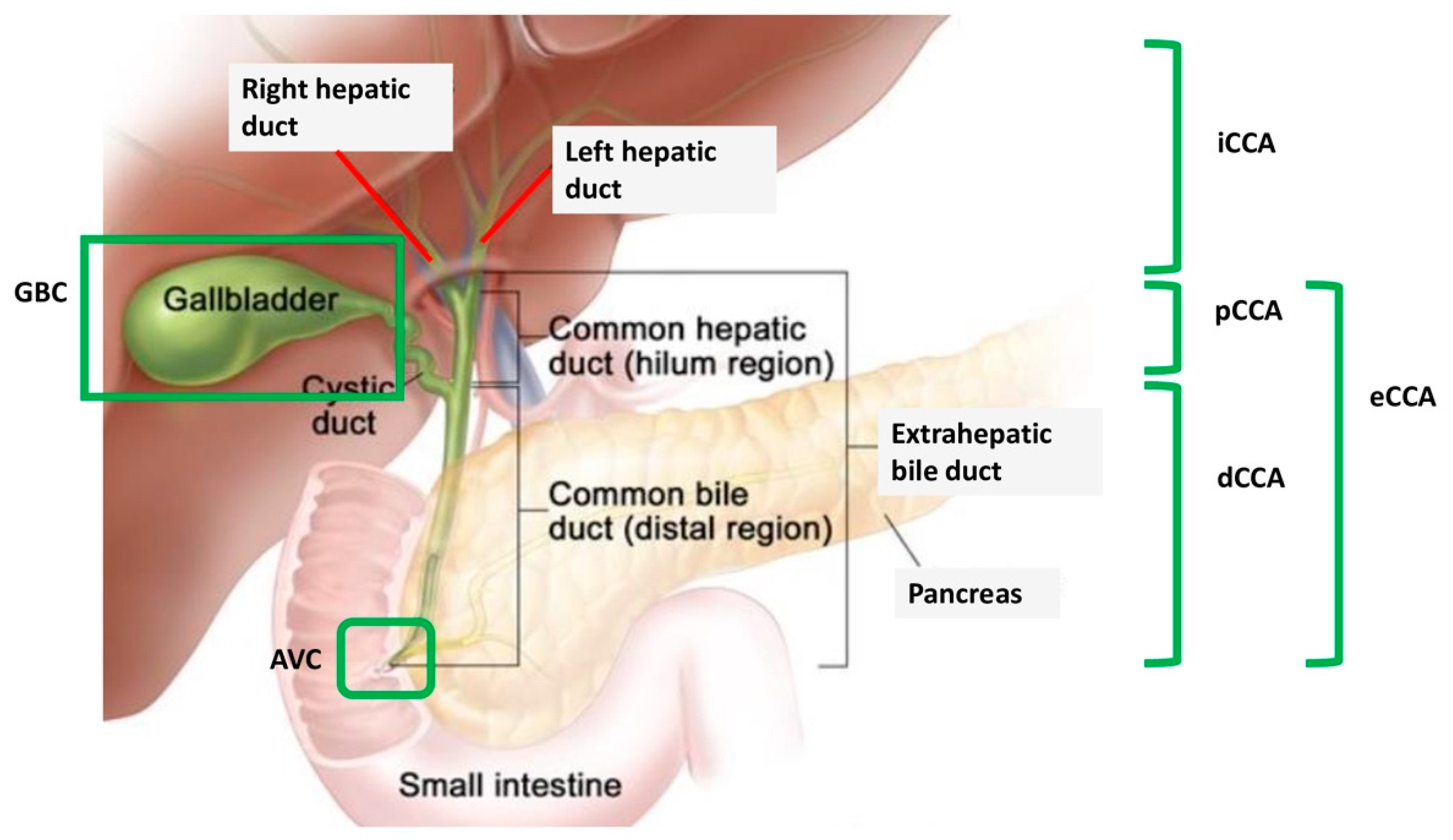
3. DDR Deficiency and BRCAm in BTC
4. PARPi in BTC
5. Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvisé, M.; Lamarca, M. Clinical Presentation, Diagnosis and Staging of Cholangiocarcinoma. Liver Int. 2019, 39, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Davidson, B.R.; Goldin, R.D.; Heaton, N.; Karani, J.; Pereira, S.P.; Rosenberg, W.M.C.; Tait, P.; Taylor-Robinson, S.D.; Thillainayagam, A.V.; et al. Guidelines for the Diagnosis and Treatment of Cholangiocarcinoma: An Update. Gut 2012, 61, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Charbel, H.; Al-Kawas, F.H. Cholangiocarcinoma: Epidemiology, risk factors, pathogenesis, and diagnosis. Curr. Gastroenterol. Rep. 2011, 13, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Zhu, A.X.; Fuchs, C.S.; Brooks, G.A. Forty-year trends in cholangiocarcinoma incidence in the US: Intrahepatic disease on the rise. Oncologist 2016, 21, 594–599. [Google Scholar] [CrossRef]
- Alsaleh, M.; Leftley, Z.; Barbera, T.A.; Sithithaworn, P.; Khuntikeo, N.; Loilome, W.; Yongvanit, P.; Cox, I.J.; Chamadol, N.; Syms, R.R.A.; et al. Cholangiocarcinoma: A Guide for the Nonspecialist. Int. J. Gen. Med. 2019, 12, 13–23. [Google Scholar] [CrossRef]
- Brandi, G.; Rizzo, A.; Dall’Olio, F.G.; Felicani, C.; Ercolani, G.; Cescon, M.; Frega, G.; Tavolari, S.; Palloni, A.; De Lorenzo, S.; et al. Percutaneous radiofrequency ablation in intrahepatic cholangiocarcinoma: A retrospective single-center experience. Int. J. Hyperth. 2020, 37, 479–485. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Valle, J.W.; Furuse, J.; Jitlal, M.; Baere, S.; Mizuno, N.; Wasan, H.; Bridgewater, J.; Okusaka, T. Cisplatin and Gemcitabine for Advanced Biliary Tract Cancer: A Meta-Analysis of Two Randomised Trials. Ann. Oncol. 2014, 25, 391–398. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Tober, N.; Nigro, M.C.; Mosca, M.; Palloni, A.; Abbati, F.; Frega, G.; De Lorenzo, S.; Tavolari, S.; et al. Second-line Treatment in Advanced Biliary Tract Cancer: Today and Tomorrow. Anticancer Res. 2020, 40, 3013–3030. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational Landscape of Intrahepatic Cholangiocarcinoma. Nat. Commun. 2014, 5, 5596. [Google Scholar]
- Rizzo, A.; Frega, G.; Ricci, A.D.; Palloni, A.; Abbati, F.; De Lorenzo, S.; Deserti, M.; Tavolari, S.; Brandi, G. Anti-EGFR Monoclonal Antibodies in Advanced Biliary Tract Cancer: A Systematic Review and Meta-analysis. In Vivo 2020, 34, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Kwong, L.N.; Javle, M. Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice. Curr. Treat. Options Oncol. 2016, 17, 58. [Google Scholar] [CrossRef]
- Golan, T.; Raitses-Gurevich, M.; Kelley, R.K.; Bocobo, A.G.; Borgida, A.; Shroff, R.T.; Holter, S.; Gallinger, S.; Ahn, D.H.; Aderka, D.; et al. Overall Survival and Clinical Characteristics of BRCA-Associated Cholangiocarcinoma: A Multicenter Retrospective Study. Oncologist 2017, 22, 804–810. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; Christensen, R.D.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Paschalis, A.; de Bono, J. Prostate Cancer 2020: “The Times They Are a’Changing”. Cancer Cell 2020, 38, 25–27. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Patel, M.; Nowsheen, S.; Maraboyina, S.; Xia, F. The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: A review. Cell Biosci. 2020, 10, 35. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA. Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef]
- Marks, E.I.; Yee, N.S. Molecular genetics and targeted therapeutics in biliary tract carcinoma. World J Gastroenterol. 2016, 22, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef] [PubMed]
- Rabenau, K.; Hofstatter, E. DNA Damage Repair and the Emerging Role of Poly(ADP-ribose) Polymerase Inhibition in Cancer Therapeutics. Clin. Ther. 2016, 38, 1577–1588. [Google Scholar] [CrossRef][Green Version]
- Min, A.; Im, S.A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef]
- Garje, R.; Vaddepally, R.K.; Zakharia, Y. PARP Inhibitors in Prostate and Urothelial Cancers. Front. Oncol. 2020, 10, 114. [Google Scholar] [CrossRef]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef]
- Xie, S.; Mortusewicz, O.; Ma, H.T.; Herr, P.; Poon, R.Y.C.; Helleday, T.; Qian, C. Timeless Interacts with PARP-1 to Promote Homologous Recombination Repair. Mol. Cell 2015, 60, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B.; Butepage, M.; Eckei, L.; Krieg, S.; Verheugd, P.; Shilton, B.H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018, 118, 1092–1136. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Messner, S.; Hassa, P.O.; Fey, M.; Hottiger, M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009, 37, 3723–3738. [Google Scholar] [CrossRef]
- Daniels, C.M.; Ong, S.E.; Leung, A.K. Phosphoproteomic approach to characterize protein monoand poly(ADP-ribosyl)ation sites from cells. J. Proteome Res. 2014, 13, 3510–3522. [Google Scholar] [CrossRef]
- Palazzo, L.; Leidecker, O.; Prokhorova, E.; Dauben, H.; Matic, I.; Ahel, I. Serine is the major residue for ADP-ribosylation upon DNA damage. Elife 2018, 7. [Google Scholar] [CrossRef]
- Leidecker, O.; Bonfiglio, J.J.; Colby, T.; Zhang, Q.; Atanassov, I.; Zaja, R.; Palazzo, L.; Stockum, A.; Ahel, I.; Matic, I. Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat. Chem. Biol. 2016, 12, 998–1000. [Google Scholar] [CrossRef]
- Leslie Pedrioli, D.M.; Leutert, M.; Bilan, V.; Nowak, K.; Gunasekera, K.; Ferrari, E.; Imhof, R.; Malmstrom, L.; Hottiger, M.O. Comprehensive ADP-ribosylome analysis identifies tyrosine as an ADP-ribose acceptor site. EMBO Rep. 2018, 19, e45310. [Google Scholar] [CrossRef]
- Martello, R.; Leutert, M.; Jungmichel, S.; Bilan, V.; Larsen, S.C.; Young, C.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 2016, 7, 12917. [Google Scholar] [CrossRef]
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol. Oncol. 2017, 147, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.N.; Eskander, R.N. PARP Inhibitors in Epithelial Ovarian Cancer. Recent Pat. Anticancer Drug Discov. 2018, 13, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Gonzalez, R.; Jacobson, M.K. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry 1987, 26, 3218–3224. [Google Scholar] [CrossRef] [PubMed]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell. Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Kunze, F.A.; Hottiger, M.O. Regulating Immunity via ADP-Ribosylation: Therapeutic Implications and Beyond. Trends Immunol. 2019, 40, 159–173. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Caldecott, K.W. Perspectives on PARPs in S Phase. Trends Genet. 2019, 35, 412–422. [Google Scholar] [CrossRef]
- Azarm, K.; Smith, S. Nuclear PARPs and genome integrity. Genes Dev. 2020, 34, 285–301. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.; Kim, D.; Yoo, C.; Kim, K.P.; Jeong, J.H.; Chang, H.M.; Lee, S.S.; Park, D.H.; Song, T.J.; Hwang, S.; et al. Therapeutic relevance of targeted sequencing in management of patients with advanced biliary tract cancer: DNA damage repair gene mutations as a predictive biomarker. Eur. J. Cancer 2019, 120, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Bekaii-Saab, T. Biliary tract cancer and genomic alterations in homologous recombinant deficiency: Exploiting synthetic lethality with PARP inhibitors. Chin. Clin. Oncol. 2020, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushmann, M.; Salem, M.E.; Battaglin, F.; et al. Molecular profile of BRCA-mutated biliary tract cancers. ESMO Open 2020, 5, e000682. [Google Scholar] [CrossRef]
- Saeed, A.; Park, R.; Al-Jumayli, M.; Al-Rajabi, R.; Sun, W. Biologics, Immunotherapy, and Future Directions in the Treatment of Advanced Cholangiocarcinoma. Clin. Colorectal. Cancer 2019, 18, 81–90. [Google Scholar] [CrossRef]
- Jenner, Z.B.; Sood, A.K.; Coleman, R.L. Evaluation of rucaparib and companion diagnostics in the PARP inhibitor landscape for recurrent ovarian cancer therapy. Future Oncol. 2016, 12, 1439–1456. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef]
- Pellegrino, B.; Mateo, J.; Serra, V.; Balmana, J. Controversies in oncology: Are genomic tests quantifying homologous recombination repair deficiency (HRD) useful for treatment decision making? ESMO Open 2019, 4, 1–5. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; Dika, I.E.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3248. [Google Scholar] [CrossRef]
- Moeini, A.; Sia, D.; Bardeesy, N.; Mazzaferro, V.; Llovet, J.M. Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W.M. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Biliary Tract Cancer: State of the Art and potential role of DNA Damage Repair. Cancer Treat. Rev. 2018, 70, 168–177. [Google Scholar] [CrossRef]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hebert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA. 2014, 111, 285–290. [Google Scholar] [CrossRef]
- Adeva, J.; Sangro, B.; Salati, M.; Edeline, J.; La Casta, A.; Bittoni, A.; Berardi, R.; Bruix, J.; Valle, J.W. Medical treatment for cholangiocarcinoma. Liver Int. 2019, 39, 123–142. [Google Scholar] [CrossRef]
- Easton, D. Cancer risks in BRCA2 mutation carriers: The breast cancer linkage consortium. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [Google Scholar]
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy—a double-edged sword. J. Appl. Genet. 2019, 60, 329–334. [Google Scholar] [CrossRef]
- Sharma, M.B.; Carus, A.; Sunde, L.; Hamilton-Dutoit, S.; Ladekarl, M. BRCA-associated pancreatico-biliary neoplasms: Four cases illustrating the emerging clinical impact of genotyping. Acta Oncol. 2016, 55, 377–381. [Google Scholar] [CrossRef]
- Xie, Y.; Jiang, Y.; Yang, X.B.; Wang, A.Q.; Zheng, Y.C.; Wan, X.S.; Sang, X.T.; Wang, K.; Zhang, D.D.; Xu, J.J.; et al. Response of BRCA1-mutated gallbladder cancer to olaparib: A case report. World J. Gastroenterol. 2016, 22, 10254–10259. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA -Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Fehling, S.C.; Miller, A.L.; Garcia, P.L.; Vance, R.B.; Yoon, K.J. The combination of BET and PARP inhibitors is synergistic in models of cholangiocarcinoma. Cancer Lett. 2020, 468, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Caramelo, O.; Silva, C.; Caramelo, F.; Frutuoso, C.; Almeida-Santos, T. The effect of neoadjuvant platinum-based chemotherapy in BRCA mutated triple negative breast cancers -systematic review and meta-analysis. Hered. Cancer Clin. Pract. 2019, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Cecere, S.; Du Bois, A.; Harter, P.; Heitz, F. Treatment of recurrent ovarian cancer. Ann. Oncol. 2017, 28, viii51–viii56. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, A.; Szylberg, Ł.; Saganek, M.; Napiontek, W.; Antosik, P.; Grzanka, D. Emerging strategies in BRCA-positive pancreatic cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Go, R.S.; Adjei, A.A. Review of the Comparative Pharmacology and Clinical Activity of Cisplatin and Carboplatin. J. Clin. Oncol. 1999, 17, 409–422. [Google Scholar] [CrossRef]
- Tutt, A.N.J.; Lord, C.J.; McCabe, N.; Farmer, H.; Turner, N.; Martin, N.M.; Jackson, S.P.; Smith, G.C.; Ashworth, A. Exploiting the DNA Repair Defect in BRCA Mutant Cells in the Design of New Therapeutic Strategies for Cancer. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 139–148. [Google Scholar] [CrossRef]
- Byrski, T.; Gronwald, J.; Huzarski, T.; Dent, R.A.; Zuziak, D.; Wi’sniowski, R.; Marczyk, E.; Blecharz, P.; Szurek, O.; Cybulski, C.; et al. Neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Heredit. Cancer Clin. Pract. 2011, 9, A4. [Google Scholar] [CrossRef]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of Neoadjuvant Cisplatin in Triple-Negative Breast Cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Byrski, T.; Huzarski, T.; Dent, R.; Marczyk, E.; Jasiowka, M.; Gronwald, J.; Jakubowicz, J.; Cybulski, C.; Wisniowski, R.; Godlewski, D.; et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res. Treat. 2014, 147, 401–405. [Google Scholar] [CrossRef]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Shroff, R.T. Biliary tract cancers: Systemic therapy for advanced disease. Chin. Clin. Oncol. 2020, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J. Hepatol. 2020, 73, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, J.; Li, R.; Han, Z.; Li, L.; Li, G.; Yang, B.; Yin, Q.; Wang, Y.; Ke, Y.; et al. Effectiveness of Olaparib Treatment in a Patient with Gallbladder Cancer with an ATM-Inactivating Mutation. Oncologist 2020, 25, 375–379. [Google Scholar] [CrossRef]
- Ricci, A.D.; Rizzo, A.; Novelli, M.; Tavolari, S.; Palloni, A.; Tober, N.; Abbati, F.; Mollica, V.; De Lorenzo, S.; Turchetti, D.; et al. Specific Toxicity of Maintenance Olaparib Versus Placebo in Advanced Malignancies: A Systematic Review and Meta-analysis. Anticancer Res. 2020, 40, 597–608. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 13, 1378–1388. [Google Scholar] [CrossRef]
- Chong, D.Q.; Zhu, A.X. The landscape of targeted therapies for cholangiocarcinoma: Current status and emerging targets. Oncotarget 2016, 7, 46750–46767. [Google Scholar] [CrossRef]
- Ding, L.; Chen, X.; Xu, X.; Qian, Y.; Liang, G.; Yao, F.; Yao, Z.; Wu, H.; Zhang, J.; He, Q.; et al. PARP1 Suppresses the Transcription of PD-L1 by Poly(ADP-Ribosyl)ating STAT3. Cancer Immunol. Res. 2019, 7, 136–149. [Google Scholar] [CrossRef]
- Friedlander, M.; Meniawy, T.; Markman, B.; Mileshkin, L.R.; Harnett, P.; Millward, M.; Lundy, J.; Freimund, A.E.; Norris, C.; Wu, J.; et al. A phase 1b study of the anti-PD-1 monoclonal antibody BGB-A317 (A317) in combination with the PARP inhibitor BGB- 290 (290) in advanced solid tumors. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Hasvold, G.; Lund-Andersen, C.; Lando, M.; Patzke, S.; Hauge, S.; Suo, Z.; Lyng, H.; Syljuasen, R.G. Hypoxia- induced alterations of G2 checkpoint regulators. Mol. Oncol. 2016, 10, 764–773. [Google Scholar] [CrossRef]
- Daly, C.S.; Flemban, A.; Shafei, M.; Conway, M.E.; Qualtrough, D.; Dean, S.J. Hypoxia modulates the stem cell population and induces EMT in the MCF-10A breast epithelial cell line. Oncol. Rep. 2018, 39, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.G.; Karam, E.; Moujaess, E.; Kourie, H.R. Poly-(ADP-ribose) polymerase inhibitors: Paradigm shift in the first-line treatment of newly diagnosed advanced ovarian cancer. Pharmacogenomics 2020, 21, 721–727. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell. Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef]
- Wang, D.; Wang, M.; Jiang, N.; Zhang, Y.; Bian, X.; Wang, X.; Roberts, T.M.; Zhao, J.J.; Liu, P.; Cheng, H. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget 2016, 7, 13153–13166. [Google Scholar] [CrossRef]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.S.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar] [CrossRef]
- Schmitz, K.J.; Lang, H.; Wohlschlaeger, J.; Sotiropoulos, G.C.; Reis, H.; Schmid, K.W.; Baba, H.A. AKT and ERK1/2 signaling in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2007, 13, 6470–6477. [Google Scholar] [CrossRef]
- Chung, J.Y.; Hong, S.M.; Choi, B.Y.; Cho, H.; Yu, E.; Hewitt, S.M. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clin. Cancer Res. 2009, 15, 660–667. [Google Scholar] [CrossRef]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Fritcher, E.G.B.; Graham, R.P.; Zhang, L.; Highsmith, W.E.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.F.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [PubMed]
- IDH-Mutant Tumors Vulnerable to PARP Inhibition. Available online: https://cancerdiscovery.aacrjournals.org/content/7/4/OF4 (accessed on 30 August 2020).
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Design | Cohort | Agent(s) | DDR Defect Screenings | Primary Endpoint |
|---|---|---|---|---|---|
| NCT03212274 | Phase II, single arm | Refractory, metastatic CCA with IDH1 or IDH2 mutation | Olaparib | no | ORR |
| NCT03207347 (UF-STO-ETI-001) | Phase II, non-randomized | CCA after prior standard systemic treatment | Niraparib | yes * | ORR |
| NCT03991832 | Phase II, non-randomized | IDH-mutated BTC after no more than 2 previous treatments | Olaparib + durvalumab | no | ORR, DCR |
| NCT03878095 | Phase II, single arm | CCA or other IDH-mutated solid tumors after prior standard treatment | Olaparib + ceralasertib | no | ORR |
| NCT03639935 | Phase II, single arm | BTC after prior standard systemic treatment | Rucaparib + nivolumab | no | Proportion of patients alive and without radiological or clinical progression at 4 months |
| NCT04042831 | Phase II, single arm | BTC with somatic/germline mutations in DDR genes after platinum-based chemotherapy | Olaparib | yes ** | ORR |
| NCT03337087 | Phase I–II, single arm | Metastatic BTC after no more than 1 line of prior therapy in the metastatic setting | Nal-IRI and 5-FU with rucaparib | yes, only for phase II (HRD or BRCA1 or BRCA2 or PALB2) | dose limiting toxicities, ORR |
| NCT04171700 | Phase II, single arm | Unresectable, locally advanced, or metastatic solid tumor after first-line treatment (including ampullary cancer) | Rucaparib | yes *** | ORR |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, A.D.; Rizzo, A.; Bonucci, C.; Tober, N.; Palloni, A.; Mollica, V.; Maggio, I.; Deserti, M.; Tavolari, S.; Brandi, G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines 2020, 7, 54. https://doi.org/10.3390/medicines7090054
Ricci AD, Rizzo A, Bonucci C, Tober N, Palloni A, Mollica V, Maggio I, Deserti M, Tavolari S, Brandi G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines. 2020; 7(9):54. https://doi.org/10.3390/medicines7090054
Chicago/Turabian StyleRicci, Angela Dalia, Alessandro Rizzo, Chiara Bonucci, Nastassja Tober, Andrea Palloni, Veronica Mollica, Ilaria Maggio, Marzia Deserti, Simona Tavolari, and Giovanni Brandi. 2020. "PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block?" Medicines 7, no. 9: 54. https://doi.org/10.3390/medicines7090054
APA StyleRicci, A. D., Rizzo, A., Bonucci, C., Tober, N., Palloni, A., Mollica, V., Maggio, I., Deserti, M., Tavolari, S., & Brandi, G. (2020). PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines, 7(9), 54. https://doi.org/10.3390/medicines7090054