Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications



Abstract
1. Introduction
2. Tumor Evolution of Prostate Cancer from Precursor Lesions
3. Genetic Abnormalities of Prostate Cancer
3.1. Intertumor and Intratumor Heterogeneity
3.2. Main Genetic Abnormalities in Prostate Cancer
3.3. Genetic Abnormalities of Metastatic Disease
3.4. Genetic Abnormalities in Neuroendocrine Prostate Cancer
4. Most Recurrent Genetic Abnormalities Observed in Prostate Cancer
4.1. TMRSS2-ERG
4.2. SPOP Mutations
4.3. CDH1 Abnormalities
4.4. Androgen Receptor Abnormalities
4.5. PTEN Gene Abnormalities
4.6. NKX3.1
4.7. MYC
4.8. RB and TP53
4.9. LRF
4.10. CDK12
4.11. PLZF
5. Racial Influences on Prostate Cancer Genomics
6. Gene Expression Profiling Studies
7. Association of Genomic Abnormalities with Patient Clinical Outcomes
8. Sensitivity of Prostate Cancer to Immunotherapy
9. Circular RNA and Prostate Cancer
10. Hormonal Regulation of Prostate Cancer
11. Abnormalities of Metabolism in Prostate Cancer
12. Prostate Stem Cells
13. Stem Cells in Benign Prostatic Hyperplasia (BPH)
14. Prostate Cancer Stem Cells
15. Novel Therapies for Prostate Cancer
16. Prostate Cancer Models
17. Emerging Topics and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Mellinger, G.T. Prognosis of prostatic carcinoma. Recent Results Cancer Res. 1977, 60, 61–72. [Google Scholar]
- Bailar, J.C.; Mellinger, G.T.; Gleason, D.F. Survival rates of patients with prostatic cancer, tumor stage, and differentiation-preliminary report. Cancer Chemother. Rep. 1966, 50, 129–136. [Google Scholar] [PubMed]
- Gleason, D.F. Histological grading and staging of prostatic carcinoma. In Urologic Pathology: The Prostate; Tannenbaum, M., Ed.; Lea and Feibiger: Philadelphia, PA, USA, 1977; p. 171. [Google Scholar]
- Epstein, J.I. Prostate cancer grading: A decade after the 2005 modified system. Modern Pathol. 2018, 31, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Zelefsky, M.J.; Sjoberg, D.D.; Nelson, J.B.; Egevad, L.; Magi-Galluzzi, C.; Vickers, A.J.; Parwani, A.V.; Reuter, V.E.; Fine, S.W.; et al. A contemporary prostate cancer grading system: A validated alternative to the Gleason score. Eur. Urol. 2016, 69, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Rubin, M.A.; Girelli, G.; Dermichelis, F. Genomic correlates to the newly proposed grading prognostic groups for prostate cancer. Eur. Urol. 2016, 69, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Tolkach, Y.; Kristiansen, G. Is high-grade prostatic intraepithelial neoplasia (HGPIN) a reliable precursor for prostate carcinoma? Implications for clonal evolution and early detection strategies. J. Pathol. 2018, 244, 389–393. [Google Scholar] [CrossRef]
- Sowalsky, A.G.; Ye, H.; Bubley, G.J.; Balk, S.P. Clonal progression of prostate cancers from Gleason grade 3 to grade 4. Cancer Res. 2013, 73, 1050–1055. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Cheville, J.C.; Murphy, S.J.; Johnson, S.H.; Zarei, S.; Kosari, F.; Sukov, W.R.; Karnes, R.J.; Vasmatzis, G. Lineage relationship of Gleason patterns in Gleason score 7 prostate cancer. Cancer Res. 2013, 73, 3275–3284. [Google Scholar] [CrossRef]
- Trock, B.J.; Fedor, H.; Gurel, B.; Jenkins, R.B.; Knudsen, B.S.; Fine, S.W.; Said, J.W.; Carter, H.B.; Lotan, T.L.; De Marzo, A.M. PTEN loss and chromosome 8 alterations in Gleason grade 3 prostate cancer cores predicts the presence of un-sampled grade 4 tumor: Implications for active surveillance. Mod. Pathol. 2016, 29, 764–771. [Google Scholar] [CrossRef]
- Hernandez, S.; Font-Tello, A.; Juanpere, N.; de Muga, S.; Lorenzo, M.; Salido, M.; Fumadò, L.; Serrano, L.; Cecchini, L.; Serrano, S.; et al. Concurrent TMPRSS2-ERG and SLC45A3-ERG rearrangements plus PTEN loss are not found in low grade prostate cancer and define an aggressive tumor subset. Prostate 2016, 76, 854–865. [Google Scholar] [CrossRef]
- VanderWeel, D.J.; Brown, C.D.; Taxy, J.B.; Gillard, M.; Hatcher, D.M.; Tom, W.R.; Stadler, W.M.; White, K.P. Low-grade prostate cancer diverges early from high grade and metastatic disease. Cancer Sci. 2014, 105, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Sowalsky, A.G.; Kissick, H.T.; Gerrin, S.J.; Schaefer, R.J.; Xia, Z.; Russo, J.W.; Arredouani, M.S.; Bubley, G.J.; Sanda, M.G.; Li, W.; et al. Gleason score 7 prostate cancer emerge through branched evolution of clonal Gleason pattern 3 and 4. Clin. Cancer Res. 2017, 23, 3823–3833. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Eeles, R.; Wedge, D.G.; Loo, P.L.; Gundem, G.; Alexandrov, L.M.; Kremeyer, B.; Butler, A.; Lynch, A.G.; Camacho, N.; et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansion in neoplastic and morphologically normal prostate tissue. Nat. Genet. 2015, 47, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Lochead, P.; Chan, A.T.; Nishihara, R.; Fuchs, C.S.; Beck, A.H.; Giovanucci, E.; Ogino, S. Etiologic field effect: Reappraisal of the field effect concept in cancer predisposition and progression. Mod. Pathol. 2015, 28, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Gerrin, S.J.; Sowalsky, A.G.; Balk, S.P.; Ye, H. Mutation profiling indicates high grade prostatic intraepithelial neoplasia as distant precursors of adjacent invasive prostatic adenocarcinoma. Prostate 2016, 76, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Shin, S.; Kim, M.S.; Baek, I.P.; Lee, S.H.; Kim, T.M.; Lee, S.H.; Chung, Y.J. Genetic progression of high grade prostatic intraepithelial neoplasia to prostate cancer. Eur. Urol. 2016, 69, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Weier, C.; Xu, M.M.; Vaghasia, A.; Gürel, B.; Gümüşkaya, B.; Esopi, D.M.; Fedor, H.; Tan, H.L.; Kulac, I.; et al. Molecular evidence that invasive adenocarcinoma can mimic prostatic intraepithelial neoplasia (PIN) and intraductal carcinoma through retrograde glandular colonization. J. Pathol. 2016, 238, 31–41. [Google Scholar] [CrossRef]
- Trabzonlu, L.; Kulac, I.; Zheng, Q.; Hicks, J.L.; Haffner, M.C.; Nelson, W.G.; Sfanos, K.S.; Ertunc, O.; Lotan, T.L.; Heaphy, C.M.; et al. Molecular Pathology of High-Grade Prostatic Intraepithelial Neoplasia: Challenges and Opportunities. Cold Spring Harb. Perspect Med. 2018, 9. [Google Scholar] [CrossRef]
- Zhou, M. High-grade prostatic intraepithelial neoplasia, PIN-like carcinoma, ductal carcinoma, and intraductal carcinoma of the prostate. Mod. Pathol. 2018, 31, S71–S79. [Google Scholar] [CrossRef]
- Paulk, A.; Giannico, G.; Epstein, J.I. PIN-like (Ductal) Adenocarcinoma of the Prostate. Am. J. Surg. Pathol. 2018, 42, 1693–1700. [Google Scholar] [CrossRef]
- Han, B.; Mehra, R.; Suleman, K.; Tomlins, S.A.; Wang, L.; Singhal, N.; Linetzky, K.A.; Palanisamy, N.; Zhou, M.; Chinnaiyan, A.M.; et al. Characterization of ETS gene aberrations in select histologic variants of prostate carcinoma. Mol. Pathol. 2009, 22, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Morais, C.L.; Herawi, M.; Toubaji, A.; Albadine, R.; Hicks, J.; Netto, G.J.; De Marzo, A.M.; Epstein, J.I.; Lotan, T.L. PTEN loss and ERG protein expression are infrequent in prostatic ductal adenocarcinomas and concurrent acinar carcinomas. Prostate 2015, 75, 1610–1619. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schweizer, M.T.; Cheng, H.H.; Tretiakova, M.S.; Vakar-Lopez, F.; Klemfuss, N.; Konnick, E.Q.; Mostaghel, E.A.; Nelson, P.S.; Yu, E.Y.; Montgomery, B.; et al. Mismatch repair deficiency may be common in ductal adenocarcinoma of the prostate. Oncotarget 2016, 7, 82504–82510. [Google Scholar] [CrossRef] [PubMed]
- Isaacsson Velho, P.; Silberstein, J.L.; Markowski, M.C.; Luo, J.; Lotan, T.L.; Isaacs, W.B.; Antonarakis, E.S. Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate 2018, 78, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Gillard, M.; Lack, J.; Pontier, A.; Gandla, D.; Hatcher, D.; Sowalsky, A.G.; Rodriguez-Nieves, J.; Vander Griend, D.; Paner, G.; VanderWeele, D. Integrative Genomic Analysis of Coincident Cancer Foci Implicates CTNNB1 and PTEN Alterations in Ductal Prostate Cancer. Eur. Urol. Focus 2019, 5, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Seipel, A.H.; Whitington, T.; Delahunt, B.; Samaratunga, H.; Mayrhofer, M.; Wiklund, P.; Grönberg, H.; Lindberg, J.; Egevad, L. Genetic profile of ductal adenocarcinoma of prostate. Human Pathol. 2017, 69, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Weier, C.; Haffner, M.C.; Mosbruger, T.; Esopi, D.M.; Hicks, J.; Zheng, Q.; Fedor, H.; Isaacs, W.B.; De Marzo, A.M.; Nelson, W.G.; et al. Nucleotide resolution analysis of TMPRSS2 and ERG rearrangements in prostate cancer. J. Pathol. 2013, 230, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Kristiansen, P.; Wiklund, P.; Grönberg, H.; Egevad, L. Tracking the origin of metastatic prostate cancer. Eur. Urol. 2015, 67, 819–822. [Google Scholar] [CrossRef]
- Taylor, R.A.; Fraser, M.; Livingstone, J.; Espiritu, S.M.; Thorne, H.; Huang, V.; Lo, W.; Shiah, Y.J.; Yamaguchi, T.N.; Sliwinski, A. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat. Commun. 2017, 8, 13671. [Google Scholar] [CrossRef]
- Kweldam, C.F.; Kummerlin, I.P.; Nieboer, D.; Verhoef, E.I.; Steyerberg, E.W.; van der Kwast, T.H.; Roobol, M.J.; van Leenders, G.J. Disease-specific survival of patients with invasive cribriform and intraductal prostate cancer at diagnostic biopsy. Mod. Pathol. 2016, 29, 630–636. [Google Scholar] [CrossRef]
- Bottcher, R.; Kweldam, C.F.; Livingstone, J.; Lalonde, E.; Yamaguchi, T.N.; Huang, V.; Yousif, F.; Fraser, M.; Bristow, R.G.; van der Kwast, T.; et al. Cribriform and intraductal prostate cancer are associated with increased genomic instability and distinct genomic alterations. BMC Cancer 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Elfandy, H.; Armenia, J.; Pederzoli, F.; Pullman, E.; Pértega-Gomes, N.; Schultz, N.; Viswanathan, K.; Vosoughi, A.; Blattner, M.; Stopsack, K.H.; et al. Genetic and Epigenetic Determinants of Aggressiveness in Cribriform Carcinoma of the Prostate. Mol. Cancer Res. 2019, 17, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Palapattu, G.S.; Salami, S.S.; Cani, A.K.; Hovelson, D.H.; Lazo de la Vega, L.; Vandenberg, K.R.; Bratley, J.V.; Liu, C.J.; Kunju, L.P.; Montgomery, J.S.; et al. Molecular Profiling to Determine Clonality of Serial Magnetic Resonance Imaging/Ultrasound Fusion Biopsies from Men on Active Surveillance for Low-Risk Prostate Cancer. Clin. Cancer Res. 2017, 23, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Klevebring, D.; Liu, W.; Neiman, M.; Xu, J.; Wiklund, P.; Mills, I.G.; Egevad, L.; Gronberg, H.; Wiklund, F. Exome sequencing of prostate cancer supports the hypothesis of independent tumor origins. Eur. Urol. 2013, 63, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Boutros, P.C.; Fraser, M.; Harding, N.J.; deBorja, R.; Trudel, D.; Lalonde, E.; Fox, N.S.; Livingstone, J.; Shiah, Y.J.; Wang, J.; et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015, 47, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wang, J.; Lampert, E.; Schlanger, S.; DePriest, A.D.; Hu, Q.; Cortes Gomez, E.; Murakam, M.; Glenn, S.T.; Conroy, J.; et al. Intratumoral and intertumoral genomic heterogeneity of multifocal localized prostate cancer impacts molecular classifications and genomic prognosticators. Eur. Urol. 2017, 71, 183–192. [Google Scholar] [CrossRef]
- Cancer Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- VanderWeele, D.J.; Finney, R.; Katayama, K.; Gillard, M.; Paner, G.; Imoto, S.; Yamaguchi, R.; Wheeler, D.; Lack, J.; Cam, M.; et al. Genomic heterogeneity within individual prostate cancer foci impacts predictive biomarkers of targeted therapy. Eur. Urol. Focus 2019, 5, 416–424. [Google Scholar] [CrossRef]
- Salami, S.S.; Hovelson, D.H.; Kaplan, J.B.; Mathieu, R.; Udager, A.M.; Curci, N.E.; Lee, M.; Plouffe, K.R.; Lazo de la Vaga, L.; Susani, M.; et al. Transcriptomic heterogeneity in multifocal prostate cancer. JCI Insight 2018, 3, e123468. [Google Scholar] [CrossRef]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstrahle, J.; Tarish, F.; Tanoglidi, A.; Vichovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer trascriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Su, F.; Zhang, W.; Zhang, D.; Zhang, Y.; Pang, C.; Huang, Y.; Wang, M.; Cui, L.; He, L.; Zhang, J.; et al. Spatial intratumoral genomic heterogeneity within localized prostate cancer revealed by single-nucleus sequencing. Eur. Urol. 2018, 74, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Lvof, M.; Zhao, S.; Axcrona, U.; Johannessen, B.; Bakken, A.C.; Carm, K.T.; Hoff, A.M.; Miklebost, O.; Meza-Zepeda, L.A.; Lie, A.K.; et al. Multifocal primary prostate cancer exhibits high degree of genomic heterogeneity. Eur. Urol. 2019, 75, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef]
- Liu, W.; Laitinen, S.; Khan, S.; Vihinen, M.; Kowalski, J.; Yu, G.; Chen, L.; Isaacs, W.B.; Visakorpi, T.; Bova, S. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 2009, 15, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Mosbruger, T.; Esopi, D.M.; Fedor, H.; Heaphy, C.M.; Walker, D.A.; Adepla, N.; Gurel, M.; Hicks, J.; Meeker, A.K.; et al. Tracking the clonal origin of lethal prostate cancer origin of lethal prostate cancer. J. Clin. Invest. 2013, 123, 4918–4922. [Google Scholar] [CrossRef] [PubMed]
- Gundem, G.; Loo, P.V.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.C.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.L.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Macintyre, G.; Wedge, D.C.; Van Loo, P.; Patel, K.; Lunka, S.; Alexandrov, L.B.; Sloggett, C.; Cmero, M.; Marass, F.; et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun. 2015, 6, 6605. [Google Scholar] [CrossRef]
- Kneppers, J.; Krijsman, O.; Melis, M.; de Jong, J.; Peeper, D.S.; Bekers, E.; van der Poel, H.; Zwart, W.; Bergman, A.M. Frequent clonal relations between metastases and non-index prostate cancer lesions. JCI Insight 2019, 4, e124756. [Google Scholar] [CrossRef]
- Carreira, S.; Romanel, A.; Goodall, J.; Grist, E.; Ferraldeschi, R.; Miranda, S.; Prandi, D.; Lorente, D.; Frenel, J.S.; Pezaro, C.; et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014, 6, 254ra125. [Google Scholar] [CrossRef]
- Bova, S.; Kallio, H.; Annali, M.; Kivinummi, K.; Hognas, G.; Hayrynen, S.; Rantapero, T.; Kivinen, V.; Isaacs, W.B.; Tolonen, T.; et al. Integrated clinical, whole-genome, and transcriptome analysis of multisampled lethal metastatic prostate cancer. Cold Spring Harb. Mol. Case Stud. 2016, 2, a000752. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumor types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Polneger, D.; Sougner, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasakaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, L.; Fuchs, S.; Krohn, A.; Masser, S.; Madei, M.; Kluth, M.; Bachmann, F.; Huland, H.; Steuber, T.; Graefen, M.; et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013, 73, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; White, T.A.; MacKenzie, A.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Milbank, J.; et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Moriney, C.; Kumar, A.; Zhang, X.; Smith, C.; Coleman, I.; Salipante, S.J.; Milbank, J.; Yu, M.; Grady, W.M.; et al. Complex MESH2 and MESH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nature Commun. 2014, 5, 4988. [Google Scholar] [CrossRef]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenan, D.; Minner, S.; Wuttig, G.; Warnatz, H.J.; Stehr, H.; Reusch, T.; et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar] [CrossRef]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feurbach, L.; Assenov, Y.; Hechmann, D.; Sidoropoulos, N.; Waszak, S.M.; Hubschmann, D.; et al. Molecular evolution of early-onset prostate cancer identifies molecular risk markers and clinical trajectories. Cancer Cell 2018, 34, 996–1011. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.; Chakravarty, D.; Han, C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Fraser, M.; Sabeinkova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, S.M.G.; Liu, L.Y.; Rubanova, Y.; Bhandari, V.; Holgersen, E.M.; Szyca, L.M.; Fox, N.S.; Chua, M.L.K.; Yamaguchi, T.N.; Heisler, L.E.; et al. The evolutionary landscape of localized prostate cancers drives clinical aggression. Cell 2018, 173, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, J.F.; Babenykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef] [PubMed]
- Wedge, D.C.; Gundem, G.; Mitchell, T.; Woodcock, D.J.; Martincorena, I.; Ghori, M.; Zamora, J.; Butler, A.; Whitaker, H.; Kote-Jarai, Z.; et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet. 2018, 50, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Sun, Y.; Dobi, A.; Srivastava, S.; Wang, W.; Srivastava, S.; Ji, Y.; Hou, J.; Zhao, G.P.; LI, Y. Systematic analysis reveals molecular charcteristics of ERG-negative prostate cancer. Sci. Rep. 2018, 8, 12868. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Moisquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.C.; Bailey, A.M.; et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018, 174, 758–769. [Google Scholar] [CrossRef]
- Lapuk, A.V.; Wu, C.; Wyatt, A.W.; McConeghy, B.J.; Brahmhatt, S.; Mo, F.; Zoubedi, A.; Anderson, S.; Bell, R.H.; Hargert, A.; et al. From sequence to molecular pathology and a mechanism driving the neuroendocrine phenotype in prostate cancer. J. Pathol. 2012, 227, 286–297. [Google Scholar] [CrossRef]
- Wu, C.; Wyatt, A.W.; McPherson, A.; Liu, D.; McConeghy, B.J.; Mo, F.; Shukin, B.; Lapuk, A.V.; Jones, S.J.; Zhao, Y.; et al. Poly-gene fusion transcripts and chromotripsis in prostate cancer. Gene Chromosomes Cancer 2012, 51, 1144–1153. [Google Scholar] [CrossRef]
- Federer-Gsponer, J.R.; Quintavalle, C.; Muller, D.C.; Dietsche, T.; Perrina, V.; Lorber, T.; Juskevicius, E.; Lienkewicz, E.; Zellweger, T.; Gasser, T.; et al. Delineation of human prostate cancer evolution identifies chromotripsis as a polyclonal event and FKBP4 as a potential driver of castration resistance. J. Pathol. 2018, 245, 74–84. [Google Scholar] [CrossRef]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Jung, S.H.; Baek, I.P.; Lee, S.H.; Choi, Y.J.; Lee, J.Y.; Lee, S.H. Regional biases in mutation screening due to intratumor heterogeneity of prostate cancer. J. Pathol. 2014, 233, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerod, J.M.; Heyde, A.; Attiyeh, M.A.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Nava Rodrigues, D.; Casiraghi, N.; Romanel, A.; Crespo, M.; Miranda, S.; Rescigno, P.; Figueiredo, I.; Riisnaes, R.; Carreira, S.; Sumanasuriya, S.; et al. RB1 heterogeneity in advanced metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Gato, D.; Thysell, E.; Tyanova, S.; Crnalic, S.; Santos, A.; Lima, T.S.; Geiger, T.; Cox, J.; Widmark, A.; Bergh, A.; et al. The proteome of prostate cancer bone metastasis reveals heterogeneity with prognostic implications. Clin. Cancer Res. 2018, 24, 5433–5444. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.L.; Greer, P.A.; Squire, J.A. Recurrent copy number alterations in prostate cancer: An in silico meta-analysis of publicly available genomic data. Cancer Genet. 2014, 207, 474–488. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymous, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Wilt, T.J.; Brawer, M.K.; Jones, K.M.; Barry, M.J.; Aronson, W.J.; Fox, S.; Gingrich, J.R.; Wei, J.J.; Gilhooly, P.; Grob, B.M.; et al. Radical prostatectomy versus observation for localized prostate cancer. N. Engl. J. Med. 2012, 367, 203–213. [Google Scholar] [CrossRef]
- Hieronymous, H.; Schultz, N.; Gopalan, A.; Carver, B.S.; Chang, M.T.; Xiao, Y.; Heguy, A.; Huberman, K.; Bernstein, M.; Assel, M.; et al. Copy number alteration burden predicts prostate cancer relapse. Proc. Natl. Acad. Sci. USA 2014, 111, 11139–11144. [Google Scholar] [CrossRef]
- Hieronymous, H.; Murali, R.; Tin, A.; Yadav, K.; Abida, W.; Moller, H.; Berney, D.; Scher, H.; Carver, B.; Scardino, P.; et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. eLife 2018, 7, e37294. [Google Scholar] [CrossRef] [PubMed]
- Camacho, N.; Val Loo, P.; Edwards, S.; Kay, J.D.; Matthwes, L.; Haase, K.; Clark, J.; Dennis, N.; Thomas, S.; Kremeyer, B.; et al. Appraising the relevance of DNA copy number loss and gain in prostate cancer using whole genome DNA sequence data. PLoS Genet. 2017, 13, e1007001. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Solimini, N.L.; Xu, Q.; Mermel, C.H.; Liang, A.C.; Cshlabach, M.R.; Luo, J.; Burrows, A.E.; Anselmo, A.N.; Bredemeyer, A.L.; Li, M.Z.; et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science 2012, 337, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Vasmatzis, G.; Kosari, F.; Murphy, S.J.; Terra, S.; Kovtun, I.V.; Harris, F.R.; Zarei, S.; Smadbeck, J.B.; Johnson, S.H.; Gaitatzes, A.G.; et al. Large chromosomal rearrangements yield biomarkers to distinguish low-risk from intermediate- and high-risk prostate cancer. Mayo Clin. Proc. 2019, 94, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Shi, L.; Cimic, A.; Romero, L.; Shi, G.; Lees, C.J.; Cline, J.M.; Seals, D.F.; Sirintrapun, J.S.; McCoy, T.P.; et al. Suppression of TAK1 promotes prostate tumorigenesis. Cancer Res. 2013, 72, 2833–2843. [Google Scholar] [CrossRef] [PubMed]
- Kluth, M.; Hesse, J.; Heinl, A.; Krohn, A.; Steurer, S.; Sirma, H.; Simon, R.; Mayer, P.S.; Schumacher, U.; Grupp, K.; et al. Genomic deletion of MAP3K7 at 6q12-22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod. Pathol. 2013, 26, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Flavin, R.; Petterson, A.; Hendrickson, W.; Fiorentino, M.; Andersson, S.O.; Finn, S.; Kunz, L.; Judson, G.L.; Lis, R.; Bailey, D.; et al. SPINK1 protein expression and prostate cancer progression. Clin. Cancer Res. 2014, 20, 4904–4911. [Google Scholar] [CrossRef]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatryroiva, O.; Simon, R.; Koop, C.; Oakes, C.; Zuecknick, M.; Lipka, D.S.; Wilischenfeldt, J.; et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Tagawa, S.T.; Park, K.; MacDonald, T.; Milowsky, M.I.; Mosquera, J.M.; Rubin, M.A.; Nanus, D.M. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J. Clin. Oncol. 2012, 30, e386–e389. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-istitutional prospective study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannpoulou, E.; Balabhapatruni, C.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, S.; Inoue, T.; Ogawa, O.; Gleave, M.E. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int. J. Urol. 2018, 25, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noordal, Z.; Cheung, C.; et al. N-NYC induces an EZH2-nucleated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016, 30, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A phase II trial of the Aurora kinase A inhibitor Alistertib for patients with castration resistant and neuroendocrine prostate cancer: Efficacy and biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, M.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ci, X.; Ahmed, M.; Hua, J.T.; Soares, F.; Lin, D.; Puca, L.; Vosoughi, A.; Xue, X.; Li, E.; et al. ONECUT2 is a driver of neuroendocrine prostate cancer. Nat. Commun. 2019, 10, 278. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Linares, J.M.; Duran, A.; Cordes, T.; L’Hermitte, A.; Badur, M.G.; Bhangoo, M.S.; Thorson, P.K.; Roslid, T.; Garcia-Olmo, D.C.; et al. Increased serine and one-carbon pathway metabolism by PKCλ/ι deficiency promotes neuroendocrine prostate cancer. Cancer Cell 2019, 35, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Tran, J.; Chen, Z.; Carracedo-Perez, A.; Alimonti, A.; Nardella, C.; Gopalan, A.; Scardino, P.T.; Gordon-Cardo, C.; Gerald, W.; et al. ETS rearrangements and prostate cancer initiation. Nature 2009, 457, E1–E3. [Google Scholar] [CrossRef]
- Park, K.; Dalton, J.T.; Narayan, R.; Barbieri, C.E.; Hancock, M.L.; Bostwick, D.G.; Steiner, M.S.; Rubin, M.A. TMPRSS2-ERG fusion predicts subsequent detection of prostate cancer patients with high-grade prostatic intraepithelial neoplasia. J. Clin. Oncol. 2014, 32, 206–211. [Google Scholar] [CrossRef]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [PubMed]
- Lara, P.N.; Heilmann, A.M.; Elvin, J.A.; Parikh, M.; de Were White, R.; Gandour-Edwards, R.; Evans, C.P.; Pan, C.X.; Schrock, A.B.; Erlich, R.; et al. TMPRSS2-ERG fusions unexpectedly identified in men initially diagnosed with nonprostatic malignancies. JCO Precis. Oncol. 2017, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Knuuttila, M.; Mehmood, A.; Maki-Jouppila, J.; Ryberg, H.; Taimen, P.; Knaapila, J.; Ettala, O.; Bostrom, P.J.; Ohlsson, C.; Venalainen, M.S.; et al. Intratumoral androgen levels are linked to TMPRSS2-ERG fusion in prostate cancer. Endocr. Relat. Cancer 2018, 25, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer rearrangements. Nat. Genet. 2010, 42, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Steurer, S.; Mayer, P.S.; Adam, M.; Krohn, A.; Koop, C.; Ospina-Klink, D.; Tehrani, A.A.; Simon, R.; Tennstedt, P.; Graefen, M.; et al. TMPRSS2-ERG fusions are strongly linked to young patient age in low-grade prostate cancer. Eur. Urol. 2014, 66, 978–981. [Google Scholar] [CrossRef] [PubMed]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymous, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Tran, J.; Gopalan, A.; Chen, Z.; Shaikh, S.; Carracedo, A.; Alimonti, A.; Nardella, C.; Varmeh, S.; Scardino, P.T.; et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet. 2009, 41, 619–624. [Google Scholar] [CrossRef]
- Mounir, Z.; Lin, F.; Lin, V.G.; Korn, J.M.; Yu, Y.; Valdez, R.; Aina, O.H.; Buchwalter, G.; Jaffe, A.B.; Korpal, M.; et al. TMPRSS2:ERG blocks neuroendocrine and luminal cell differentiation to maintain prostate cancer proliferation. Oncogene 2015, 34, 3815–3825. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef]
- Sandoval, G.J.; Pulice, J.L.; Pakula, H.; Schenone, M.; Takeda, D.Y.; Pop, M.; Boulay, G.; Williamson, K.E.; MacBride, M.J.; Pan, J.; et al. Binding of TMPRS22-ERG to BAF chromatin remodeling complexes mediates prostate oncogenesis. Mol. Cell 2018, 71, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Blee, A.M.; He, Y.; Yang, Y.; Ye, Z.; Yan, Y.; Pan, Y.; Ma, T.; Dugdale, J.; Kuehn, J.; Kohli, M.; et al. TMPRS22-ERG controls luminal epithelial lineage and antiandrogen sensitivity in PTEN and TP53-mutated prostate cancer. Clin. Cancer Res. 2018, 24, 4551–4565. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Xavier, C.P.; Sukumar, G.; Tan, S.H.; Ravindranath, L.; Seraj, N.; Kumar, V.; Sreenath, T.; McLeod, D.G.; Petrovics, G.; et al. Identification of a small molecule that selectively inhibits ERG-positive cancer cell growth. Cancer Res. 2018, 78, 3659–3671. [Google Scholar] [CrossRef] [PubMed]
- Baena, E.; Shao, S.; Linn, D.E.; Glass, K.; Hamblen, M.J.; Fuijwara, Y.; Kim, J.; Nguyen, M.; Zhang, X.; Godinho, F.J.; et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013, 27, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chi, P.; Rockowitz, S.; Iaquinta, P.J.; Shamu, T.; Shukla, S.; Gao, D.; Sirota, I.; Carver, B.S.; Wongvipat, J.; et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis response to PTEN loss. Nat. Med. 2013, 19, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Krohn, A.; Freudenthaler, F.; Harasimowicz, S.; Kluth, M.; Fuchs, S.; Burkhardt, L.; Stahl, P.C.; Tsourlakis, M.; Bauer, M.; Tennstedt, P.; et al. Heterogeneity and chronology of PTEN deletion and ERG fusion in prostate cancer. Mod. Pathol. 2014, 27, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Fallahabadi, Z.R.; Noori Dalooi, M.R.; Mahdian, R.; Behjati, F.; Shokrgozar, M.A.; Abolhasani, M.; Asgari, M.; Shahrokh, H. Frequency of PTEN alterations, TMPRSS2-ERG fusion and their association in prostate cancer. Gene 2016, 575, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Mosquera, J.M.; Garofalo, A.; Oh, C.; Baco, M.; Amin-Mansour, A.; Rabasha, B.; Bahl, S.; Mullane, S.A.; Robinson, B.D.; et al. Exome sequencing of African-American prostate cancer reveals loss-of-function ERF mutations. Cancer Discov. 2017, 7, 973–983. [Google Scholar] [CrossRef]
- Bose, R.; Karthaus, W.R.; Armenia, J.; Abida, W.; Iaquinta, P.J.; Zhang, Z.; Wongvipat, J.; Wasmuth, E.V.; Shah, N.; Sullivan, P.S.; et al. ERF mutations reveal a balance of ETS factors controlling prostate oncogenesis. Nature 2017, 546, 671–675. [Google Scholar] [CrossRef]
- Attard, G.; Cooper, C.S.; de Bono, J.S. Steroid hormone receptors in prostate cancer: A hard habit to break? Cancer Cell 2009, 16, 458–462. [Google Scholar] [CrossRef]
- Wang, X.; Qiao, Y.; Asangani, I.A.; Poliakov, A.; Cieslik, M.; Pichiaya, S.; Chakravarthi, B.; Cao, X.; Jing, X.; Wang, C.X.; et al. Development of peptidomimetic inhibitors of the ERG gene fusion product in prostate cancer. Cancer Cell 2017, 31, 532–548. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Tan, S.H.; Xavier, C.P.; Katta, S.; Huang, W.; Ravindranath, L.; Jamal, M.; Li, H.; Srivastava, M.; Srivatsan, E.S.; et al. Synergistic activity with NOTCH inhibition and androgen ablation in ERG-Positive prostate cancer cells. Mol. Cancer Cell 2017, 15, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Bratsalavsky, G.; Fisher, H.; Byler, T.; Iacob, I.; Chung, I.; Elvin, I.A.; Vergiolio, I.A.; Ramkissoon, S.; Suh, J.; Allan, E.; et al. Difference of genomic signatures and opportunities for targeted and immunotherapies in castrate resistant TMPRSS2:ERG fusion positive and TPRSS2:ERG wild type refractory acinal (CRPC) and neuroendocrine prostate cancer (CRNEPC). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Geng, C.; He, B.; Xu, L.; Barbieri, C.E.; Eedunuri, V.K.; Chew, S.A.; Zimmerman, M.; Bond, R.; Shou, J.; Li, C.; et al. Prostate cancer associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein. Proc. Natl. Acad. Sci. USA 2013, 110, 6997–7002. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Rajapakshe, K.; Shah, S.S.; Shou, J.; Eedunuri, V.K.; Foley, C.; Fiskus, W.; Rajendran, M.; Chew, S.A.; Zimmermann, M.; et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014, 74, 5631–5643. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Wang, C.; Deug, Y.; Yu, L.; Huang, H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014, 6, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Kaochar, S.; Li, M.; Rajapakshe, K.; Dong, J.; Foley, C.; Dong, B.; Zhang, L.; Kwon, O.J.; Shah, S.S.; et al. SPOP regulates prostate epithelial cell proliferatrion and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene 2017, 36, 4767–4777. [Google Scholar] [CrossRef]
- Theurillat, J.P.; Udeshi, N.D.; Errington, W.J.; Svinika, T.; Baca, S.C.; Pop, M.; Wild, P.J.; Blattner, M.; Groner, A.C.; Rubin, M.A.; et al. Prostate cancer-ubiquitylone analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346, 85–89. [Google Scholar] [CrossRef]
- Fong, K.W.; Zhao, J.C.; Song, B.; Yu, J. TRIM28 protects TRIM24 from SPOP-mediated degradation and promotes prostate cancer progression. Nat. Commun. 2018, 9, 5007. [Google Scholar] [CrossRef]
- Groner, A.C.; Cato, L.; de Tibolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 is an oncogenic transcriptional activator prostate cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, M.; Zhu, Y.; Dai, X.; Dang, F.; Ren, J.; Ren, S.; Shulga, Y.V.; Beca, F.; Gan, W.; et al. SPOP promotes Nanog destruction to suppress stem cell traits and prostate cancer progression. Dev. Cell 2018, 48, 329–344. [Google Scholar] [CrossRef]
- Liu, D.; Thakar, M.; Aishalalfa, M.; Erho, N.; Shoag, J.; Jenkins, R.B.; Karnes, R.J.; Shaffer, A.M.; Rubin, M.A.; Trock, B.; et al. Impact of the SPOP mutant subtype on the interpretation of clinical parametrs in prostate cancer. JCO Precis. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Shoag, J.; Liu, D.; Blattner, M.; Sboner, A.; Park, K.; Deonarine, L.; Robinson, B.D.; Mosquera, J.M.; Chen, Y.; Rubin, M.A.; et al. SPOP mutation drives prostate neoplasia without stabilizing oncogenic transcription factor ERG. J. Clin. Invest. 2018, 128, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef]
- Boysen, G.; Rodrigues, D.N.; Rescigno, P.; Seed, G.; Dolling, D.; Riisnaes, R.; Crespo, M.; Zafeiriou, Z.; Suamanasuriya, S.; Bianchini, D.; et al. SPOP-mutated/CHD1-deleted lethal prostate cancer and abiraterone sensitivity. Clin. Cancer Res. 2018, 24, 5585–5593. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Gan, W.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.; Zhang, J.; Chen, T.; Shimizu, K.; et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Net. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef]
- Wang, X.; Jin, J.; Wan, F.; Zhao, L.; Chu, H.; Chen, C.; Liao, G.; Liu, J.; Yu, Y.; Teng, H.; et al. AMPK promotes SPOP-mediated NANOG degradation to regulate prostate cancer cell stemness. Mol. Cell 2019, 48, 345–360. [Google Scholar] [CrossRef]
- Bouchard, J.J.; Otero, J.H.; Scott, D.C.; Szulc, D.C.; Martin, E.W.; Sabri, N.; Granata, D.; Mrazahn, M.R.; Lindorff-Larsen, K.; Salvatella, X.; et al. Cancer mutations of the tumor suppressor SPOP disrupt the formation of active, phase-separated compartments. Mol. Cell 2018, 72, 19–36. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Ten, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via Cul3SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Ren, S.; Wei, G.H.; Liu, H.; Wang, L.; Hou, Y.; Zhu, S.; Peng, L.; Zhang, Q.; Cheng, Y.; Su, H.; et al. Whole-genome and transcriptome sequencing of prostate cancer identify new genetic alterations driving disease progression. Eur. Urol. 2018, 73, 322–339. [Google Scholar] [CrossRef]
- Zhou, J.; Li, J.; Serafim, R.B.; Ketchum, S.; Ferreira, C.G.; Liu, J.C.; Coe, K.A.; Price, B.D.; Yusufzai, T. Human CHD1 is required for early DNA-damage signaling and is uniquely regulated by its N terminus. Nucleic Acid Res. 2018, 8, 3891–3995. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.U.; Rider, L.; Nieto, C.; Romero, L.; Karimpour-Fard, A.; Loda, M.; Lucia, M.S.; Wu, M.; Shi, L.; Cimic, A.; et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res. 2015, 75, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Tereshchenko, I.V.; Zhong, H.; Chekmareva, M.A.; Kane-Goldsmith, N.; Santanam, U.; Petrosky, W.; Stein, M.N.; Gapesan, S.; Singer, E.A.; Moore, D.; et al. ERG and CHD1 heterogeneity in prostate cancer: use of confocal microscopy in assessment of microscopic foci. Prostate 2014, 74, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Lu, X.; Wang, G.; Lan, Z.; Liao, W.; Li, J.; Liang, X.; Chen, J.R.; Shah, S.; Shang, X.; et al. Synthetic essentiality of chromatin remodeling factor CHD1 in PTEN deficient cancer. Nature 2017, 542, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.N.; Boysen, G.; Sumanasuriya, S.; Seed, G.; Marzo, A.M.; de Bono, J. The molecular undepinnings of prostate cancer: impacts on management and pathology practice. J. Pathol. 2017, 241, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Kari, V.; Mansur, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016, 17, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, W.; et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann. Oncol. 2017, 28, 1495–1507. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.E.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Loipez, R.; Nava Rodrigues, D.; Robinson, D.; Omin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castratrion-resistant prostate cancer: A randomized, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 17, 975–986. [Google Scholar] [CrossRef]
- Goodall, J.; Mato, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Guedes, L.B.; Antonarakis, E.S.; Schweizer, M.T.; Mirkheshti, N.; Ajmutari, F.; Park, J.C.; Giavaris, S.; Hicks, J.; Eisenberger, M.A.; De Marzo, A.M.; et al. MSH2 loss in primary prostate cancer. Clin. Cancer Res. 2017, 23, 6863–6874. [Google Scholar] [CrossRef] [PubMed]
- Nava Rodrigues, D.; Rescigno, P.; Liu, D.; Yuan, W.; Carreira, S.; Lambrios, M.B.; Seed, G.; Mateo, J.; Riisnanes, R.; Mullane, S.; et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Invest. 2018, 128, 4441–4453. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Shaukat, F.; Isacsson Velho, P.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical features and therapeutic outcomes in men with advanced prostate cancer and DNA mismatch repair gene mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Kaur, H.B.; Alharbi, A.M.; Pritchard, C.C.; Epstein, J.I. DNA damage repair alterations are frequent in prostatic adenocarcinomas with focal pleomorphic giant cell features. Histopathology 2019, 74, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen signaling in prostate cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a30452. [Google Scholar] [CrossRef]
- Chang, K.H.; Ercole, C.E.; Sharifi, N. Androgen metabolism in prostate cancer: From molecular mechanisms to clinical consequences. Br. J. Cancer 2014, 111, 1249–1254. [Google Scholar] [CrossRef]
- Vikakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef]
- Koivisto, P.; Kononen, J.; Palmberg, C.; Tammela, T.; Hyytinen, E.; Isola, J.; Treapman, J.; Cleutjens, K.; Noordzij, A.; Visakorpi, T.; et al. Androgen receptor gene amplification: A possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997, 57, 314–319. [Google Scholar]
- Linja, M.J.; Savinainen, K.J.; Saramaki, O.R.; Tammela, T.L.; Vassela, R.L.; Visakorpi, T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001, 61, 3550–3555. [Google Scholar]
- Chen, C.D.; Weisbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessela, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Spisak, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csbal, I.; Solymosi, N.; Szalklasi, Z.; et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell 2018, 174, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carnot-Zhang, J.; Whelan, C.W.; Haradvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoadesw, J.; et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018, 174, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Li, R.; Kuri, B.; Lotan, Y.; Roehborn, C.G.; Liu, J.; Vessella, R.; Nelson, P.S.; Kapur, P.; Guo, X.; et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell 2013, 154, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of androgen receptor in prostate cancer: A review. World J. Mens Health 2018. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Goto, T.; Akamatsu, S.; Yamasaki, T.; Inoue, T.; Ogawa, O.; Kobayashi, T. Castration-resistant prostate cancer refractory to second-generation androgen receptor axis-targeted agents: Opportunities and challenges. Cancers 2018, 10, 345. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegfert, A.; LeBihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarker4s of therapeutic resistance in castration-resistant prostate cancer. Prostate Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef]
- Annala, M.; Vandekekhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating tumor DNA genomics correlate with resistance to abiraterone ans enzalutamide in prostate cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef]
- Sonpavde, G.; Agarwal, N.; Pond, G.R.; Nagy, R.J.; Nussenzveig, R.H.; Hahn, A.W.; Sartor, O.; Gourdin, T.S.; Nondagopal, L.; Ledet, E.M.; et al. Circulating tumor DNA alterations in patients with metastatic castration-resistant prostate cancer. Cancer 2019, 125, 1459–1469. [Google Scholar] [CrossRef]
- Lallous, N.; Volik, S.V.; Awrey, S.; Leblanc, E.; Murillo, J.; Singh, K.; Azad, A.A.; Wyatt, A.W.; LeBihan, S.; Chi, K.N.; et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016, 17, 10. [Google Scholar] [CrossRef]
- Chen, E.J.; Sowalsky, A.G.; Gao, S.; Cai, C.; Voznensky, O.; Schaefer, R.; Loda, M.; True, L.D.; Ye, H.; Troncoso, P.; et al. Abiraterone in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 2015, 21, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimoint, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Romanel, A.; Gasi Tandefelt, D.; Conteduca, V.; Jayaram, A.; Casiraghi, N.; Wetterskog, D.; Salvi, S.; Amadori, D.; Zafeiriou, Z.; Rescigno, P.; et al. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 2015, 7, 312re10. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.J.; Chen, Y.H.; Carducci, M.; Liu, G.; Jarrad, D.F.; Eisenberger, M.; Wang, Y.N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomized controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Rodriguez-Antolin, A.; Aleskeev, B.Y.; Orgaroglu, M.; Ye, D.; Fewerband, S.; Protheroe, A.; De Porree, P.; et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.O.; Dearneley, D.P.; Ritchie, A.W.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Li, Z.; Bishop, A.C.; Alyamani, M.; Garcia, J.A.; Dreicer, R.; Bunch, D.; Liu, J.; Upadhyay, S.K.; Auchus, R.J.; Shafiri, N. Conversion of abiraterone to D4A drives anti-tumor activity in prostate cancer. Nature 2015, 523, 347–351. [Google Scholar] [CrossRef]
- Attard, G.; Reid, A.H.; Yap, T.A.; Raynaud, F.; Dowsett, M.; Settatree, S.; Barrett, M.; Parker, C.; Martins, V.; Folkerd, E.; et al. Phase I clinical trial of a selective inhibitor of cyp17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef]
- Chi, K.N.; Protheroe, A.; Rodriguez-Antolin, A.; Facchini, G.; Suttman, H.; Matsubara, N.; Ye, Z.; Keam, B.; Damiao, R.; Li, T.; et al. Patient-reported outcomes following abiraterone acetate plus prednisone added to androgen deprivation therapy in patients with newly diagnosed metastatic castration-naïve prostate cancer (LATITUDE): An international, randomized phase 3 trial. Lancet Oncol. 2018, 19, 194–206. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, N.; Matsubara, N.; Rodrigiuez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomized, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Sydes, M.R.; Spears, M.R.; Mason, M.D.; Clarke, N.W.; Dearnaley, D.P.; de Bono, J.S.; Attard, G.; Chowdhury, S.; Cross, W.; Gillesseen, S.; et al. Adding abiraterone or docetaxel to long-term hormone therapy for prostate cancer: Directly randomized data from the STAMPEDE multi-arm, multi-stage platform protocol. Ann. Oncol. 2018, 29, 1235–1248. [Google Scholar] [CrossRef]
- Tannock, J.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Oudard, S.; Théodore, C.; James, N.D.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Petrylak, J.P.; Tangen, C.M.; Hussain, M.H.A.; Lara, P.N.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef]
- Ryan, C.M.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Carles, J.; et al. Abiraterone in metastatic prostate cancer without previous cxhemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naïve men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomized, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- De Bono, J.S.; Smith, M.R.; Saad, F.; Rathkopf, D.E.; Mulders, P.F.A.; Small, E.J.; Shore, N.D.; Fizazi, K.; De Porre, P.; Kheoh, T.; et al. Subsequent chemotherapy and treatment patterns after abiraterone acetate in patients with metastatic castration-resistant prostate cancer: Post-hoc analysis of COU-AA-302. Eur. Urol. 2017, 71, 656–664. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Tapli, M.E.; Sternberg, C.N.; Miller, K.; de Witt, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Ratkopf, D.E.; Loriot, Y.; Higano, C.S.; Iversen, P.; Batthacarya, S.; Carles, J.; Chowdury, S.; Davis, I.D. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Ratkopf, D.E.; Loriot, Y.; Higano, C.S.; Iversen, P.; Evans, C.P.; Kim, C.S.; Kimura, G.; Miller, K.; et al. Enzalutamide in men with chemotherapy-naïve metastatic castration-resistant prostate cancer: Extended analysis of the phase 3 PREVAIL study. Eur. Urol. 2017, 71, 151–154. [Google Scholar] [CrossRef]
- Siemens, D.R.; Klotz, L.; Heidenreich, A.; Chowdhury, S.; Villers, A.; Baron, B.; Van Os, S.; Hassabou, N.; Wang, F.; Lin, P.; et al. Efficacy and safety of enzalutamide vs. Bicalutamide in younger and older patients with metastatic castration resistant prostate cancer in the TERRAIN trial. J. Urol. 2018, 199, 147–154. [Google Scholar] [CrossRef]
- Penson, D.F.; Armstrong, A.J.; Concepcion, R.; Agarwal, N.; Olsson, C.; Karsh, L.; Dunshee, C.; Wang, F.; Wu, K.; Krivoshik, A.; et al. Enzalutamide versus bicalutamide in castration-resistant prostate cancer: The STRIVE trial. J. Clin. Oncol. 2016, 34, 298–2106. [Google Scholar] [CrossRef]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, K.; et al. Enzalutamide in men with nonmetastatic. Castration-resistant prostate cancer. N. Engl. J. Med. 2018, 378, 2465–2478. [Google Scholar] [CrossRef]
- Tombol, B.; Saad, F.; Penson, D.; Hussain, M.; Sternberg, C.N.; Morlock, R.; Ramaswamy, K.; Ivanescu, C.; Attard, G. Patient-reported outcomes following enzalutamide or placebo in men with non-metastatic, castration-resistant prostate cancer (PROSPER): A multicenter, randomized, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 556–569. [Google Scholar] [CrossRef]
- Khalaf, D.J.; Sunderland, K.; Eigl, B.J.; Kollmannsberger, C.K.; Ivanov, N.; Finch, D.L.; Oja, C.; Vergidis, J.; Zulfiqar, M.; Gleave, M.E.; et al. Health-relatyed quality of life for abiraterone plus prednisone versus enzalutamide in patients with metastatic castration-resistant prostate cancer: Results from a phase II randomized trial. Eur. J. Urol. 2019, 75, 940–947. [Google Scholar] [CrossRef]
- Smith, M.; Parker, C.; Saad, F.; Miller, K.; Tombal, B.; Ng, Q.S.; Boegemann, M.; Matveev, V.; Piulats, J.M.; Zucca, L.E.; et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): A randomized, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2019, 20, 408–419. [Google Scholar] [CrossRef]
- Parker, C.C.; James, N.D.; Brawley, C.D.; Clarke, N.W.; Hoyle, A.P.; Ali, A.; Ritchie, A.W.S.; Attard, G.; Chowdhury, S.; Cross, W.; et al. Radiotherapy to the primary tumor for newly diagnosed, mertastatic prostate cancer (STAMPEDE): A randomized controlled phase 3 trial. Lancet 2018, 392, 2353–2366. [Google Scholar] [CrossRef]
- Attard, G.; Borre, M.; Gurney, H.; Loriot, Y.; Andresen-Danil, C.; Kalleda, R.; Pham, T.; Taplin, M.E.; PLATO Collaborators. Abiraterone alone or in combination with enzalutamide in metastatic castration-resistant prostate cancer with rising prostate-specific antigen during enzalutamide treatment. J. Clin. Oncol. 2018, 36, 2639–2646. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide treatment and metastasis-free survival in porstate. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Saad, F.; Cella, D.; Basch, E.; Hadaschick, R.A.; Mainwaing, P.N.; Oudard, S.; Graff, J.N.; McQuarrie, K.; Li, S.; Hudgens, S.; et al. Effect of apalutamide on health-related quality of life in patients with non-metastatic castration-resistant prostate cancer: An analysis of the SPARTAN randomized, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1404–1416. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Higano, C.S. Enzalutamide, Apalutamide, or Dorolutamide; are apples or bananas best for patients? Nat. Rev. Urol. 2019, 16, 335–336. [Google Scholar] [CrossRef]
- Kwegir-Afful, A.K.; Ramalimgan, S.; Purushottamachar, P.; Ramamurthy, V.P.; Njar, V.C. Galaterone and VNPT55 induce proteosomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenograft in vivo. Oncotarget 2015, 6, 27440–27460. [Google Scholar]
- McClurg, U.L.; Azizyan, M.; Dransfield, D.T.; Namved, N.; Chit, N.; Nakjang, S.; Robson, C.N. The novel anti-androgen candidate galaterone, targets deubiquitinating enzymes, USP12 and USP46, to control prostate cancer growth and survival. Oncotarget 2018, 9, 24992–25007. [Google Scholar] [CrossRef]
- Montgomery, B.; Eisenberger, M.A.; Rettig, M.B.; Chu, F.; Pili, R.; Stephenson, J.J.; Vogelzang, N.J.; Koletsky, A.J.; Norquist, L.T.; Edenfield, W.J.; et al. Androgen receptor modulation optimized for response (ARMOR) phase I and II studies: Galaterone for the treatment of castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 1356–1363. [Google Scholar] [CrossRef]
- Yang, Y.C.; Banuyelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Tageting androgen receptor activation function-1 with EPI to evercome resistance mechanisms in castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef]
- McKay, R.R.; Ye, H.; Xie, W.; Lis, R.; Calagua, C.; Zhang, Z.; Trinh, O.D.; Chang, S.L.; Harshman, L.C.; Ross, A.E.; et al. Evaluation of intense androgen deprivation before prostatectomy: A randomized phase II trial of enzalutamide and leuprolide with or without abiraterone. J. Clin. Oncol. 2019, in press. [Google Scholar] [CrossRef]
- Rosenthal, S.A.; Hu, C.; Sartor, O.; Gomella, L.G.; Amin, M.B.; Purdy, J.; Michalski, J.M.; Garzotto, M.G.; Pervez, N.; Balogh, A.G.; et al. Effect of chemotherapy with docetaxel with androgen suppression and radiotherapy for localized high-risk prostate cancer: The randomized phase III NRG oncology RTOG 0521 trial. J. Clin. Oncol. 2019, 37, 923–931. [Google Scholar] [CrossRef]
- Sowalsky, A.G.; Ye, H.; Bhasin, M.; Van Allen, E.M.; Loda, M.; Lis, R.T.; Montaser-Koushari, L.; Calagua, C.; Ma, F.; Russo, J.W.; et al. Neoadjuvant-intensive androgen deprivation therapy selects for prostate tumor foci with diverse subclonal oncogenic alterations. Cancer Res. 2018, 78, 4716–4730. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chang, L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalafa, M.; Speers, C.; Cooperberg, M.; Kim, W.; Ryan, C.J.; et al. Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef]
- Bill-Axelson, A.; Holmberg, L.; Garmo, H.; Taari, K.; Busch, C.; Nordling, S.; Haggman, M.; Andersson, S.O.; Andrén, O.; Steineck, G.; et al. Radical prostatectomy or watchful waiting in prostate cancer-29-year follow-up. N. Engl. J. Med. 2018, 379, 2319–2329. [Google Scholar] [CrossRef]
- Rodriguez-Bravo, V.; Pippa, R.; Song, W.M.; Cordon, M.C.; Dominguez-Andres, A.; Fujiwara, N.; Woo, J.; Koh, A.P.; Koh, A.P.; Ertel, A.; et al. Nuclear pores promote lethal prostate cancer by increasing POM121-driven E2F1, MYC, and AR nuclear import. Cell 2018, 174, 1200–1215. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.E.; Kalpainen, A.; Corella, A.N.; et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef]
- Brennen, W.M.; Isaacs, J.T. Cellular origin of androgen receptor pathway-independent prostate cancer and implications for therapy. Cancer Cell 2017, 9, 399–401. [Google Scholar] [CrossRef]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Humpreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammed, T.A.; Chen, Y.; Fedor, H.L. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 37, 1028–1038. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Xiao, W.; Yan, L.; Guan, W.; Hu, Z.; Wu, L.; Huang, Q.; Wang, J.; Xu, H.; et al. Androgen-receptor splice variant-7-positive prostate cancer: A novel molecular subtype with markedly worse androgen-deprivation therapy outcomes in newly diagnosed patients. Mod. Pathol. 2018, 31, 198–208. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Halabi, S.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danile, D.C.; Healy, P.; Ananad, M.; Rothwell, C.J.; et al. Prospective multicenter validation of androgen receptor splice variant 7 and hormone therapy resistance in high-risk castration-resistant prostate cancer: The PROPHECY study. J. Clin. Oncol. 2019, 37, 1120–1129. [Google Scholar] [CrossRef]
- Kohli, M.; Ho, Y.; Hillman, D.W.; Van Etten, J.L.; Henzler, C.; Yang, R.; Sperger, J.M.; Li, Y.; Tseng, E.; Hon, T.; et al. Androgen receptor variant AR-V9 is coexpressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clin. Cancer Res. 2017, 23, 4704–4715. [Google Scholar] [CrossRef]
- Paschalis, A.; Sharp, A.; Welti, J.C.; Neeb, A.; Raj, G.V.; Luo, J.; Plymate, S.R.; deBono, J. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–675. [Google Scholar] [CrossRef]
- Henzler, G.; Li, Y.; Yang, R.; McBride, T.; Ho, Y.; Sprenger, C.; Liu, G.; Coleman, I.; Lakely, B.; Li, R.; et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun. 2016, 7, 13668. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, F.; Xu, S.; Cui, X.; Hussain, A.; Fazli, L.; Gleave, M.; Dong, X.; Qi, J. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E4584–E4593. [Google Scholar] [CrossRef]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Nava Rodrigues, D.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Invest. 2019, 129, 192–208. [Google Scholar] [CrossRef]
- Antonorakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef]
- Hodara, E.; Morrison, G.; Cunha, A.; Zaifeld, D.; Xu, T.; Xu, Y.; Dampsey, P.W.; Pagano, P.C.; Bischoff, F.; Khurana, A.; et al. Multiparametric liquid biopsy analysis in metastatic prostate cancer. JCI Insight 2019, 4, 125529. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Mizuno, K.; Yamassaki, T.; Miyazaki, Y.; Makino, Y.; Okasho, K.; Li, X.; Utsunomiya, N.; Goto, T.; Kobayashi, T.; et al. Clinical utility of androgen receptor gene aberrations in circulating cell-free DNA as biomarker for treatment of castration-resistant prostate cancer. Sci. Rep. 2019, 9, 4030. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Mostert, B.; van der Vlugt-Daane, M.; Kraan, J.; Beaufort, C.M.; Van, M.; Prager, W.J.C.; De Laere, B.; Belle, N.; Hamberg, P.; et al. An in-depth evaluation of the validity and logistics surrounding the testing of AR-V7 mRNA expression in circulating tumor cells. J. Mol. Diagn. 2018, 20, 316–325. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, D.; Thomas-Ahner, J.M.; Lu, C.; Zhao, P.; Zhang, Q.; Geraghty, C.; Yan, P.S.; Hankey, W.; Sunkel, B.; et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. USA 2018, 115, 6810–6815. [Google Scholar] [CrossRef]
- Cai, L.; Tsai, Y.H.; Wang, P.; Wang, J.; Li, D.; Fan, H.; Zhao, Y.; Bareja, R.; Lu, R.; Wilson, E.M.; et al. ZFX mediates non-canonical oncogenic functions of the androgen receptor splice variant 7 in castrate-resistant prostate cancer. Mol. Cell 2018, 72, 341–354. [Google Scholar] [CrossRef]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, Y.; Li, W.; Uo, T.; et al. Arv7 repressees tumor-suppressor genes in catration-resistant prostate cancer. Cancer Cell 2019, 35, 401–413. [Google Scholar] [CrossRef]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-kB2/p52:c-Myc:hnRNPA1 pathway regulates expression of androgen receptor splice variants and enzalutamide sensitivity in prostate cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef]
- Tummala, R.; Lou, W.; Gao, A.C.; Nadiminty, N. Quercetin targets nhRNPA1 to overcome enzalutamide resistance in prostate cancer cells. Mol. Cancer Ther. 2017, 16, 2770–2779. [Google Scholar] [CrossRef]
- Carabet, L.A.; Leblanc, E.; Lallous, N.; Morin, H.; Ghaidi, F.; Lee, J.; Rennie, P.S.; Cherkasov, A. Computer-aided discovery of small molecules targeting RNA splicing activity of hnRNP A1 in castration-resistant prostate cancer. Molecules 2019, 14, 763. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef]
- Onstenk, W.; Sieuwerts, A.M.; Kraan, J.; Van, M.; Nieuweboer, A.J.; Mathjssen, R.H.; Hamberg, P.; Meulenbeld, H.J.; De Laere, B.; Drix, L.Y.; et al. Efficacy of Cabazitaxel in castration-resistant prostate cancer is independent of the presence of AR-V7 in circulating tumor cells. Eur. Urol. 2015, 68, 939–945. [Google Scholar] [CrossRef]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the validity of nuclear-localized androgen receptor splice variant 7 in circulating tumor cells as predictive biomarker for castration-resistant prostate cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Tagawa, S.T.; Galletti, G.; Worroll, D.; Ballman, K.; Vanhuyse, M.; Sonpavde, G.; North, S.; Albany, C.; Tsao, C.K.; et al. Randomized, noncompareative, phase II trial or early switch from docetaxel to cabazitaxel or vice versa, with integrated biomarker analysis, in men with chemotherapy-naïve, metastatic, castration-resistant prostate cancer. J. Clin. Oncol. 2017, 35, 3181–3188. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Anatonarakis, E.S.; Giyrezi, A.; Galletti, G.; Kim, S.; Worrol, D.; Stewart, J.; Zaher, A.; Sztrowski, T.P.; Ballman, K.V.; et al. Expression of AR-V7 and Arv567es in circulating tumor cells correlates with outcomes to taxane therapy in men with metastatic prostate cancer treated in TAXYNERGY. Clin. Cancer Res. 2019, 25, 1880–1886. [Google Scholar] [CrossRef]
- Thadani-Mulero, M.; Portella, L.; Sun, S.; Sung, M.; Matov, A.; Vessella, R.L.; Corey, E.; Nanus, D.M.; Plymate, S.R.; Giannakakou, P. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res. 2014, 74, 2270–2282. [Google Scholar] [CrossRef]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef]
- Aggarwal, R.R.; Quigley, D.A.; Huang, J.; Zhang, L.; Beer, T.M.; Rettig, M.B.; Reiter, R.E.; Gleave, M.E.; Thomas, G.V.; Foye, A.; et al. Whole genome and transcriptional analysis of treatment-emergent small cell neuroendocrine prostate cancer demonstrates intra-class heterogeneity. Mol. Cancer Res. 2019, 17, 1235–1240. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef]
- Hamid, A.A.; Gray, K.P.; Shaw, G.; MacConaill, L.; Evan, C.; Bernard, B.; Loda, M.; Corcoran, N.; VanEllen, E.; Choudhury, A.; et al. Compound genomic alterations of TP53, PTEN, and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur. Urol. 2019, 76, 89–97. [Google Scholar] [CrossRef]
- Lee, S.H.; Johnson, D.; Luong, R.; Sun, Z. Crosstalking between androgen and PI3K/AKT signaling pathways in prostate cancer cells. J. Biol. Chem. 2015, 290, 2759–2768. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Rodriguy, D.N.; Riisnaes, R.; Miranda, S.; Figueirido, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN protein loss and clinical outcome for castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef]
- Ding, Z.; Wu, C.J.; Chu, G.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.; Wu, X.; Lis, R.; et al. SMAD4-dependent barrier contrains prostate cancer growth and metastatic progression. Nature 2011, 470, 269–273. [Google Scholar] [CrossRef]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Jackson, W.C.; Suresh, K.; Tumati, V.M.; Allen, S.G.; Dess, R.T.; Salami, S.S.; George, A.; Kaffenberger, S.D.; Miller, S.C.; Hewarn, J.W.D.; et al. Intermediate endpointrs after postprostectomy radiotherapy: 5-year distant metastasis to predict overall survival. Eur. Urol. 2018, 74, 413–419. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN-and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, D.; Ding, H.F.; Kim, J.; Zhang, J.; Hai, T.; Yan, C. Loss of ATF3 promotes AKT activation and prostate cancer development in a PTEN knockout mouse model. Oncogene 2015, 34, 4975–4984. [Google Scholar] [CrossRef]
- Schwartz, S.; Woongipat, J.; Trigwell, C.B.; Hanco, U.; Corver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback suppression of PI3KA signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef]
- Ikeda, S.; Elkin, S.K.; Tomson, B.N.; Carter, J.L.; Kurzrock, R. Next-generation sequencing of prostate cancer: Genomic and pathway alterations, potential actionability patterns, and relative rate of use of clinical-grade testing. Cancer Biol. Ther. 2019, 20, 219–226. [Google Scholar] [CrossRef]
- Millis, S.Z.; Jardim, D.L.; Albacker, L.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Kurzrock, R. Phosphatidylinositol 3-kinase pathway genomic alterations in 60,991 diverse solid tumors informs targeted therapy opportunities. Cancer 2019, 125, 1185–1199. [Google Scholar] [CrossRef]
- Pearson, H.B.; Li, J.; Meniel, V.S.; Fennell, C.M.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; Macpherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of PIK3CA mutation as a genetic driver of prostate cancer that cooperates with PTEN loss to accelerate progression and castration-resistant growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR signaling and the PI3K pathway in prostate cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.S.; Kim, D.Y.; So, I.; Jeon, J.H. PI3K pathway in prostate cancer: All resistant roads lead to PI3K. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 198–206. [Google Scholar] [CrossRef]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the clinical development of PI3K inhibitors: Strategies to improve their impact in solid tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef]
- Yang, J.; Niue, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Kim, S.M.; Nguyen, T.T.; Ravl, A.; Kubinick, P.; Finicle, B.T.; Jayashankar, V.; Malacrida, L.; Hou, J.; Robertson, J.; Gao, D.; et al. PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov. 2018, 8, 866–883. [Google Scholar] [CrossRef]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Llkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1αMediated metabolic switch. Oncogene 2014, 33, 5251–5261. [Google Scholar] [CrossRef]
- Penfold, L.; Woods, A.; Muckett, P.; Nikitin, A.Y.; Kent, T.R.; Zhang, S.; Graham, R.; Pollard, A.; Carling, D. CAMKK2 promotes prostate cancer independently of AMPK via increased lipogenesis. Cancer Res. 2018, 78, 6747–6761. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Rachagani, S.; Muniyan, S.; Siddiqui, J.A.; Cruz, E.; Sharma, S.; Krishnan, R.; Killips, B.J.; Sheinin, Y.; Lele, S.M.; et al. FDPS cooperates with PTEN loss to promote prostate cancer progression through modulation of small GTPases/AKT axis. Oncogene 2019, 38, 5265–5280. [Google Scholar] [CrossRef]
- Goldstein, J.; Goyal, R.; Roland, J.T.; Gellert, L.L.; Clark, P.E.; Hameed, O.; Giannico, G.A. MAGI-2 is a sensitive and specific marker of prostatic adenocarcinoma: A comparison with AMACR. Am. J. Clin. Pathol. 2016, 146, 294–302. [Google Scholar] [CrossRef]
- Goldstein, J.; Borowsky, A.D.; Goyal, R.; Roland, J.T.; Arnold, S.A.; Gellert, L.L.; Clark, P.E.; Hameed, O.; Giannico, G.A. MAGI-2 in prostate cancer: An immohistochemical study. Hum. Pathol. 2016, 52, 83–91. [Google Scholar] [CrossRef]
- David, S.D.N.; Arnold Egloff, S.A.; Goyal, R.; Clark, P.E.; Phillips, S.; Gellert, L.L.; Hameed, O.; Giannico, G.A. MAGI2 is an independent predictor of biochemical recurrence in prostate cancer. Prostate 2018, 78, 616–622. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, Z.A. Transcriptional regulation of the Nkx3.1 gene in prostate luminal stem cell specification and cancer initiation via its 3’ genomic region. J. Biol. Chem. 2017, 292, 13521–13530. [Google Scholar] [CrossRef]
- Dutta, A.; Le Magnen, C.; Mitrofanon, A.; Uoyang, X.; Califano, A.; Abate-Shen, C. Identification of an NKX3.1-G9a-UTY transcriptional regulatory network that controls prostate differentiation. Science 2016, 352, 1576–1580. [Google Scholar] [CrossRef]
- Le Magnen, C.; Virk, R.K.; Dutta, A.; Kim, J.Y.; Panja, S.; Lopez-Bujanda, Z.A.; Califano, A.; Drake, C.G.; Mitrofanova, A.; Abate-Shen, C. Cooperation of loss of NKX3.1 and inflammation in prostate cancer initiation. Dis. Model Mechan. 2018, 11, 35139. [Google Scholar] [CrossRef]
- Gurel, B.; Ali, T.Z.; Montgomery, E.A.; Begum, S.; Hicks, J.; Goggins, M.; Eberhart, C.G.; Clark, D.P.; Bierbich, C.J.; Epstein, I.I. NKX3.1 as a marker of prostatic origin in metastatic tumors. Am. J. Surg. Pathol. 2010, 34, 1097–1105. [Google Scholar] [CrossRef]
- Yang, C.C.; Chung, A.; Ku, C.Y.; Brill, L.M.; Williams, R.; Wolf, D.A. Systems analysis of the prostate tumor suppressor NKX3.1 supports role in DNA repair and luminal cell differentiation. F100Res. 2014, 3, 115. [Google Scholar] [CrossRef][Green Version]
- Lei, Q.; Jiao, J.; Xin, L.; Chang, S.J.; Wang, S.; Gao, J.; Gleave, M.E.; Witte, O.N.; Liu, X.; Wu, H.; et al. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell 2006, 9, 367–372. [Google Scholar] [CrossRef]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk SNP-mediated promoter-enhancer switching drives prostate cancer through lncRNA PCAT19. Cell 2018, 174, 564–575. [Google Scholar] [CrossRef]
- Gao, P.; Xia, J.; Sipeki, C.; Dong, X.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell 2018, 174, 576–589. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Keizman, D.; Zhong, Z.; Gurel, B.; Lotan, T.L.; Hicks, J.L.; Fedor, H.L.; Carducci, M.A.; De Marzo, A.M.; Eisenberger, M.A. An immunohistochemical signature comprising PTEN, MYC, and Ki67 predicts progression in prostate cancer patients receiving adjuvant docetaxel after prostatectomy. Cancer 2012, 118, 6063–6071. [Google Scholar] [CrossRef]
- Iwata, T.; Schultz, D.; Hicks, J.; Hubbard, G.K.; Mutton, L.N.; Lotan, T.L.; Betehl, C.; Lotz, M.T.; Yegnasubramanian, S.; Ne Dang, C.V.; et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of NKX3.1 in mouse luminal epithelial cells. PLoS ONE 2010, 5, e9427. [Google Scholar] [CrossRef]
- Zhangh, C.; Zhang, S.; Zhang, Z.; He, J.; Xu, Y.; Liu, S. ROCK has a crucial role in regulating prostate tumor growth through interaction with c-Myc. Oncogene 2014, 33, 5582–5591. [Google Scholar] [CrossRef]
- Priolo, C.; Pyne, S.; Rose, J.; Regan, E.R.; Zadra, G.; Photopoulos, C.; Cacciatore, S.; Schultz, D.; Scaglia, N.; McDunn, J.; et al. AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Cancer Res. 2014, 74, 7198–7204. [Google Scholar] [CrossRef]
- Wee, Z.N.; Li, Z.; Lee, P.L.; Lim, Y.P.; Yu, Q. EZH2-mediated inactivation of IFN-gamma-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep. 2014, 8, 204–216. [Google Scholar] [CrossRef]
- Koh, C.M.; Gurel, B.; Sutcliffe, S.; Aryee, M.J.; Schultz, D.; Iwata, T.; Uemura, M.; Zeller, K.I.; Anele, U.; Zheng, Q.; et al. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am. J. Pathol. 2011, 178, 1824–1834. [Google Scholar] [CrossRef]
- Baena-Del Valle, J.; Zheng, Q.; Esopi, D.; Rubenstein, M.; Hubbard, G.K.; Moncallano, M.C.; Hrusakewycz, A.; Vaghsia, A.; Yegnasubramanian, S.; Wheelon, S.J.; et al. MYC drives overexpression of telomerase RNA (hTR/TERC) in prostate cancer. J. Pathol. 2018, 244, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Barfold, S.J.; Urbanucci, A.; Itkonen, H.M.; Fazli, L.; Hicks, J.J.; Thiede, B.; Rennie, P.S.; Yegnasubramanian, S.; De Marzo, A.; Mills, I.G. c-Myc antagonizes the transcriptional activity of the androgen receptor in prostate cancer affecting key gene networks. EBioMedicine 2017, 18, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnov, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Arnoldussen, Y.J.; Storm, M.; Tesokova, M.; Neuseth, H.Z.; Zhoo, S.; Fazli, S.; Rennie, P.; Risberg, B.; Waehre, H.; et al. Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO Mol. Med. 2015, 7, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Matejcic, M.; Saunders, E.J.; Daadaev, T.; Brook, M.N.; Waug, K.; Sheng, X.; Olama, A.A.A.; Schumascher, F.R.; Ingles, S.A.; Govindasami, K.; et al. Germilne variation at 8q24 and prostate cancer risk in men of European ancestry. Nat. Commun. 2018, 9, 4616. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, A.E.; Rahl, P.B.; Shi, J.; Jakidis, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Ciesilk, M.; Yang, R.; Escara-Wilke, J.; Wilders-Romans, K.; Dhomireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef]
- Coleman, D.J.; Gao, L.; Schwartzmah, J.; Koskola, J.E.; Sampson, D.; Derrick, D.S.; Urrutia, J.; Balter, A.; Buchard, J.; King, C.J.; et al. Maintenance of MYC expressionpromotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Sci. Rep. 2019, 9, 3823. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration- resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7128–7138. [Google Scholar] [CrossRef]
- Kregel, S.; Malik, R.; Asangani, I.A.; Wilder-Romans, K.; Rajhendiran, T.; Xiao, L.; Vo, J.N.; Soni, T.; Cieslik, M.; Fernandez-Selas, E.; et al. Functional and mechanistic interrogation of BET bromodomain degraders for the treatment of metastatic castration resistant prostate cancer. Clin. Cancer Res. 2019, 25, 4038–4048. [Google Scholar] [CrossRef]
- Chaytor, L.; Simcock, M.; Nakjang, S.; Heath, R.; Walker, L.; Robson, C.; Jones, D.; Gaughan, L. The pioneering role of GATA2 in androgen receptor variant regulation is controlled by bromodomain and extraterminal proteins in castrate-resistant prostate cancer. Mol. Cancer Ther. 2019, 17, 1264–1278. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morissey, C.; Zhang, X.; Comstock, C.E.; Witkiewicz, A.K.; et al. The retinoblastoma tumor suppressor controls androgen signalling and human prostate cancer progression. J. Clin. Invest. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.K.; Akakura, S.; Peresie, J.; Goodrich, D.W.; Foster, B.A.; Gelman, I.H. A transgenic mouse model for early prostate metastasis lymph nodes. Cancer Res. 2014, 74, 945–953. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Seshadri, M.; Goodrich, M.M.; Labbe, D.P.; Cortes Gomez, E.; Wang, J.; Long, H.W.; Xu, B.; et al. Rb1 and TRP53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antindrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Bonnell, M.; Karthaus, W.; Hoover, E.; Chen, C.C.; Wonguipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, C.; Boopathi, E.; Liu, Y.; Erbel, A.; Bhardway, A.; Addya, S.; Williams, N.; Cincent, J.J.; Cotria, P. RB loss promotes prostate cancer metastasis. Cancer Res. 2017, 77, 982–995. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Sun, R.; Chen, L.H.; Diplas, B.H.; Moure, C.J.; Wang, W.; Hansen, L.J.; Tao, Y.; Chen, X.; Chen, C.J.; et al. Mutant allele quantification reveals a genetic basis for TP53 mutation-driven castration resistance in prostate cancer cells. Sci. Rep. 2018, 8, 12507. [Google Scholar] [CrossRef] [PubMed]
- De Laere, B.; Oeyens, S.; Mayrhofer, M.; Whitington, T.; van Dam, P.J.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; et al. TP53 outperforms other androgen receptor biomarkers to predict abiraterone or enzalutamide outcome in metastatic castration resistant prostate cancer. Clin. Cancer Res. 2019, 25, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Mahon, K.L.; Stricker, P.; Kentch, J.; Grogan, J.; Delprado, W.; Turner, J.; Horvath, L.; Quinn, D.I. P53 as a predictor of clinical outcome in localized prostate cancer. J. Clin. Oncol. 2018, 36, 57. [Google Scholar] [CrossRef]
- Kaur, H.B.; Lu, T.; Guedes, L.B.; Maldonado, L.; Reitz, L.; Barbe, J.R.; De Marzo, A.M.; Tomlins, J.A.; Sfanos, K.S.; Eisenberger, M.; et al. TP53 missense mutation is associated with increased tumor-infiltrating T-cells in primary prostate cancer. Hum. Pathol. 2019, 87, 95–102. [Google Scholar] [CrossRef]
- Wang, G.; Lunardi, A.; Zhang, J.; Chen, Z.; Ala, U.; Webster, K.A.; Tay, Y.; Gonzalez-Billlabeilla, E.; Egia, A.; Shaffer, D.R.; et al. Zbtb7a suppresses prostate cancer through repression of a Sox9-dependent pathway for cellular senescence bypass and tumor invasion. Nat. Genet. 2013, 45, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.C.; Capper, A.; Ning, J.; Knight, E.; de Bono, J.; Swain, A. SOX9 is a driver of aggressive prostate cancer by promoting invasion, cell fate and cytoskeleton alterations and epithelial to mesenchymal transition. Oncotarget 2018, 9, 7604–7615. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Ye, H.; He, H.H.; Gerrin, S.J.; Chen, S.; Tanenbaum, B.A.; Cai, C.; Sowalsky, A.G.; He, L.; Wang, H.; et al. SOX9 drives WNT pathway activation in prostate cancer. J. Clin. Invest. 2016, 126, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Ciesik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018, 173, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Nenseth, H.Z.; Saatcioglu, F. Role of PLZF as a tumor suppressor in prostate cancer. Oncotarget 2017, 8, 71317–71324. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Qu, S.; Tesikova, M.; Wang, L.; Kristian, A.; Maelkandsmo, G.M.; Kong, H.; Zhang, T.; Jeronimo, C.; Teixeira, M.R.; et al. Molecular circuit involving KLK4 integrates androgen and mTOR signaling in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E2572–E2581. [Google Scholar] [CrossRef]
- Xiao, G.Q.; Unger, P.; Yang, Q.; Kinoshita, Y.; Siungh, K.; McMahon, L.; Nastiuk, K.; Sha, K.; Krolewsk, J.; Burstein, D.; et al. Loss of PLZF expression in prostate cancer by immunohistochemistry correlates with tumor aggressiveness and metastasis. PLoS ONE 2015, 10, e0121318. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Botta, G.; Gao, S.; Li, T.; VanAllen, E.M.; Treacy, D.J.; Cai, C.; He, H.H.; Sweeney, C.J.; Brown, M.; et al. PLZF, a tumor suppressor genetically lost in metastatic castration-resistant prostate cancer, is a mediator of resistance to androgen deprivation therapy. Cancer Res. 2015, 75, 1944–1948. [Google Scholar] [CrossRef]
- Stopsack, K.H.; Gerke, T.; Tyekucheva, S.; Mazzu, Y.Z.; Lee, G.S.; Chakrabarty, G.; Abida, W.; Mucci, L.A.; Kantoff, P.W. Low expression of the androgen-induced tumor suppressor gene PLZF in lethal prostate cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 707–714. [Google Scholar] [CrossRef]
- Khani, F.; Mosquera, J.M.; Park, K.; Blattner, M.; O’Reilly, C.; MacDonald, T.Y.; Chen, Z.; Srivastava, A.; Tewari, A.K.; Barbieri, C.E.; et al. Evidence for molecular differences in prostate cancer between African American and Caucasian Men. Clin. Cancer Res. 2014, 20, 4925–4934. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Yu, Y.; Varambally, S.; Mehra, R.; Perner, S.; Demichelis, F.; Helgeson, B.E.; Laxman, B.; Morris, D.S.; et al. The role of SPINK1 in EGR rearrangement-negative prostate cancers. Cancer Cell 2008, 13, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Faisal, F.A.; Kaur, H.B.; Tosoian, J.J.; Tomlins, S.A.; Schaeffer, E.M.; Lotan, T.L. SPINK1 expression is enriched in African American prostate cancer but is not associated with altered imnmune infiltration or oncologic outcomes post-prostatectomy. Prostate Cancer Prostatic Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Petrovics, G.; Li, H.; Stumpel, T.; Tan, S.H.; Young, D.; Katta, S.; Li, Q.; Ying, K.; Klocke, B.; Ravindranath, L.; et al. A novel genomic alteration of LSAMP associates with aggressive prostate cancer in African American men. EBioMedicine 2015, 2, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, K.J.; Paris, P.L.; Hoffmann, T.J.; Cardin, N.J.; Kazma, R.; Mefford, J.A.; Simko, J.P.; Ngo, V.; Chen, Y.; Levin, A.M.; et al. Mutational landscape of aggressive prostate tumors in African American men. Cancer Res. 2016, 76, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W. Towards greater inclusion in cancer genomics studies. Cancer Res. 2018, 78, 6726–6727. [Google Scholar] [CrossRef] [PubMed]
- Jaratlerdsiri, W.; Chan, E.; Gong, T.; Petersen, D.; Kalsbeek, A.; Venter, P.A.; Stricker, P.D.; Bornman, R.; Hayes, V.M. Whole-genome sequencing reveals elevated tumor burden and initiating driver mutations in African men with treatment-naïve, high-risk prostate cancer. Cancer Res. 2018, 78, 6736–6746. [Google Scholar] [CrossRef] [PubMed]
- Tonon, L.; Fromont, G.; Boyault, S.; Thomas, E.; Ferrari, A.; Sertier, A.S.; Kielbassa, J.; Le Texier, V.; Kamoun, A.; Elarouci, A.; et al. Mutational profile of aggressive, localized prostate cancer from African Carribean men versus European ancestry men. Eur. Urol. 2019, 75, 11–15. [Google Scholar] [CrossRef]
- Kherandish, P.; Chinegwundoh, F. Ethnic differences in prostate cancer. Br. J. Cancer 2011, 105, 481–485. [Google Scholar] [CrossRef]
- Mahal, B.A.; Berman, R.A.; Taplin, M.E.; Huang, F.W. Prostate cancer-specific mortality across gleason scores in black vs. nonblack men. JAMA 2018, 320, 2479–2481. [Google Scholar] [CrossRef]
- Mahal, B.A.; Alshalalfa, M.; Spratt, D.E.; Davicioni, E.; Zhao, S.G.; Feng, F.Y.; Rebbeck, T.R.; Nguyen, P.L.; Huang, F.W. Prostate cancer genomic-risk differences between African-American and white men across gleason scores. Eur. Urol. 2019, 75, 1038–1040. [Google Scholar] [CrossRef]
- Yamoah, K.; Johnson, M.H.; Choeumg, V.; Faisal, F.A.; Yousefi, K.; Haddad, Z.; Ross, A.E.; Alihalafa, M.; Den, R.; Lal, P.; et al. Novel biomarker signature that may predict aggressive disease in African American men with prostate cancer. J. Clin. Oncol. 2015, 33, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Lung, P.Y.; Shell, A.B.; Gad, S.A.; Bustamante, J.J.; Ali, H.I.; Rim, J.S.; Deep, G.; Zhang, J.; Abd Elmageed, Z.Y. Dysregulated gene expression predicts tumor aggressivenesss in African-American prostate cancer patients. Sci. Rep. 2018, 8, 16335. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J.; Vidal, A.C.; Howard, L.E.; Terris, M.K.; Cooperberg, M.R.; Amling, C.L.; Kane, C.J.; Aronson, W.J. Shared Equal Access Regional Cancer Hospital (SEARCH) Database Staudy Group. Race and risk of metastases and survival after radical prostatectomy: Results from the SEARCH database. Cancer 2017, 123, 4199–4206. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.C.; Howard, L.E.; De Hoedt, A.; Kane, C.J.; Terris, M.K.; Aronson, W.J.; Cooperberger, M.R.; Amling, C.L.; Lechpammer, S.; Flanders, S.C.; et al. Does race predict the development of metastases in men who receive androgen-deprivation therapy for a biochemical recurrence after radical prostatectomy? Cancer 2019, 125, 434–441. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.V.; Whittington, R.; Malkowicz, S.B.; Schultz, D.; Blank, K.M.; Broderick, G.A.; Tomasewski, J.E.; Renshaw, A.A.; Kaplan, I.; Beard, C.J.; et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998, 280, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Thorat, M.A.; Andriole, G.; Brawley, O.W.; Ciulig, Z.; Eeles, R.A.; Ford, L.G.; Hamdy, F.C.; Holmberg, L.; Llic, D.; et al. Prevention and early detecxtion of prostate cancer. Lancet Oncol. 2014, 15, e484–e492. [Google Scholar] [CrossRef]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Fosetr, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef]
- Cuzicz, J.; Berney, D.M.; Fisher, G.; Mesher, D.; Moller, H.; Reid, J.E.; Perry, M.; Park, J.; Yonus, A.; Gutin, A.; et al. Prognostic value of a cell cycle progression signature for prostate cancer death in a conservatively managed needle biopsy cohort. Br. J. Cancer 2012, 106, 1095–1099. [Google Scholar] [CrossRef]
- Klein, E.A.; Cooperberg, M.R.; Magi-Galluzzi, C.; Simko, J.P.; Falzarano, S.M.; Maddala, T.; ChaN, J.M.; Li, J.; Cowan, J.E.; Tsiatis, A.C.; et al. A 17-gene assay to predict prostate cancer aggressiveness in the context of Gleason grade heterogeneity, tumor multifocality, a biopsy undersampling. Eur. Urol. 2014, 66, 550–560. [Google Scholar] [CrossRef]
- Erho, N.; Crisan, A.; Vergara, I.A.; Mitra, A.P.; Ghadessi, M.; Buerki, C.; Bergstriath, E.J.; Kollmeyer, T.; Fink, S.; Haddad, Z.; et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS ONE 2013, 24, e66855. [Google Scholar] [CrossRef]
- Karnes, R.J.; Bergstrahl, E.J.; Davicioni, E.; Ghadessi, M.; Buerki, C.; Mitra, A.P.; Crisan, A.; Erho, N.; Vergara, I.A.; Lam, L.L.; et al. Validation of a genomic classifier that predicts metastasis following radical prostectomy in at risk population. J. Urol. 2013, 190, 2047–2053. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Yousefi, K.; Haddad, Z.; Choeurng, V.; Buerki, C.; Stephenson, A.J.; Li, J.; Kattan, M.W.; Magi-Galluzzi, C.; Davicioni, E. A genomic classifier improves prediction of mestatic disease within 5 years after surgery in node-negative high-risk prostate cancer patients managed by radical prostatectomy without adjuvant therapy. Eur. Urol. 2015, 67, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.E.; Johnson, M.H.; Yousefi, K.; Davicioni, E.; Netto, G.J.; Marchionni, L.; Fedor, H.L.; Glavarts, S.; Choerung, V.; Buerki, C.; et al. Tissue-based genomic augments post-prostatectomy risk stratification in a natural history cohort of intermediate-and high-risk men. Eur. Urol. 2016, 69, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Yousefy, K.; Deheshi, S.; Ross, A.E.; Den, R.B.; Schaeffer, E.M.; Trock, B.J.; Zhang, J.; Glass, A.G.; Dicker, A.P.; et al. Individual patient-level meta-analysis of the performance of the Decipher Genomic Classifier in high-risk men after prostatectomy to predict development of metastatic disease. J. Clin. Oncol. 2017, 35, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Shoag, J.E.; Tosoian, J.J.; Salami, S.S.; Barbieri, C.E. Unraveling prostate cancer genomics, pathology, and magnetic resonance imaging visibility. Eur. Urol. 2019, 76, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Erho, N.; Chan, J.M.; Feng, F.Y.; Fishbane, N.; Zhao, S.G.; Simko, J.P.; Cowan, J.E.; Lehrer, J.; Alshalalfa, M.; et al. The diverse genomic landscape of clinically low-risk prostate cancer. Eur. Urol. 2018, 74, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Zhang, J.; Santiago-Jiménez, M.; Dess, E.R.T.; Davis, J.W.; Den, R.B.; Dicker, A.P.; Kane, C.J.; Pollack, A.; Stoyanova, R.; et al. Development and validation of a novel integrated clinical-genomic risk group classification for lacalized prostate cancer. J. Clin. Oncol. 2018, 36, 581–590. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chang, S.L.; Spratt, D.E.; Erho, N.; Yu, M.; Ashba, H.A.; Ashalalfa, M.; Speers, C.; Tomlins, S.A.; Davicioni, E.; et al. Development and validation of a 24-gene predictor of response to postoperative radiotherapy in prostate cancer: A matched, retrospective analysis. Lancet Oncol. 2016, 17, 1612–1620. [Google Scholar] [CrossRef]
- Van Den Eeden, S.K.; Lu, R.; Zhang, N.; Quesenberry, C.P.; Shan, J.; Han, J.S.; Tsiatis, A.C.; Leimpeter, A.D.; Lawrence, H.J.; Febbo, P.G.; et al. A biopsy-based 17-gene genomic prostate score as a predictor of metastases and prostate cancer death in surgically treated men with clinically localized disease. Eur. Urol. 2018, 73, 129–138. [Google Scholar] [CrossRef]
- Cullen, J.; Rosner, I.L.; Brand, T.C.; Zhang, N.; Tsiatis, A.C.; Moncur, J.; Ali, A.; Chen, Y.; Knezevic, D.; Maddala, T.; et al. A biopsy-based 17-gene genomic prostate score predicts recurrence after radical prostectomy and adverse surgical pathology in a racially diverse population of men with clinically low- and intermediate-risk prostate cancer. Eur. Urol. 2015, 68, 123–131. [Google Scholar] [CrossRef]
- Lapointe, J.; Li, C.; Higgings, J.; de Rijn, M.; Bair, E.; Montgomery, K.; Egevald, M.; Rayford, W.; Bergerheim, U.; Ekman, P.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Li, C.; Giacomini, C.; Salari, K.; Huang, S.; Wang, P.; Ferrari, M.; Hernandez-Boussard, T.; Brooks, J.; Pollack, J. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res. 2007, 67, 8504–8510. [Google Scholar] [CrossRef] [PubMed]
- Markert, E.K.; Mizuno, H.; Vazquez, A.; Levine, A.J. Molecular classification of prostate cancer using curated expression signatures. Proc. Natl. Acad. Sci. USA 2011, 108, 21276–21281. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Rye, M.B.; Bertilsson, H.; Drablos, F.; Angelsen, A.; Bathen, T.; Tessem, M.B. Gene signatures ESC, MYC and ERG-fusion are early markers of a potentially dangerous subtype of prostate cancer. BMC Med. Genom. 2014, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yin, X.; Shen, P.; Sun, G.; Yang, Y.; Liu, J.; Chen, N.; Zeng, H. The association between SPINK1 and clinical outcomes in patients with prostate cancer: A systematic review and meta-analysis. Onco Targets Ther. 2017, 10, 3123–3130. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Cyrta, J.; Murphy, D.A.; Walczak, E.G.; Ran, L.; Agrawal, P.; Xie, Y.; Chen, Y.; Wang, S.; Zhan, Y.; et al. Aberrant activation of a gastrointestinal transcriptional circuit in prostate cancer mediates castration resistance. Cancer Cell 2017, 32, 792–806. [Google Scholar] [CrossRef]
- Bhatia, V.; Yadav, A.; Tiwari, R.; Nigam, S.; Goel, S.; Carskadon, S.; Gupta, N.; Goel, A.; Palanisamy, N.; Ateeq, B. Epigenetic silencing of miRNA-338-5p and miRNA-421 drives SPINK1-positive prostate cancer. Clin. Cancer Res. 2019, 25, 2755–2768. [Google Scholar] [CrossRef]
- Smith, B.A.; Sokolov, A.; Uzunangelov, V.; Baertsch, R.; Newton, Y.; Graim, K.; Mathis, C.; Cheng, D.; Stuart, J.M.; Witte, O.N. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 2015, 112, E6544–E6552. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vichery, T. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chen, W.S.; Das, R.; Chang, S.L.; Tomlins, S.A.; Chou, J.; Quigley, D.A.; Dang, H.X.; Barnard, T.J.; Mahal, B.A.; et al. Clinical and genomic implications of luminal and basal subtypes across carcinomas. Clin. Cancer Res. 2019, 25, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Knudsen, B.S.; Er, N.; Alshalalfa, M.; Takhar, M.; Ashab, H.; Davicioni, E.; Karnes, J.; Klein, E.A.; Den, R.B.; et al. Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res. 2016, 76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; Wu, T.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.L.; Massic, C.E.; Ramos-Montoya, A.; Zecchini, Y.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef]
- Chen, Z.; Lan, X.; Tomas-Ahner, J.M.; Wu, D.; Liu, X.; Ye, Z.; Wang, L.; Sunkel, B.; Grenade, C.; Chen, J.; et al. Agonist and antagonist switch DNA motifs recognized by human androgen receptor in prostate cancer. EMBO J. 2015, 34, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Stelloo, S.; Nevedomskaya, E.; van der Poel, H.; de Jong, J.; van Leenders, G.; Jenster, G.; Wessels, L.; Bergam, A.M.; Zwart, W. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol. Med. 2015, 7, 1450–1463. [Google Scholar] [CrossRef]
- Dudani, J.S.; Ibrahim, M.; Kirkparick, J.; Warren, A.D.; Bhatia, S.N. Classification of prostate cancer using a protease activity nanosensor library. Proc. Natl. Acad. Sci. USA 2018, 115, 8954–8959. [Google Scholar] [CrossRef]
- Perera, M.; Papa, N.; Roberts, M.; Williams, M.; Udovicich, C.; Vela, I.; Chrisrtidis, D.; Bolton, D.; Hofman, M.S.; Lawrentschuk, N.; et al. Gallium-68 prostate-specific membrane antigen positron emission tomography in advanced prostate cancer-updated diagnostic utility, sensitivity, specificity and distribution of prostate-specific membrane antigen-avid lesions: A systematic review and meta-analysis. Eur. Urol. 2019, in press. [Google Scholar]
- Lee, J.K.; Bangayan, N.J.; Chai, T.; Smith, B.A.; Pariva, T.E.; Yun, S.; Vashisht, A.; Zhang, Q.; Park, J.W.; Carey, E.; et al. Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E4473–E4482. [Google Scholar] [CrossRef]
- Stylianou, N.; Lehman, M.L.; Wang, C.; Fard, A.T.; Rockstroh, A.; Fazli, L.; Jovanovic, L.; Ward, M.; Sadowski, M.C.; Kashyap, A.S. A molecular portrait of epithelial-mesenchymal plasticity in prostate cancer associated with clinical outcome. Oncogene 2019, 38, 913–934. [Google Scholar] [CrossRef]
- Heselmeyer-Haddad, K.; Berroa Garcia, L.; Bradley, A.; Ortiz-Melendez, C.; Lee, W.J.; Christensen, R.; Prindiville, S.A.; Calzone, K.A.; Soballe, P.W.; Hu, Y.; et al. Single-cell genetic analysis reveals insights into clonal development of prostate cancers and indicates loss of PTEN as a marker of poor prognosis. Am. J. Pathol. 2014, 184, 2671–2686. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Heumann, A.; Rico, S.D.; Hicks, J.; Lecksell, K.; Koop, C.; Sauter, G.; Schlomm, T.; Simon, R. PTEN loss detection in prostate cancer: Comparison of PTEN immunohistochemistry and PTEN FISH in a large retrospective prostatectomy cohort. Oncotarget 2017, 8, 65566–65576. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Carvalho, F.L.; Peskoe, S.B.; Hicks, J.L.; Good, J.; Fedor, H.; Humphreys, E.; Han, M.; Platz, E.A.; Squire, J.A. PTEN loss is associated with upgrading of prostate cancer from biopsy to radical prostectomy. Mod. Pathol. 2015, 28, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Alshalalfa, M.; Davicioni, E.; Erho, N.; Yousefi, K.; Zhao, S.; Haddad, Z.; Den, R.B.; Dicker, A.P.; Trock, B.J.; et al. Characterization of 1577 primary prostate cancers reveals novel biological and clinicopathologic insights into molecular subtypes. Eur. Urol. 2015, 68, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Galletti, G.; Matov, A.; Beltran, H.; Fontugne, J.; Miguel Mosquera, J.; Cheung, C.; MacDonald, T.Y.; Sung, M.; O’Toole, S.; Kench, J.G.; et al. ERG induces taxane resistance in castration-resistant prostate cancer. Nat. Commun. 2014, 5, 5548. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, E.; Ishkanian, A.S.; Sykes, J.; Fraser, M.; Ross-Adams, H.; Erho, N.; Dunning, M.J.; Halim, S.; Lamb, A.D.; Moon, N.C.; et al. Tumor genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: A retrospective cohort study. Lancet Oncol. 2014, 15, 1521–1532. [Google Scholar] [CrossRef]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Schuuman, K.; Valle-Encinas, E.; Lobo, J.; Kriigsman, O.; Peeper, D.S.; Chang, S.L.; Feng, F.Y.C.; et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat. Commun. 2018, 9, 4900. [Google Scholar] [CrossRef]
- Aytes, A.; Giacobbe, A.; Mitrofanova, A.; Ruggero, K.; Cyrta, J.; Arriaga, J.; Palomero, L.; Farran-Matas, S.; Rubin, M.A.; Shen, M.M.; et al. NSD2 is a conserved driver of metastatic prostate cancer progression. Nat. Commun. 2018, 9, 5201. [Google Scholar] [CrossRef]
- Cheng, L.C.; Tan, V.M.; Ganesan, S.; Drake, J.M. Integrating phosphoproteomics into the clinical management of prostate cancer. Clin. Transl. Med. 2017, 6, 9. [Google Scholar] [CrossRef]
- Yang, W.; Freeman, M.R.; Kyprianou, N. Personalisation of prostate cancer therapy through phosphoproteomics. Nat. Rev. Urol. 2018, 15, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.M.; Graham, N.A.; Lee, J.K.; Stoyanova, T.; Faltermeier, C.M.; Sud, S.; Titz, B.; Huang, J.; Pienta, K.J.; Graeber, T.G.; et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and intrapatient heterogeneity of therapeutic kinase targets. Proc. Natl. Acad. Sci. USA 2013, 110, E4762–E4769. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.M.; Paull, E.Q.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome integration reveals patient-specific networks in prostate cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Gato, D.; Wikstrom, P.; Tyanova, S.; Lavallee, C.; Thysell, E.; Carlsson, J.; Hagglof, C.; Cox, J.; Andren, O.; Stattin, P.; et al. The proteome of primary prostate cancer. Eur. Urol. 2016, 69, 942–952. [Google Scholar] [CrossRef]
- Thysell, E.; Vidman, L.; Ylitalo, E.B.; Jernberg, E.; Crnalic, S.; Iglesias-Gato, D.; Flores-Morales, A.; Stattin, P.; Egevad, L.; Widmark, A.; et al. The proteome of prostate cancer bone metastasis reveals heterogeneity with prognostic implications. Mol. Oncol. 2019. [Google Scholar] [CrossRef]
- Latonen, L.; Afyounian, E.; Jylha, A.; Nattinen, J.; Aapola, U.; Annala, M.; Kiyinnumi, K.K.; Tammela, T.T.L.; Beueman, R.W.; Uusitalo, H.; et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 2018, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Huang, V.; Livingstone, J.; Wang, J.; Fox, N.S.; Kurganovs, N.; Ignatchenko, V.; Fritsch, K.; Donmez, N.; Heisler, L.E.; et al. The proteogenomic landscape of curable prostate cancer. Cancer Cell 2019, 35, 414–427. [Google Scholar] [CrossRef]
- Day, J.R.; Jost, M.; Reynolds, M.A.; Groskopf, J.; Rittenhouse, H. PCA3: From basic molecular science to the clinical lab. Cancer Lett. 2011, 301, 1–6. [Google Scholar] [CrossRef]
- Cui, Y.; Cao, W.; Li, Q.; Shen, H.; Liu, C.; Dheng, J.; Xu, J.; Shao, Q. Evaluation of prostate cancer antigen 3 for detecting prostate cancer: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 25776. [Google Scholar] [CrossRef]
- Lee, G.L.; Dobi, A.; Srivastava, S. Diagnostic performance of the PCA3 urine test. Nat. Rev. Urol. 2011, 8, 123–124. [Google Scholar] [CrossRef]
- Koo, K.; Mainwaring, P.N.; Tomlins, S.A.; Trau, M. Merging new-age biomarkers and naniodiagnostics for precision prostate cancer management. Nat. Rev. Urol. 2019, 16, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Presner, J.R.; Iyer, M.K.; Suhu, A.; Assangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Devicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Presner, J.R.; Zhao, S.; Erho, N.; Schipper, M.; Iyer, M.K.; Dhanasakaran, S.M.; Magi-Galluzzi, C.; Mehra, R.; Sahu, A.; Siddiqui, J.; et al. RNA biomarkers associatyed with metastatic progression in prostate cancer: A multi-institutional high-throughput analysis of SChLAP1. Lancet Oncol. 2014, 15, 1469–1480. [Google Scholar] [CrossRef]
- Mehra, R.; Udager, M.A.; Ahearn, T.U.; Cao, X.; Feng, F.Y.; Lada, M.; Petlman, J.S.; Kantoff, P.; Mucci, L.A.; Chinnayan, A.M. Overexpression of the long non-coding RNA SChLAP1 independently predicts lethal prostate cancer. Eur. Urol. 2016, 70, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.L.K.; Lo, W.; Pintilie, M.; Murgic, J.; Lalonda, E.; Bhandari, V.; Mahamud, O.; Gopalan, A.; Kweldam, C.F.; van Lenders, G.J.L.H.; et al. A prostate cancer “nimbous” genomic instability and Schlap1 dysregulation underpin aggression of intraductal and cribiform subpathologies. Eur. Urol. 2017, 72, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Aggarwal, R.; Zhang, L.; Zhao, S.G.; Thomas, G.V.; Beer, T.M.; Quigley, D.A.; Foye, A.; Playdle, D.; Huang, J.; et al. Genomic drivers of poor prognosis and enzalutamide resistance in metastatic castration-resistant prostate cancer. Eur. Urol. Oncol. 2019, 2, 405–412. [Google Scholar]
- Baxevanis, C.N.; Fortis, C.P.; Perez, S.A. Prostate cancer: Any room left for immunotherapies? Immnutherapy 2019, 11, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB promotes cancer immunity by inhibiting NF-kB activation and PD-L1 expression. Mol. Cell 2019, 73, 22–35. [Google Scholar] [CrossRef]
- Prestipino, A.; Zeiser, R. Clinical implications of tumor-intrinsic mechanisms regulating PD-L1. Sci. Transl. Med. 2018, 11, 4810. [Google Scholar] [CrossRef]
- Gevensleben, H.; Dietrich, D.; Golletz, C.; Steiner, S.; Jung, M.; Thiesler, T.; Majores, M.; Stein, J.; Uhl, B.; Muller, S.; et al. The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer. Clin. Cancer Res. 2016, 22, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Gevensleben, H.; Holmes, E.E.; Goltz, D.; Dietrichj, J.; Sailer, V.; Ellinger, J.; Dietrich, D.; Kristiansen, G. PD-l1 promoter methylation is a prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients following radical prostatectomy. Oncotarget 2016, 29, 79943–79955. [Google Scholar] [CrossRef] [PubMed]
- Ness, N.; Andersen, S.; Khanehkenari, M.R.; Nordbakken, C.V.; Valkov, A.; Paulsen, E.E.; Nordby, Y.; Bremnes, R.M.; Donnem, T.; Busund, L.T.; et al. The prognostic role of immune checkpoint markers programmed cell death protein 1 (PD-1) and programmed death ligand 1 (PD-1) in a large, multicenter prostate cancer cohort. Oncoratget 2017, 8, 26789–26801. [Google Scholar] [CrossRef] [PubMed]
- Calagua, C.; Russo, J.; Sun, Y.; Schaefer, R.; Lis, R.; Zhang, Z.; Mahonev, K.; Bublev, G.J.; Loda, M.; Taplin, M.E.; et al. Expression of PD-L1 in hormone-naïve and treated prostate cancer patients receiving neoadjuvant abiraterone acetate plus prednisone and leuprolide. Clin. Cancer Res. 2017, 23, 6812–6822. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Guner, G.; Taheri, D.; Netto, G.J.; Palsgrove, D.N.; Zheng, Q.; Guedes, L.B.; Kim, K.; Tsai, H.; Esopi, D.M.; et al. Comprehensive evaluation of programmed death-ligand 1 expression in primary and metastatic prostate cancer. Am. J. Pathol. 2018, 188, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, Q.; Zhou, Y.; He, M.; Chen, J.; Gao, Y.; Wang, X. The clinicopathologic and prognostic significance of programmed cell death ligand 1 (PD-L1) expression in patients with prostate cancer: A systematic review and meta-analysis. Front. Pharmacol. 2019, 9, 1494. [Google Scholar] [CrossRef]
- Zhao, S.G.; Lehrer, J.; Chang, S.L.; Das, R.; Erho, N.; Liu, Y.; Sjostrom, M.; Den, R.B.; Freedland, S.J.; Klein, E.A.; et al. The immune landscape of prostate cancer and nomination of PD-L2 as a potential therapeutic target. J. Natl. Cancer Inst. 2019, 111, 301–310. [Google Scholar] [CrossRef]
- Ylatalo, E.R.; Thysell, E.; Jernberg, E.; Lundholm, M.; Crnalic, S.; Egevad, L.; Statin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Subgroups of castration- resistant prostate cancer bone metastases defined through an invcerse relationship between androgen receptor activity and immune response. Eur. Urol. 2017, 71, 776–787. [Google Scholar] [CrossRef]
- Venturini, N.J.; Drake, C.G. Immunotherapy for prostate cancer. Cold Spring Harb. Persp. Med. 2019, 9. [Google Scholar] [CrossRef]
- Small, E.J.; Schellhammer, P.F.; Higasno, C.S.; Redfern, C.H.; Nemunaitis, J.J.; Valone, F.H.; Verjee, S.S.; Jones, L.A.; Hershberg, R.M. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J. Clin. Oncol. 2006, 24, 3089–3094. [Google Scholar] [CrossRef]
- Dutcher, G.; Bilen, M.A. Therapeutic vaccines for genitourinary malignancies. Vaccines 2018, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicenter, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomised, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naïve castration-resistant prostate cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Boudadi, K.; Suzman, D.L.; Anagnostou, V.; Fu, W.; Luber, B.; Wang, H.; Niknafs, N.; White, J.R.; Silberstein, J.L.; Sullivan, R.; et al. Ipilimumab plus nivolumab and DNA-repait defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 2018, 19, 28561–28571. [Google Scholar]
- Graff, J.N.; Alumkal, J.J.; Drake, C.G.; Thomas, G.V.; Redmond, W.L.; Farhad, M.; Cetnar, J.P.; Ey, F.S.; Bergan, R.C.; Slottke, R.; et al. Early evidence if anti-PD-1 activity in enhzalutamide-resistant prostate cancer. Oncotarget 2016, 7, 52810–52817. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.D.; Goh, J.C.; Ojamaa, K.; Rodriguez, J.M.P.; Drake, C.G.; Holmes, C.J.; Wu, H.; Poehlein, C.H.; Antonarakis, E.S. KEYNOTE-199: Pembrolizumab (pembro) for docetaxel-refractory metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2018, 36, 5007. [Google Scholar] [CrossRef]
- Garmat, M.; McNeel, D.G. Androgen deprivation and immunotherapy for the treatment of prostate cancer. Endocr. Rel. Cancer 2017, 24, T297–T310. [Google Scholar] [CrossRef]
- Graff, J.N.; Alumkal, J.J.; Thompson, R.F.; Moran, A.; Thomas, G.V.; Wood, M.A.; Drake, C.G.; Slottke, R.; Beer, T.M. Pembrolizumab (Pembro) plus enzalutamide (Enz) in metastatic castration resistant prostate cancer (mCRPC): Extended follow up. J. Clin. Oncol. 2018, 36, 5047. [Google Scholar] [CrossRef]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; Troncoso, P.; et al. VISTA in an inhibitory immune checkpointy that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Beonrdis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castrationb-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S. A new molecular taxonomy to predict immune checkpoint inhibitor sensitivity in prostate cancer. Oncologist 2019, 24, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hampelmann, J.A.; Lockwood, C.M.; Konnick, E.Q.; Schweizer, M.T.; Anatonarakis, E.S.; Lotan, T.L.; Montgomery, B.; Nelson, P.S.; Klemfun, N.; Salipante, S.J.; et al. Microsatellite instability in prostate cancer by PCR or next-generation sequencing. J. Immunother. Cancer 2018, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C.; Olmos, D.; Saunders, E.; Leongamomiert, D.; Tymrakiewicz, M.; Mahmud, N.; Dadaev, T.; Govindasami, K.; Guy, M.; et al. Germile BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 2013, 3, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C.; Leongamomiert, D.; Saunders, E.; Tymrakiewicz, M.; Dadaev, T.; Govindasami, K.; Guy, M.; Ellis, L.; Frost, D.; et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localized prostate cancer. Eur. Urol. 2015, 66, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.B.; Helfand, B.; Mamawala, M.; Wu, Y.; Landis, P.; Yu, H.; Wiley, K.; Na, R.; Shi, Z.; Petkewicz, J.; et al. Germline mutations in ATM and BRCA1/2 are associated with grade reclassification in men on active surveillance for prostate cancer. Eur. Urol. 2019, 75, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Fraser, M.; Rebello, R.J.; Boutros, P.C.; Murphy, D.G.; Bristow, R.G.; Risbridger, G.P. The influence of BRCA2 mutation on localized prostate cancer. Nat. Rev. Urol. 2019, 16, 281–290. [Google Scholar] [CrossRef]
- Annala, M.; Struss, W.J.; Warner, E.W.; Beja, K.; Vandekerkhove, G.; Wong, A.; Khalaf, D.; Seppala, I.L.; So, A.; Lo, G.; et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair-deficient prostate cancer. Eur. Urol. 2017, 72, 34–42. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Liang, C.; Wang, H.; Chen, Y.; Silberstein, J.L.; Piana, D.; Lai, Z.; Chen, Y.; et al. Germline DNA-repair gene mutations and outcomnes in men with metastatic castration-resistant prostate cancer receiving first-line abiraterone and enzalutamide. Eur. Urol. 2018, 74, 218–225. [Google Scholar] [CrossRef]
- Mateo, J.; Cheng, H.H.; Beltran, H.; Dolling, D.; Xu, W.; Pritchard, C.C.; Mossop, H.; Rescigno, P.; Perez-Lopez, R.; Sailer, V.; et al. Clinical outcome of prostate cancer patients with germline DNA repair mutations: Retrospective analysis from an international study. Eur. Urol. 2018, 73, 667–693. [Google Scholar] [CrossRef]
- Castro, E.; Romero-Laorden, N.; Del Pozo, A.; Lozano, R.; Medina, A.; Puents, J.; Piulats, J.M.; Lorente, D.; Saez, M.I.; Morales-Barrera, R.; et al. PROREPAIR-B: A prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2019, 37, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.H.; Sokolova, A.O.; McNatty, A.L.; Cheng, H.H.; Eisenberger, M.A.; Bryce, A.H.; Schweizer, M.T.; Antonarakis, E.S. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur. Urol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.H.; Fu, W.; Wang, H.; Baras, A.S.; Lotran, T.L.; Antonoreakis, E.S. Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: Association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis. 2019, 22, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, L.; Chen, L.L. The biogenesis, functions, and challenges of circular RNAs. Mol. Cell 2018, 71, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The landscape of circular RNA in cancer. Cell 2019, 176, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, V.; Xu, X.; Livingstone, J.; Soares, F.; Jeon, J.; Zeng, Y.; Hua, J.T.; Petricca, J.; Guo, H.; et al. Widespread and functional RNA circularization in localized prostate cancer. Cell 2019, 176, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Wan, X.; Zhang, Y.; Zhang, P.; Zhang, Y.; Zhang, X.; Qi, X.; Wu, H.; Huang, J.; Li, Y. Androgen-responsive circular RNA circSMARCA5 is up-regulated and promotes cell proliferation in prostate cancer. Biochem. Biophys. Res. Commun. 2017, 493, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.S.; Cai, Y.; Feng, T.; Yang, D.; Yuan, S.; Yang, X.; He, S.; Li, Z.; Wang, Y.; Tang, Y.; et al. Upregulated circular RNA circ-102004 that promotes cell proliferation in prostate cancer. Int. J. Biol. Macromol. 2019, 122, 1235–1243. [Google Scholar]
- Niu, Y.; Altuwaijri, S.; Lai, K.P.; Wu, C.T.; Ricke, W.A.; Messing, E.M.; Yao, J.; Yeh, S.; Chang, C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12182–12187. [Google Scholar] [CrossRef]
- Niu, Y.; Altuwaijri, S.; Yeh, S.; Lai, K.P.; Yu, S.; Chuang, K.H.; Huang, S.P.; Lardy, H.; Chang, C. Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc. Natl. Acad. Sci. USA 2008, 105, 12188–12193. [Google Scholar] [CrossRef]
- Memarzadeh, S.; Cai, H.; Janzen, D.; Xin, L.; Lukacs, R.M.; Riedinger, M.; Zong, Y.; DeGaendt, K.; Verhoeven, G.; Huang, J.; et al. Role of autonomous androgen receptor signaling in prostate cancer initiation is dichotomous and depends on the oncogenic signal. Proc. Natl. Acad. Sci. USA 2011, 108, 7962–7967. [Google Scholar] [CrossRef] [PubMed]
- Vander Griend, D.; D’Antonio, J.; Gurel, B.; Antony, L.; DeMarzo, A.M.; Isaacs, J.T. Cell autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate 2010, 70, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Cowin, P.A.; Cunha, G.R.; Pera, M.; Tramson, A.O.; Pedersen, J.; Risbridger, G.P. Formation of human prostate tissue from embryonic stem cells. Nat. Methods 2006, 3, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.Y.; Shi, G.B.; Lara, H.M.; Hu, D.P.; Ho, S.M.; Madueke, I.C.; Kajdacsy-Balla, A.; Prins, G.S. Estrogen-initiated transformation of prostate epithelium derived from normal human prostate stem-progenitor cells. Endocrinology 2011, 152, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Rouet, V.; Bogorad, R.; Kayser, C.; Kessal, K.; Genestie, C.; Bardier, A.; Grattan, D.R.; Kelder, B.; Kopchick, J.J.; Kelly, P.A.; et al. Local prolactin is a target to prevent expansion of basal/stem cells in prostate tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 199–204. [Google Scholar] [CrossRef]
- Sackmann-Sala, L.; Chiche, A.; Mosquera-Garrote, N.; Boutillon, F.; Cordier, C.; Pourmir, I.; Pascual-Mathey, L.; Kessal, K.; Pigat, N.; Camparo, P.; et al. Prolactin-induced prostate tumorigenesis links sustained Stat5 signaling with the amplification of basal/stem cells and emergence of putative luminal progenitors. Am. J. Pathol. 2014, 184, 3105–3119. [Google Scholar] [CrossRef]
- Thomas, C.; Zoubaldi, A.; Koruma, H.; Fazil, L.; Lamoureux, F.; Beraldi, E.; Monia, B.P.; Mac Leod, A.R.; Thuroff, J.W.; Gleave, M.E. Transcription factor Stat5 knockdown enhances receptor androgen degradation and delays castration-resistant prostate cancer progression in vivo. Mol. Cancer Ther. 2011, 10, 347–359. [Google Scholar] [CrossRef]
- Gu, L.; Liao, Z.; Hoang, D.T.; Dagvadorj, A.; Gupta, S.; Blackmon, S.; Ellsworth, E.; Talati, P.; Leiby, B.; Zinda, M.; et al. Pharmacologic inhibition of Jak2-Stat5 signaling by Jak2 inhibitor AZD1480 potently suppresses growth of both primary and castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 5658–5674. [Google Scholar] [CrossRef]
- Hoang, D.T.; Gu, L.; Liao, Z.; Shen, F.; Talati, P.G.; Koptyra, M.; Tan, S.H.; Ellsworth, E.; Gupta, S.; Montie, H.; et al. Inhibition of Stat5a/b enhances proteasomal degradation of androgen receptor liganded by antiandrogens in prostate cancer. Mol. Cancer Ther. 2015, 14, 713–726. [Google Scholar] [CrossRef]
- Talati, P.G.; Gu, L.; Ellsworth, E.M.; Girondo, M.A.; Trerotola, M.; Leiby, B.; Dagvadori, A.; McCue, P.A.; Lallas, C.D.; Trabulsi, E.J.; et al. Jak2-Stat5a/b signaling induces epithelial-to-mesenchymal transition and stem-like cell properties in prostate cancer. Am. J. Pathol. 2015, 185, 2505–2522. [Google Scholar] [CrossRef]
- Mirtti, T.; Leiby, B.E.; Abdulghani, J.; Aaltonen, E.; Pavela, M.; Mamtani, A.; Alanen, K.; Egevad, L.; Granfors, T.; Josefsson, A.; et al. Nuclear Stat5a/b predicts early recurrence and prostate cancer-specific death in patients treated by radical prostatectomy. Hum. Pathol. 2013, 44, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Haddad, B.R.; Gu, L.; Mirtti, T.; Dagvadori, A.; Vogiatzi, P.; Hoang, D.T.; Bajaj, R.; Leiby, B.; Ellsworth, E.; Blackmon, S.; et al. STAT5A/B gene locus undergoes amplification during human prostate cancer progression. Am. J. Pathol. 2013, 182, 2264–2275. [Google Scholar] [CrossRef] [PubMed]
- Maranto, C.; Udhane, V.; Hoang, D.T.; Gu, L.; Alexeev, V.; Malas, K.; Cardenas, K.; Brody, J.R.; Rodeck, U.; Bergom, C.; et al. STAT5A/B blockade sensitizes prostate cancer to radiation through inhibition of RAD51 and DNA repair. Clin. Cancer Res. 2018, 24, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, P. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Roudier, P.; True, L.D.; Higano, C.S.; Vesselle, H.; Ellius, W.; Lange, P.; Vessella, R.L. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum. Pathol. 2003, 34, 646–653. [Google Scholar] [CrossRef]
- Shah, R.B.; Mehra, R.; Chinnayan, A.M.; Shen, R.; Ghosh, D.; Zhou, M.; Macvicar, G.R.; Varambally, S.; Harwood, J.; Bismar, T.A.; et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res 2004, 64, 9209–9216. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Rycai, K.; Deng, Q.; Jeter, C.; Liu, C.; Honorio, S.; Li, H.; Davis, T.; Suraneni, M.; et al. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget 2015, 6, 23959–23986. [Google Scholar] [CrossRef]
- Li, Q.; Deng, Q.; Chao, H.P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer AR heterogeneity to distinct castration and ezalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef] [PubMed]
- Finones, R.R.; Yeargin, J.; Lee, M.; Kaur, A.P.; Cheng, C.; Sun, P.; Wu, C.; Nguyen, C.; Wang-Rodriguez, J.; Meyer, A.N.; et al. Early human prostate adenocarcinomas harbor androgen-independent cancer cells. PLoS ONE 2013, 8, e74438. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Giunchi, F.; Fiorentino, M.; Loda, M. The matabolic landscape of prostate cancer. Eur. Oncol. 2019, 2, 28–36. [Google Scholar]
- Zandra, G.; Photopoulos, C.; Loda, M. The fat side of prostate cancer. Biochim. Biophys. Acta 2013, 1831, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Loda, M. Metabolic vulnerabilities of prostate cancer: Diagnostic and therapeutic opportunities. Cold Spring Harb. Perspect. Med. 2018, 8, a030569. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Centenera, M.M.; Swinnen, J.V. Androgen control of lipid metabolism in prostate cancer: Novel insights and future applications. Endocr. Telat. Cancer 2016, 23, R219–R227. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compatmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Nassar, Z.D.; Zhang, A.Y.; Hesseini-Beheshti, E.; Centenera, M.M.; Schreuder, M.; Lin, H.M.; Aishah, A.; Varney, B.; Liu-Fu, F.; et al. Extracellular fatty acids are the major contributor to lipid synthesis in prostate cancer. Mol. Cancer Res. 2019, 17, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Montgomewry, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resitant tumor growth. Cancer Res. 2008, 67, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhana, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef]
- Han, W.; Gao, S.; Barrett, D.; Ahmed, M.; Han, D.; Macoska, J.A.; He, H.H.; Cai, C. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 2018, 37, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Cheng, L.; Mao, F.; Zhang, Z.; Farah, E.; Bosler, J.; Bai, Y.; Ahmad, N.; Kuang, S.; Li, L.; et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 2018, 293, 14328–14341. [Google Scholar] [CrossRef]
- Gordon, J.A.; Noble, J.W.; Midha, A.; Derakhshan, F.; Wang, G.; Adomat, H.H.; Guns, E.S.T.; Lin, Y.Y.; Ren, S.; Collins, C.C.; et al. Upregulation of scavenger receptor B1 is required for steroidogenic and non-steroidogenic cholesterol metabolism in prostate cancer. Cancer Res. 2019, 79, 3320–3331. [Google Scholar] [CrossRef] [PubMed]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zeng, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.K.; Shojaie, A.; Panzitt, K.; Sonavane, R.; Venghatakrishnan, H.; Manikkam, M.; Zaslavsky, A.; Putuiuri, V.; Vasu, V.T.; Zhang, Y.; et al. Inhibition of the hexosamine biosynthetic pathway promotes castration-resistant prostate cancer. Nat. Commun. 2016, 7, 11612. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermasen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth anf tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Lin, C.; Rajapakshe, K.; Dong, J.; Shi, Y.; Tsouko, E.; Mukhopadhyay, R.; Jasso, D.; Dawood, W.; Corfa, C.; et al. Glutamine transporters are targets of multiple oncogenic signaling pathways in prostate cancer. Mol. Cancer Res. 2017, 15, 1017–1028. [Google Scholar] [CrossRef]
- Zacharias, N.M.; McCullough, C.; Shanmugavelandy, S.; Lee, J.; Lee, Y.; Dutta, P.; McHenry, J.; Nguyen, L.; Norton, W.; Jones, L.W.; et al. Metabolic differences in glutamine utilization lead to metaboliuc vulnerabilities in prostate cancer. Sci. Rep. 2017, 7, 16159. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Aragwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Invest. 2018, 128, 4472–4484. [Google Scholar] [CrossRef]
- Gonthier, K.; Poluri, R.T.K.; Weidmann, C.; Tadros, M.; Audet-Walsh, E. Reprogramming of isocitrate dehydrogenase expression and activity by the androgen receptor in prostate cancer. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Geng, H.; Xue, C.; Mendonca, J.; Sun, X.X.; Liu, Q.; Reardon, P.N.; Chen, Y.; Qian, K.; Hua, V.; Chen, A.; et al. Interplay between hypoxia and androgen controls a metabolic switch conferring resistance to androgen/AR-targeted therapy. Nat. Commun. 2018, 9, 4972. [Google Scholar] [CrossRef]
- Jayaprakash, P.; AI, M.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; Shah, K.; et al. Targeted hypoxiua reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Invest. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Henry, G.H.; Malewska, A.; Joseph, D.B.; Malladi, V.S.; Lee, J.; Torrealba, J.; Mauck, R.J.; Gahan, J.C.; Raj, G.V.; Roehborn, C.G.; et al. A cellular anatomy of the normal adult human prostate and prostatic urethra. Cell Rep. 2018, 25, 3530–3542. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.T. Control of cell proliferation and cell death in the normal and neoplastic prostate: A stem cell model. In Bening Prostatic Hyperplasia; Rodgers, C.H., Coffey, D.S., Cunha, G., Grayshack, J.T., Henman, R., Horton, R., Eds.; Department of Health and Human Services: Washington, DC, USA, 1987; pp. 85–94. [Google Scholar]
- Litvinov, I.V.; De Marzo, A.M.; Isaacs, J.T. Is the Achille’s heel for prostate cancer cancer therapy a gain of function in androgen receptor signaling? J. Clin. Endocrinol. Metab. 2003, 88, 2972–2982. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsujimura, A.; Koikawa, Y.; Salm, S.; Takao, T.; Coetzee, S.; Moscatelli, D. Proximal location of mouse prostate epithelial stem cells: A model of prostatic homeostasis. J. Cell Biol. 2002, 157, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Shen, M.M. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene 2011, 30, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hamana, T.; Liu, J.; Wang, C.; An, L.; You, P.; Chang, J.Y.; Xu, J.; McKeehan, W.L.; Wang, F. Prostate sphere-forming stem cells are derived from the P63-expressing basal compartment. J. Biol. Chem. 2015, 290, 17745–17752. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Shen, M.M. Prostate stem cells and cancer stem cells. CSH Perspect. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Habib, F.K.; Maitland, N.J.; Neal, D.E. Identification and isolation of human prostate epithelial stem cells based on alpha(2)beta(1)-integrin expression. J. Cell Sci. 2001, 114, 3865–3872. [Google Scholar] [PubMed]
- Richardson, G.D.; Robson, C.N.; Lang, S.H.; Neal, D.E.; Maitland, N.J.; Collins, A.T. CD133, a novel marker for human prostatic epithelial stem cells. J. Cell Sci. 2004, 117, 3539–3545. [Google Scholar] [CrossRef] [PubMed]
- Garraway, I.P.; Sun, W.; Tran, C.P.; Perner, S.; Zhang, B.; Goldstein, A.S. Human prostate sphere-forming cells represent a subset of basal epithelial cells capable of glandular regeneration in vivo. Prostate 2010, 70, 491–501. [Google Scholar] [CrossRef]
- Goldstein, A.S.; Witte, O.N. A plethora of progenitors in the post-natal prostate. EMBO Rep. 2012, 13, 1036–1037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leong, K.G.; Wang, B.E.; Johnson, L.; Gao, W.Q. Generation of a prostate from a single adult stem cell. Nature 2008, 456, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.S.; Lawson, D.A.; Cheng, D.; Sun, W.; Garraway, I.P.; Witte, O.N. Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proc. Natl. Acad. Sci. USA 2008, 105, 20882–20887. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, R.V.; Memarzandeh, S.; Wu, H.; Witte, O.N. Bmi-1 is a crucial regulator of prostate stem cell renewal and malignant transformation. Cell Stem Cell 2010, 7, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, H.H.; Chu, M.; Liu, Y.; Zhang, C.; Liu, G.; Yang, X.; Yang, R.; Gao, W.Q. Symmetrical and asymmetrical division analysis provides evidence for a hierarchy of prostate epitrhelial cell lineages. Nat. Commun. 2014, 5, 4756. [Google Scholar]
- Wang, B.E.; Wang, X.; Long, J.E.; Easthman-Anderson, J.; Firestein, R.; Junttilia, M.R. Castration-resistant Lgr5(+) cells are long-lived stem cells required for prostatic regeneration. Stem Cell Rep. 2015, 4, 768–779. [Google Scholar] [CrossRef]
- Toivanen, R.; Mohan, A.; Shen, M.M. Basal progenitors contribute to repair of the propstate epitheliumn following induced luminal anoikis. Stem Cell Rep. 2016, 6, 660–667. [Google Scholar] [CrossRef]
- Moad, M.; Hannezo, E.; Buczacki, S.J.; Wilson, L.; El-Sherif, A.; Sims, D.; Pickard, R.; Wright, N.A.; Williamson, S.C.; Tumbull, D.M.; et al. Multipotent basal stem cell cells, maintained in localized proximal niches, support directed long-ranging epithelial flows in human prostates. Cell Rep. 2017, 20, 1609–1622. [Google Scholar] [CrossRef]
- Hu, W.Y.; Hu, D.P.; Xie, L.; Li, Y.; Majumdar, S.; Nonn, L.; Hu, H.; Snioda, T.; Prins, G.S. Isolation and functional interrogation of adult human prostate epithelial stem cells at single cell resolution. Stem Cell Res. 2017, 23, 1–12. [Google Scholar] [CrossRef]
- Wang, X.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef]
- Ousset, M.; Van Keymulen, A.; Bouvencourt, E.; Sharma, N.; Achouri, Y.; Simons, B.D.; Blanpain, C. Multipotent and unipotent progenitors contribute to prostate postnatal development. Nat. Cell Biol. 2012, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.I.; Bergren, S.K.; Badoni, K.K.; McKiermann, J.M.; Benson, M.C.; Hibshoosh, H.; et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, W.; Iaquinta, P.; Drost, J.; Gracanin, A.; van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthet, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.J.; Valdez, J.; Zhang, L.; Zhang, B.; Wei, X.; Su, Q.; Ittmann, M.M.; Creighton, C.J.; Xin, L. Increased Notch signaling inhibits anoikis and stimulates proliferation of prostate luminal epithelial cells. Nat. Commun. 2014, 5, 4416. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.S.; Stoyanova, T.; Witte, O.N. Primitive origins of prostate cancer: In vivo evidence for prostate-regenerating cells and prostate-initiating cells. Mol. Oncol. 2010, 4, 385–396. [Google Scholar] [CrossRef]
- Kwon, D.J.; Zhang, L.; Xin, L. Stem cell antigen-1 identifies a distinct androgen-independent murine prostatic luminal cell lineage with bipotent potential. Stem Cells 2016, 34, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Grogan, T.R.; Hieronymous, H.; Hashimoto, T.; Mottahedeh, J.; Cheng, D.; Zhang, L.; Huang, K.; Stoyanova, T.; Park, J.W.; et al. Low CD38 identifies progenitor-like inflammation-associated luminal cells that can initiate human prostate cancer and predict poor outcome. Cell Rep. 2016, 17, 2596–2606. [Google Scholar] [CrossRef]
- Motthedeh, J.; Haffner, M.C.; Grogan, T.R.; Hashomoto, T.; Crowell, P.D.; Beltran, H.; Sboner, A.; Bareja, R.; Esopi, D.; Isaacs, W.B.; et al. CD38 is methylated in prostate cancer and regulates extracellular NAD. Cancer Metab. 2018, 6, 13. [Google Scholar] [CrossRef]
- Chmielewski, J.P.; Bowlby, S.C.; Wheeler, F.B.; Shi, L.; Gui, G.; Davis, A.L.; Howard, T.D.; D’Agostino, R.B.; Miller, R.D.; Sirintrapun, S.J.; et al. CD38 inhibits prostate cancer metabolism and proliferation by reducing cellular NAD+ pools. Mol. Cancer Res. 2018, 16, 1687–1700. [Google Scholar] [CrossRef]
- Zhang, D.; Jeter, C.; Gang, S.; Tracz, A.; Lu, Y.; Shen, J.; Tang, D.G. Histone 2b-GFP label-retaining prostate luminal cells possess progenitor cell properties and are intrinsically resistant to castration. Stem Cell Rep. 2018, 10, 228–242. [Google Scholar] [CrossRef]
- Barros-Silva, J.; Linn, D.E.; Steiner, I.; Guo, G.; Ali, A.; Pakula, H.; Ashton, G.; Peset, I.; Brown, M.; Clarke, N.W.; et al. Single-cell analysis identifies LY6D as marker linking castration-resistant prostate luminal cells to prostate progenitors and cancer. Cell Rep. 2018, 25, 3504–3518. [Google Scholar] [CrossRef] [PubMed]
- Na, R.; Helfand, B.T.; Chen, H.; Conran, C.A.; Crawford, S.E.; Hayward, S.W.; Tammela, T.L.J.; Hoffman-Bolton, J.; Zheng, S.L.; Walsh, P.C.; et al. A genetic variant near GATA3 implicated in inherited susceptibility and etiology of benign prostatic hyperplasia (BPH) and lower urinary tract symptoms (LUTS). Prostate 2017, 77, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Sigursson, J.K.; Stefansdottir, L.; Agnarsson, B.A.; Isaksson, H.J.; Stefansson, O.A.; Gudjonsson, S.A.; Gudbjartsson, D.F.; Masson, G.; Frigge, M.L.; et al. Genome-wide associations for benign prostatic hyperplasia reveal a genetic correlation with serum levels of PSA. Nat. Commun. 2018, 9, 4568. [Google Scholar] [CrossRef] [PubMed]
- Hellwege, J.N.; Stallings, S.; Torstenson, E.S.; Carroll, R.; Borthwick, K.M.; Brillian, M.H.; Crosslin, D.; Gordon, A.; Hripcsak, G.; Jarvik, G.P.; et al. Heritability and genome-wide association study of benign prostatic hyperplasia (BPH) in the eMERGE network. Sci. Rep. 2019, 9, 6077. [Google Scholar] [CrossRef] [PubMed]
- Lin, V.K.; Wang, S.Y.; Vazquez, D.V.; XU, C.; Zhang, S.; Tang, L. Prostatic stromal cells derived from benign prostatic hyperplasia specimens possess stem cell like property. Prostate 2007, 67, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Brennen, W.N.; Zhang, B.; Kulac, I.; Kisteman, L.N.; Anthony, L.; Wang, H.; Wang, H.; Meeker, A.K.; De Marzo, A.M.; Garraway, I.P.; et al. Mesenchymal stem cell infiltration during neoplastic transformation of the human prostate. Oncotarget 2017, 8, 46710–46727. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, L.; Tintani, F.; Xie, H.; Li, C.; Cui, Z.; Wan, M.; Zu, X.; Qi, L.; Cao, X. Aberrant transforming growth factor-β activation recruits mesenchymal stem cells during prostatic hyperplasia. Stem Cells Transl. Med. 2017, 6, 394–404. [Google Scholar] [CrossRef]
- Yamamoto, H.; Masters, J.R.; Dasgupta, P.; Chandra, A.; Popert, R.; Freeman, R.; Ahmed, A. CD49f is an efficient marker of monolayer and spheroid colony-forming cells of the benign and malignant human prostate. PLoS ONE 2012, 7, e46979. [Google Scholar] [CrossRef]
- Imura, M.; Kojima, Y.; Kubota, Y.; Hamakawa, T.; Yasui, T.; Sasaki, S.; Hayashi, Y.; Kohri, K. Regulation of cell proliferation through a KIT-mediated mechanism in benign prostatic hyperplasia. Prostate 2012, 72, 1506–1512. [Google Scholar] [CrossRef]
- Stoyanova, T.; Goldstein, A.S.; Cai, H.; Drake, J.M.; Huang, J.; Witte, O.N. Regulated proteolysis of Trop2 drives epithelail hyperplasia and stem cell self-renewal via beta-catenin signaling. Genes Dev. 2012, 26, 2271–2285. [Google Scholar] [CrossRef]
- Brennen, W.N.; Isaacs, J.T. Mesenchymal stem cells and the embryonic reawakening theory of BPH. Nat. Rev. Urol. 2018, 15, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Mei, W.; Lin, X.; Kapoor, A.; Gu, Y.; Zhao, K.; Tang, D. The contributions of prostate cancer stem cells in prostate cancer initiation and metastasis. Cancers 2019, 11, 434. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Yuan, J.; Wills, M.; Kasper, S. Prostate cancer cells with stem cell characteristics reconstitute the original tumor in vivo. Cancer Res. 2007, 67, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, M.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Ramirez, N.; Voller, M.; Watterwald, A. In vitro propagation and characterization of neoplastic stem/progenitor-like cells from human prostate cancer tissue. Prostate 2009, 15, 1683–1693. [Google Scholar] [CrossRef]
- Wei, A.V.; Xin, L. CD133 does not enrich for the stem cell activity in vivo in adult mouse prostates. Stem Cell Res. 2016, 16, 597–606. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kalantari, E.; Asgari, M.; Nikpanah, S.; Salarieh, N.; Asadi Lari, M.H.; Madid, Z. Co-expression of putative cancer stem cell markers CD44 and CD133 in prostate carcinomas. Pathol. Oncol. Res. 2017, 23, 793–802. [Google Scholar] [CrossRef]
- Kanwal, R.; Shulka, S.; Walker, E.; Gupta, S. Acquisition of tumorigenic potential and therapeutic resistance in CD133+ subpopulation of prostate cancer cells exhibiting stem cell-like characteristics. Cancer Lett. 2018, 4309, 25–33. [Google Scholar] [CrossRef]
- Reyes, E.E.; Gillard, M.; Duggan, R.; Wrobleski, K.; Kregel, S.; Isikbay, M.; Kach, J.; Brechka, H.; Vander Weele, D.J.; Szmulewitz, R.Z.; et al. Molecular analysis of CD133-positive circulating tumor cells from patients with metastatic castration-resistant prostate cancer. J. Transl. Sci. 2015, 1, 21. [Google Scholar]
- Eaton, C.L.; Colombel, M.; van der Pluijm, G. Evaluation of the frequency of putative prostate cancer stem cells in primary and metastatic prostate cancer. Prostate 2010, 70, 875–882. [Google Scholar] [CrossRef]
- Van den Hoogen, C.; van der Horst, G.; Cheung, H.; Buijs, J.; Lippitt, J.; Guzman-Ramirez, N.; Hamdy, F.; Eaton, C.L.; Thalmann, G.; Cecchini, M.; et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010, 70, 5163–5173. [Google Scholar] [CrossRef] [PubMed]
- Le Magnen, C.; Bubendorf, L.; Rentsch, C.A.; Mengus, C.; Gasponer, J.; Zell Weger, T.; Rieken, M.; Thelmann, G.N.; Cecchini, M.C.; Germann, M.; et al. Characterization and clinical relevance of ALDH bright populations in prostate cancer. Clin. Cancer Res. 2013, 19, 5361–5371. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Q.; Liu, X.; Liu, C.; Liu, R.; Rycaj, K.; Zhang, D.; Liu, B.; Jeter, C.; Calhoun-Davis, T.; et al. Defining a population of stem-like human prostate cancer cells that can generate and propagate castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 4505–4516. [Google Scholar] [CrossRef] [PubMed]
- Cojoc, M.; Peitzsch, C.; Kurth, I.; Trautmann, F.; Kunz-Schugart, L.A.; Telegeev, G.D.; Stakhoksky, E.A.; Walker, J.R.; Simin, K.; Lyle, S.; et al. Aldehyde dehydrogenase is regulated by β-catenin/TCF and promotes radioresistance in prostate cancer progenitor cells. Cancer Res. 2015, 75, 1482–1494. [Google Scholar] [CrossRef] [PubMed]
- Colombel, M.; Eaton, C.L.; Harudy, F.; Ricci, E.; van der Pluijm, G.; Cecchini, M.; Mege-Lechevalliern, F.; Clezardin, P.; Thalmann, G. Increased expression of putative cancer stem cell markers in primary prostate cancer is associated with progression of bone metastases. Prostate 2012, 72, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Van Leenders, G.; Sookhall, R.; Tenbel, W.; de Ridder, C.; Reneman, C.; Sacchetti, A.; Vissers, K.; van Weerden, W.; Jenster, G. Activation of c-MET induces a stem-like phenotype in human prostate cancer. PLoS ONE 2011, 6, e26753. [Google Scholar] [CrossRef] [PubMed]
- Nishida, S.; Hiroashi, Y.; Torigoe, T.; Inoue, R.; Kitamura, H.; Takahashi, A.; Masumai, N.; Tsukamoto, T.; Sato, N. Prostate cancer stem-like cells/cancer-initiating cells have an autocrine system of hepatocyte growth factor. Cancer Sci. 2013, 104, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; Ingram, A.J.; Tang, D. Propagation of human prostate cancer stem-like cells occurs through EGFR-mediated ERK activation. PLoS ONE 2013, 8, e61716. [Google Scholar] [CrossRef]
- Wei, C.; Guomin, W.; Ruizhe, Q. Cancer-stem like cells in human prostate carcinoma cells DU145: The seeds of the cell line? Cancer Biol. Ther. 2007, 6, 763–768. [Google Scholar] [CrossRef]
- Hurt, E.M.; Kawasaki, B.T.; Klarmann, G.J.; Thomas, S.B.; Farrar, W.L. CD44+CD24-prostate cells are clearly cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br. J. Cancer 2008, 98, 756–765. [Google Scholar] [CrossRef]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Zhou, J.; Claypool, K.; Tang, D.G. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005, 65, 6207–6219. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, H.; Tang, S. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tyumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Tang, D.G. Hierarchical organization of prostate cancer cells in xenograft tumors: The CD44+apha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007, 67, 6796–6805. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Takahashi, S.; Urano, T.; Takayama, K.; Sugihara, T.; Obinata, D.; Yamada, Y.; Kumagai, J.; Kuma, H.; Oschi, Y.; et al. Expression of androgen and estrogen signaling components and stem cell markers to predict cancer progression and cancer-specific survival in patients with metastatic prostate cancer. Clin. Cancer Res. 2014, 20, 4625–4635. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Korsten, H.; Ziel-van der Made, A.; Ma, X.; van der Kwast, T.; Trapman, J. Accumulating progenitor cells in the luminal epithelial cell layer are candidate tumor initiating cells in a Pten knockout mouse prostate cancer model. PLoS ONE 2009, 4, e5662. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.D.; McKissic, S.A.; Logan, M.; Roh, M.; Franco, O.E.; Wang, J.; Doubinskaia, I.; van der Meer, R.; Hayward, S.W.; Eischen, C.M.; et al. Nkx3.1 and Myc crossregulate shared target genes in mouse and human prostate tumorigenesis. J. Clin. Invest. 2012, 122, 1907–1919. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; De Marzo, A.M.; Meeker, A.K.; Nelson, W.G.; Yegnasubramanian, S. Transcription-induced DNA double strtand breaks: both oncogenic force and potential therapeutic target? Clin. Cancer Res. 2011, 17, 3858–3864. [Google Scholar] [CrossRef]
- Wang, Z.; Toivanen, R.; Bergren, S.K.; Chambon, P.; Shen, M.M. Luminal cells are favored as the cell of origin for prostate cancer. Cell Rep. 2014, 8, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.J.; Zhang, L.; Ihmann, M.M.; Xiu, L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with basal cell origin. Proc. Natl. Acad. Sci. USA 2013, 111, E592–E600. [Google Scholar] [CrossRef] [PubMed]
- Mulhollaud, D.J.; Xin, L.; Morim, A.; Lawson, D.; Witte, O.; Wu, H. Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-Null prostate cancer model. Cancer Res. 2009, 69, 8555–8562. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a cell of origin for human prostate cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat. Cell Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.; Zhang, B.; Zhang, L.; Ittmann, M.; Xin, L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell 2012, 21, 253–265. [Google Scholar] [CrossRef]
- Lu, T.L.; Huana, Y.F.; You, L.R.; Chao, N.C.; Su, F.Y.; Chang, J.L.; Chen, C.M. Conditionally ablated PTEN in prostate basal cells promotes basal-to-luminal differentiation and causes invasive prostate cancer in mice. Am. J. Pathol. 2013, 182, 975–991. [Google Scholar] [CrossRef]
- Parisotto, M.; Grelet, E.; Bizzi, R.E.; Dai, Y.; Terzic, J.; Eckert, D.; Gargowitsch, L.; Bornert, J.M.; Metzger, D. PTEN deletion in luminal cells of mature prostate induces replication stress and senescence. J. Exp. Med. 2018, 215, 1749–1763. [Google Scholar] [CrossRef]
- Sackmann Sala, L.; Boutillon, F.; Menara, G.; De Goyon-Pélard, A.; Leprévost, M.; Codzamanian, J.; Lister, N.; Pencik, J.; Clark, A.; Cagnard, N.; et al. A rare castration-resistant progenitor cell population is highly enriched in Pten-null prostate tumors. J. Pathol. 2017, 243, 51–64. [Google Scholar] [CrossRef]
- Cai, H.; Memarzadeh, S.; Stoyanova, T.; Beharry, Z.; Kraft, A.S.; Witte, O.N. Collaboration of KRas and androgen receptor signaling stimulates EZH2 expression and tumor propagating cells in prostate cancer. Cancer Res. 2012, 72, 4672–4681. [Google Scholar] [CrossRef]
- Matsika, A.; Srinivasan, B.; Day, C.; Mader, S.A.; Kiernan, D.M.; Broomfield, A.; Fu, J.; Hooper, J.D.; Kench, J.G.; Samarartunga, H. Cancer stem cell markers in prostate cancer: An immunohistochemical study of ALDH1, SOX2 and EZH2. Pathology 2015, 47, 622–628. [Google Scholar] [CrossRef]
- Labbé, D.P.; Sweeney, C.J.; Brown, M.; Galbo, P.; Rosario, S.; Wadoskuy, K.M.; Ku, S.Y.; Sjostrom, M.; Alshalalkfa, M.; Erho, N.; et al. TOP2A and EZH2 provide early detection of an aggressive prostate cancer subgroup. Cancer Res. 2017, 23, 7072–7083. [Google Scholar] [CrossRef] [PubMed]
- Gorodetska, I.; Lukiyanchulk, V.; Peitzsch, C.; Kozetska, I.; Dubrovska, A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Litcch, J.D.; Zhao, J.C.; YU, J. Polycomb-and methylation-indepoendent roles of EZH2 as a transcription activator. Cell Rep. 2018, 25, 2808–2820. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, Z.; Cheng, L.; Wang, R.; Chen, X.; Kong, Y.; Feng, F.; Ahmad, N.; Li, L.; Liu, X. Inhibition of enhancer of zeste homolog 2 (EZH2) overcomes enzalutamide-resistance in castration-resistance prostate cancer. J. Biol. Chem. 2019, 294, 9911–9923. [Google Scholar] [CrossRef] [PubMed]
- Fong, K.W.; Zhao, J.C.; Kim, J.; Li, S.; Wang, Y.A.; Song, B.; Riftie, L.; Hu, M.; Yang, X.; Perbal, B.; et al. Polycomb-mediated disruption of an androgen receptor feedback loop drives castration-resistant prostate cancer. Cancer Res. 2017, 77, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Taplin, M.E.; Hussain, A.; Shore, N.D.; Bradley, B.; Trojer, P.; Lebedinsky, C. A phase 1b/2 of CPI-1205, a small molecule inhibitor of EZH2, combined with enzalutamide (E) or abiraterone/rpednisone (A/P) in patients with metastatic castration resistant prostate cancer (mCRPC). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Rajasekhar, V.; Studer, L.; Gerald, W.; Socci, N.D.; Scher, H. Tumor-initiating stem-like cells in human prostate cancer exhibit increased NF-kB signaling. Nat. Commun 2011, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Copeland, B.T.; Bowman, M.J.; Ashman, L.K. Genetic ablation of the teraspanin CD151 reduces spontaneous metastatic spread of prostate cancer in the TRAMP model. Mol. Cancer Res. 2013, 11, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.G.; Arnold, S.A.; Jiang, M.; Palmer, T.D.; Ketova, T.; Merkel, A.; Pickup, M.; Samaras, S.; Shyr, Y.; Moses, H.L.; et al. ALCAM/CD166 is a TGF-β-responsive marker and functional regulator of prostate cancer metastasis to bone. Cancer Res. 2014, 74, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, X.; Laffin, B.; Chen, X.; Choy, G.; Jeter, C.R.; Calhoun-Davis, T.; Li, H.; Palapattu, G.S.; Pang, S.; et al. The PSA-/lo prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell 2012, 10, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kelner, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Hwang, C.I.; Corney, D.C.; Flesken-Nikitin, A.; Jiang, L.; Oner, G.M.; Munroe, R.J.; Schimenti, J.C.; Hermeking, H.; Nikitin, A.Y. miR-34 cooperates with p53 in suppression of prostate cancer by point regulation of stem cells compartment. Cell Rep. 2014, 6, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Cai, J.; Li, H.; He, C.; Zhou, C.; Dong, Y.; Chen, L.; Zhang, B.; Wang, Y.; Zhang, Y.; et al. p53-dependent CD51 expression contributes to characteristics of cancer stem cells in prostate cancer. Cell Death Dis. 2018, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.; Heddleston, J.; Pinna, C.; Huband, A.; Stadler, B.; Choi, M.; Bar, M.; et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Li, X.; Xu, Y.; Zhang, S.; Mou, W.; Liu, Y.; Liu, Y.; Lv, D.; Liu, C.H.; Tan, X.; et al. SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. J. Mol. Cell. Biol. 2011, 3, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehses, H.; Shen, J.J.; Thang, D.G. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Liu, B.; Lu, Y.; Chao, H.P.; Zhang, D.; Liu, X.; Chen, X.; Li, Q.; Rycaj, K.; Calhoun-Davis, T.; et al. NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov. 2016, 2, 16041. [Google Scholar] [CrossRef]
- Hepburn, A.C.; Steele, R.E.; Veeratterapillay, R.; Wilson, L.; Kounatidou, E.E.; Barnard, A.; Berry, P.; Cassidy, J.R.; Moad, M.; El-Sherif, A.; et al. The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene 2019, 38, 4412–4424. [Google Scholar] [CrossRef]
- Cai, C.; Wang, H.; He, H.H.; He, L.; Ma, F.; Mucci, L.; Wang, Q.; Fiore, C.; Sowalsky, A.G.; Loda, M.; et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J. Clin. Invest. 2013, 123, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Sikka, S.C. Interplay between SOX9, Wnt/β-catenin and androgen receptor signaling in castration-resistant prostate cancer. Mol. Sci. 2019, 20, 2066. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kim, Y.S.; Fanar, W.; Hurt, E. Long-term recovery of irradiated prostate cancer increases cancer stem cells. Prostate 2012, 72, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Kyjacova, L.; Hubackova, S.; Krejcikova, K.; Strauss, R.; Hanzikova, H.; Dzijak, R.; Imrichova, T.; Simoiva, J.; Reinius, M.; Barte, K.J.; et al. Radiotherapy-induced plasticity of prostate cancer mobilizes stem-like non-adherent, Erk signaling-dependent cells. Cell Death Diff. 2015, 22, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Kowolik, C.M.; Lim, M.; Zuro, D.; Hui, S.K.; Riggs, A.D.; Horne, D.A. SMCA1 is associated with radioresistance in prostate cancer and acts by regulating epithelial-mesenchymal transition and cancer stem-like properties. Mol. Carcinog. 2019, 19, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barreco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of Notch-and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Chitkara, D.; Mehzarin, R.; Behrman, S.W.; Wake, R.W.; Mahato, R.I. Chemoresistance in prostate cancer cells is regulated by miRNAs and Hedgehog pathway. PLoS ONE 2012, 7, e40021. [Google Scholar] [CrossRef] [PubMed]
- Statkiewicz, M.; Maryan, N.; Lipiec, A.; Grecka, E.; Grigorowicz, M.A.; Omiotek, M.; Gorska, A.; Mikula, M.; Malecki, M. The role of the SHH gene in prostate cancer cell resistance to paclitaxel. Prostate 2014, 74, 1142–1152. [Google Scholar] [CrossRef]
- Liu, C.; Li, Z.; Bi, L.; Zhou, B.; Xu, C.; Huang, J.; Xu, K. NOTCH1 signaling promotes chemoresistance via regulainting ABCC1 expression in prostate cancer stem cells. Mol. Cell. Biochem. 2014, 393, 265–270. [Google Scholar] [CrossRef]
- Qiu, S.; Deng, L.; Bao, Y.; Jin, K.; Tu, X.; Li, J.; Liao, X.; Liu, Z.; Yang, L.; Wei, Q. Reversal docetaxel resistance in prostate cancer by Notch signaling inhibition. Anticancer Drugs 2018, 29, 871–879. [Google Scholar] [CrossRef]
- Siddique, H.R.; Parray, A.; Tarapore, R.; Wang, L.; Mukhtar, H.; Karnes, R.J.; Deng, Y.; Kornety, B.R.; Saleem, M. BMI1 polycomb group protein acts as a master switch for groth and death of tumor cells: Regulates TCF4-transcriptional factor-induced BCL2 signaling. PLoS ONE 2013, 8, e60664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sha, J.; Yang, G.; Huang, X.; Bo, J.; Huang, Y. Activation of Notch pathway is linked with epithelial-mesenchymal transition in prostate cancer. Cell Cycle 2017, 16, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, K.; Cheng, C.; Ji, Z.; Wang, X.; Wang, M.; Chu, M.; Tang, D.G.; Zhu, H.H.; Gao, Q. Numb-/low enriches a castration-resistant prostate cancer subpopulation associated with enhanced Notch and Hedgehog signaling. Clin. Cancer Res. 2017, 23, 6744–6756. [Google Scholar] [CrossRef] [PubMed]
- Farah, E.; Li, C.; Cheng, L.; Kong, Y.; Lanman, N.A.; Pascuzzi, P.; Lorenz, G.R.; Zhang, Y.; Ahmad, N.; Li, L.; et al. NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J. Biol. Chem. 2019, 294, 8543–8554. [Google Scholar] [CrossRef] [PubMed]
- Ammirante, M.; Luo, J.L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 454, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Ammirante, M.; Kuraishy, A.; Shalapour, S.; Strasner, A.; Ramirez-Sanchez, C.; Zhang, W.; Shebaik, A.; Karin, M. An IKKα-E2F1-BMI1 cascade activated by infiltrating B cells controls prostate regeneration and tumor recurrence. Genes Dev. 2013, 27, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Zhang, T.; Liu, C.; Badeaux, M.A.; Liu, B.; Liu, R.; Jeter, C.; Chen, X.; Vlonov, A.V.; Tang, D.G. miRNa-128 suppresses prostate cancer by inhibiting BMI-1 to inhibit tumor-initiasting cells. Cancer Res. 2014, 74, 4183–4195. [Google Scholar] [CrossRef]
- Bansal, N.; Bartucci, M.; Yusuff, S.; Davis, S.; Flaherty, K.; Huselid, E.; Patrizii, M.; Jones, D.; Cao, L.; Sydorenko, N.; et al. BMI-1 targeting interferes with patient-derived tumor-initiating cell survival and tumor growth in prostate cancer. Clin. Cancer Res. 2016, 22, 6176–6191. [Google Scholar] [CrossRef]
- Zhu, S.; Zhao, D.; Yan, L.; Jiang, W.; Kim, J.S.; Gu, B.; Liu, Q.; Wang, R.; Xia, B.; Zhao, J.C.; et al. BMI1 regulates androgen receptor in prostate cancer independently of the polycomb repressive complex 1. Nat. Commun. 2018, 9, 500. [Google Scholar] [CrossRef]
- Yoo, Y.A.; Roh, M.; Naseem, A.F.; Lysy, B.; Desouki, M.M.; Unno, K.; Abulkadir, S.A. Bmi1 marks distinct castration-resistant luminal progenitor cells competent for prostate regeneration and tumor initiation. Nat. Commun. 2016, 7, 12943. [Google Scholar] [CrossRef]
- Yoo, Y.A.; Vatapalli, R.; Lysy, B.; Mok, H.; Desouki, M.M.; Abulkadir, S.A. The role of castration-resistant Bmi1+Sox2+ cells in driving recurrence in prostate cancer. J. Natl. Cancer Inst. 2019, 111, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kroon, P.; Berry, P.; Stower, M.; Rodrigues, G.; Mann, V.M.; Simms, M.; Bhasin, D.; Chettian, S.; Li, C.; Li, P.K.; et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013, 73, 5288–5298. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Oyan, A.M.; Liu, R.; Hua, Y.; Zhang, J.; Houlanf, R.; Popa, M.; Liu, X.; Brokstad, K.A.; Simon, R.; et al. Generation of prostate tumor-initiating cells is associated with elevation of reactive oxygen species and IL-6/STAT3 signaling. Cancer Res. 2013, 73, 7090–7100. [Google Scholar] [CrossRef] [PubMed]
- Schoereder, A.; Herrann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Muller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell pgenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Wang, X.; Ma, L.; Chen, L.; Xiao, M.; Huang, L.; Cao, Y.; Bai, J.; Ma, D.; Zhou, J.; et al. Inhibition of STAT3 signaling targets both tumor-initiating and differentiated cell populations in prostate cancer. Oncotarget 2014, 5, 8416–8428. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, C.; Liu, F.; Li, H.Z.; Peng, G.; Gao, X.; Dong, K.Q.; Wang, H.R.; Kong, D.P.; Qu, M.; et al. Reciprocal network between cancer stem-like cells and macrophages facilitates the progression and androgen deprivation therapy resistance of prostate cancer. Clin. Cancer Res. 2018, 24, 4612–4626. [Google Scholar] [CrossRef]
- Dallavalle, C.; Albino, D.; Civenni, G.; Merulla, J.; Ostano, P.; Mello-Grand, M.; Rossi, S.; Losa, M.; D’Ambrosio, G.; Sessa, F.; et al. MicroRNA-424 impairs ubiquitination to activate STAT3 and promote prostate tumor progression. J. Clin. Invest. 2016, 126, 4585–4602. [Google Scholar] [CrossRef]
- Krzyzanowska, A.; Don-Doncow, N.; Marginean, F.E.; Gaber, A.; Watson, R.W.; Hellsten, R.; Bjartell, A. Expression of tSTAT3, pSTAT727 and pSTAT705 in the epithelial cells of hormone-naïve prostate cancer. Prostate 2019, 79, 784–797. [Google Scholar] [CrossRef]
- Don-Doncow, N.; Marginean, F.; Coleman, I.; Nelson, P.S.; Ehrnstrom, R.; Krzyzanowska, A.; Morissey, C.; Hellsten, R.; Bjartell, A. Expression of STAT3 in prostate cancer metastases. Eur. Urol. 2017, 71, 313–316. [Google Scholar] [CrossRef]
- Culig, Z.; Puhr, M. Interleukin-6 and prostate cancer: Current developments and unsolved questions. Mol. Cell Endrocrinol. 2018, 462, 25–30. [Google Scholar] [CrossRef]
- Yoshioka, T.; Otero, J.; Chen, Y.; Kim, Y.M.; Koutcher, J.A.; Satagopan, J.; Reuter, V.; Corver, B.; de Stanchina, E.; Enomoto, K. β4 integrin signaling induces expansion of prostate tumor progenitors. J. Clin. Invest. 2013, 123, 682–699. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.T.; Liu, C.B.; Li, B.Y. Glycogen synthase kinase 3 and prostate cancer: An update. Zhongua Nan Ke Xue 2017, 23, 178–182. [Google Scholar]
- Kroon, J.; in’t Veld, L.S.; Buijs, J.T.; Cheung, H.; van der Horst, G.; van der Pluijm, J. Glycogen synthase kinase-3beta inhibition depletes the population of prostate cancer stem/progenitor-like cells and attenuates metastatic growth. Oncotarget 2014, 15, 8986–8994. [Google Scholar]
- Lalonde, E.; Alkallas, R.; Chua, M.L.K.; Hider, S.; Meng, A.; Zheng, J.; Jao, C.Q.; Picard, V.; Orain, M.; Hovington, H.; et al. Translating a prognostic DNA genomic classifier into the clinic: retrospective validation in 563 localized prostate tumors. Eur. Urol. 2017, 72, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, M.; Warde, P.; Menard, C.; Chung, P.; Toi, A.; Ishkanian, A.; McLean, M.; Pintilie, M.; Sykes, J.; Gosporadowicz, M.; et al. Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin. Cancer Res. 2012, 18, 2108–2114. [Google Scholar] [CrossRef] [PubMed]
- Marhold, M.; Tomasich, E.; El-Gazzar, A.; Heller, G.; Spittler, A.; Horvat, R.; Krainer, M.; Horak, P. HIF-1 alpha regulates mTOR signaling and viability of prostate cancer stem cells. Mol Cancer Res 2015, 13, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhang, L.; Zhou, Z.; Kwon, O.J.; Zhang, Y.; Nguyen, H.; Dumpit, R.; True, L.; Nelson, P.; Dong, B.; et al. Spatially restricted stromal Wnt signaling restrains prostate epithelial progenitor growth through direct and indirect mechanisms. Cell Stem Cell 2019, 24, 1–16. [Google Scholar] [CrossRef]
- Mutillo-Garzòn, V.; Kypta, R. WNT signaling in prostate cancer. Nat. Rev. Urol. 2017, 14, 683–696. [Google Scholar] [CrossRef]
- Yun, E.J.; Zhou, J.; Lin, C.J.; Hernandez, E.; Fazli, L.; Gleave, M.; Hsich, J.T. Targeting cancer stem cells in catsration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 670–679. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Atallah, N.M.; Pacsuzzi, P.E.; Gupta, S.; Liu, X. Inhibition of the Wnt/β-catenin pathway overcomes resistance to Enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, H.; Liu, A.; Li, J.; Liu, C.; Sun, B.; Chen, L.; Gao, Y.; Xu, D.; Su, C. Cell division cycle 20 (CDc20) drives prostate cancer progression via stabilization of β-catenin in cancer stem-like cells. EBioMedicine 2019, 42, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Augello, M.A.; Liu, D.; Doenaruine, L.D.; Robinson, B.D.; Huang, D.; Stelloo, S.; Blattner, M.; Doane, A.S.; Wong, E.; Wong, E.; et al. CHD1 loss alters AR binding at lineage-specific enhnacers and modulates distinct transcriptional programs to drive prostate tumorigenesis. Cancer Cell 2019, 35, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.S.; Drake, J.M.; Burnes, D.L.; Finley, D.S.; Zhang, H.; Reiter, R.E.; Huang, J.; Witte, O.N. Purification and direct transformation of epithelial progenitor cells from primary human prostate. Nat. Protoc. 2011, 6, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, R.U.; Goldstein, A.S.; Lawson, D.A.; Cheng, D.; Witte, O.N. Isolation, cultivation and characterization of adult murine prostate stem cells. Nat. Protoc. 2010, 5, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Risbridger, G.P. Cross-species stromal signaling programs human embryonic stem cell differentiation. Differentiation 2014, 87, 76–82. [Google Scholar] [CrossRef]
- Toivanen, R.; Berman, D.M.; Wang, H.; Pedersen, J.; Fridenberg, M.; Meeker, A.K.; Ellem, S.J.; Risbridiger, G.P.; Taylor, R.A. Brief report: A bioassay to identify primary human prostate cancer repopulating cells. Stem Cells 2011, 29, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.G.; Taylor, R.; Toivanen, R.; Pedersen, J.; Norden, S.; Pook, D.W.; Frydenberg, M.; Australian Cancer BioResource; Papariris, M.M.; Niranjan, B.; et al. A preclinical xenograft model of prostate cancer using human tumors. Nat. Protoc. 2013, 8, 836–848. [Google Scholar] [CrossRef]
- Namekawa, T.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Application of prostate cancer models for preclinical study: Advantages and limitations of cell lines, patient-derived xenografts, and three-dimensional culture of patient-derived cells. Cells 2019, 8, E74. [Google Scholar] [CrossRef]
- Risbridger, G.P.; Taylor, R.A. Patient-derived prostate cancer: From basic science to the clinic. Horm. Cancer 2016, 7, 236–240. [Google Scholar] [CrossRef]
- Risbridger, G.P.; Toivanen, R.; Taylor, R.A. Preclinical models of prostate cancer: Patient-derived xenografts, organoids, and other explant models. Cold Spring Harb. Perspect. Med. 2018, 8, ao30356. [Google Scholar] [CrossRef]
- Toivanen, R.; Fridenberg, M.; Murphy, D.; Pedersen, J.; Ryan, A.; Pook, D.; Berman, D.M.; Australian Cancer BioResource; Taylor, R.A.; Risbridger, G.P.; et al. A preclinical xenograft model identifies castration-tolerant cancer-repopulating cells in localized prostate tumors. Sci. Transl. Med. 2013, 5, 187ra71. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Ma, Z.; Yeh, C.R.; Luo, J.; Lin, T.H.; Lai, K.P.; Yamashita, S.; Liang, L.; Tian, J.M.; Li, L.; et al. New therapy targeting differential androgen receptor signaling in prostate cancer stem/progenitor vs. non-stem/progenitor cells. J. Mol. Cell. Biol. 2013, 5, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.G.; Obinata, D.; Sandhu, S.; Selth, L.A.; Wong, S.Q.; Porter, L.H.; Lister, N.; Pook, D.; Pezaro, C.J.; Goode, D.L.; et al. Patient-derived models of Abiraterone-and Enzalutamide-resistant prostate cancer reveal sensitivity to ribosome-directed therapy. Eur. Urol. 2018, 74, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Porder, L.H.; Hashimoto, K.; Lawrence, M.G.; Pazaro, C.; Clouston, D.; Wang, H.; Papargiris, M.; Thorne, H.; Li, J.; Kcon, F.; et al. Intraductal carcinoma of the prostate can evade androgen deprivation, with emergence of castrate-tolerant cells. BJU Int. 2018, 12, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Seiler, D.; Zheng, J.; Liu, G.; Wang, S.; Yamashiro, J.; Reiter, R.E.; Huang, J.; Zeng, G. Enrichment of putative prostate cancer stem cells after androgen deprivation: Upregulation of pluripotency trasactivators concurs with resistance to androgen deprivation in LNCaP cell lines. Prostate 2013, 73, 1378–1390. [Google Scholar] [CrossRef]
- Razzak, M. Genetics: ZBTB7A suppresses castration-resistant prostate cancer. Nat. Rev. Clin. Oncol. 2013, 10, 427. [Google Scholar] [CrossRef]
- Lunardi, A.; Ala, U.; Epping, M.T.; Salmena, L.; Clohessy, J.G.; Webster, K.A.; Wang, G.; Mazzucchelli, R.; Bainconi, M.; Stack, E.C.; et al. A co-clinical approach identifies mechansims and potential therapies for androgen deprivation resistance in prostate cancer. Nature Genet. 2013, 45, 747–755. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Loriot, Y.; Beraldi, E.; Zhang, F.; Wyatt, A.W.; Nakowi, N.A.; Mo, F.; Zhou, T.; Kim, Y.; Monia, B.P. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cance cell growth. Clin. Cancer Res. 2015, 21, 1675–1681. [Google Scholar] [CrossRef]
- Xino, L.; Tien, J.C.; Vo, J.; Tan, M.; Parolia, A.; Zhang, Y.; Wang, L.; Qiao, Y.; Shakla, S.; Wang, X. Epigenetic reprogramming with antisence oligonucleotides enhances the effectiveness of androgen receptor inhibition in catsrtaion-resistant prostate cancer. Cancer Res. 2018, 78, 5731–5740. [Google Scholar]
- Bianchini, D.; Omlin, A.G.; Pezaro, C.J.; Mukherji, D.; Lorente Estelle, D.; Ferraldeschi, A.Z.R.; Crespo, M.; Buchbinder, A.; Attard, G.; Scher, H.I.; et al. First-in-human phase I study of ENZ-4176, a locked nucleic acid antisense oligonucleotide (LNA-ASO) to androgen receptor (AR) mRNA in patients with castration-resistant prostate cancer (CRPC). J. Clin. Oncol. 2013, 109, 2579–2586. [Google Scholar]
- Mohammed, O.S.; Nyquist, M.D.; Schweitzer, M.T.; Balk, S.P.; Corey, E.; Plymate, S.; Nelson, P.S.; Mostaghel, E.A. Supraphysiological testaosterone therapy in the treatment of prostate cancer: Models, mechanism and questions. Cancers 2017, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, M.T.; Antonorakis, E.S.; Wang, H.; Ajibove, A.S.; Spitz, A.; Cao, H.; Luo, J.; Haffner, M.C.; Yegnasubramanian, S.; Carducci, M.A.; et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer. Results from a pilot clinical study. Sci.Transl. Med. 2015, 7, 269ra2. [Google Scholar] [CrossRef] [PubMed]
- Teplin, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; De Carli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer fater progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 19, 76–86. [Google Scholar]
- Dorff, T.B.; Fanti, S.; Farolfi, A.; Reiter, R.E.; Sadun, T.Y.; Sartor, O. The evolving role of prostate-specific membrane antigen-based diagnostics and therapeutics in prostate cancer. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Kumar, A.R.; Murphy, D.G.; et al. [177Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Targeted Alpha Therapy Working Group; Parker, C.; Lwington, V.; Shore, N.; Kratochwil, C.; Levy, M.; Linden, O.; Noordzij, W.; Park, J.; Saad, F. Targeted alpha therapy, an emerging class of cancer agents: A review. Jama Oncol. 2018, 4, 1765–1772. [Google Scholar] [PubMed]
- Denham, J.W.; Joseph, D.; Lamb, D.S.; Spry, N.A.; Duchesne, G.; Matthews, J.; Atkinson, C.; Tai, K.H.; Christie, D.; Kenny, L.; et al. Short-term androgen suppression and radiotherapy versus intermediate-arm androgen suppression and radiotherapy, with or without zolendronic acid, in men with locally advanced prostate cancer (TROG 03.04 RADAR): 10 year results from a randomized, phase 3, factorial trial. Lancet Oncol. 2019, 20, 267–281. [Google Scholar]
- Roach, M.; Moughan, J.; Lawton, C.; Dicker, A.P.; Zeitzer, K.; Gore, E.M.; Kwok, Y.; Seider, M.J.; Hsu, I.C.; Hartford, A.C.; et al. Sequence of hormonal therapy and radiotherapy field size in unfavourable, localized prostate cancer (NRG/RTOG 9413): Long-term results of a randomized, phase 3 trial. Lancet Oncol. 2018, 19, 1504–1515. [Google Scholar] [CrossRef]
- Tiki, D.; Chen, M.H.; Wu, J.; Huland, H.; Graefen, M.; Braccioforte, M.; Moran, B.J.; D’Amico, A.V. Surgery vs. radiotherapy in the management of biopsy Gleason score 9-10 prostate cancer and the risk of mortality. JAMA Oncol. 2019, 5, 213–220. [Google Scholar]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middhe, S.; Autio, K.A.; Vargas, K.A.; Rathkof, D.; Morris, M.J.; Danila, D.C.; Slavin, S.F. Analysis of prevalence of microsatellite instabilities of prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logethetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Annala, M.; Taavittsainen, S.; Vandekerkhove, G.; Bacon, J.; Beja, K.; Chi, K.N.; Nykter, M.; Wyatt, A.W. Frequent mutation of the FOXA1 untranslated region in prostate cancer. Commun. Biol. 2018, 1, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Garcia-Bassets, I.; Benner, C.; Li, W.; Su, X.; Zhou, Y.; Qiu, J.; Liu, W.; Kaikkonen, M.U.; Ohgi, K.A.; et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Yu, J. Current perspectives on FOXA1 regulation of androgen receptor signaling and prostate cancer. Genes Dis. 2015, 2, 144–151. [Google Scholar] [CrossRef]
- Jin, H.J.; Zhao, J.C.; Wu, L.; Yu, J. Coopereativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 2014, 5, 3972. [Google Scholar] [CrossRef]
- Jin, H.J.; Zhao, J.C.; Ogden, I.; Bergan, R.C.; Yu, J. Androgen receptor-independent function of FOXA1 in prostate cancer metastasis. Cancer Res. 2013, 73, 3725–3736. [Google Scholar] [CrossRef]
- Kim, J.; Jin, H.J.; Zhao, J.C.; Yang, Y.A.; Li, Y.; Yang, X.; Dong, X.; Yu, J. FOXA1 inhibits prostate cancer neuroendocrine differentiation. Oncogene 2017, 36, 4072–4080. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Park, S.H.; Xhao, J.C.; Fong, K.W.; Li, S.; Lee, Y.; Yang, Y.A.; Sridhar, S.; Lu, X.; Abulkadir, S.A.; et al. Taregting FOXA1-mediated repression of TGF-β signaling suppresses castration-resistant prostate cancer progression. J. Clin. Invest. 2019, 129, 569–582. [Google Scholar] [CrossRef]
- Zhao, J.C.; Fong, K.W.; Jin, H.J.; Yang, Y.A.; Kim, J.; Yu, J. FOXA1 cats upstream of GATA2 and AR in hormonal regulation of gene expression. Oncogene 2016, 35, 4335–4344. [Google Scholar] [CrossRef]
- Rodriguez-Bravo, V.; Carceles-Cordon, M.; Hoshida, Y.; Cordon-Cardo, C.; Galsky, M.D.; Domingo-Domenech, J. The role of GATA2 in lethal cancer aggressiveness. Nat. Rev. Urol. 2017, 14, 38–48. [Google Scholar] [CrossRef]
- Umetani, M.; Nakao, H.; Doi, T.; Iwasaki, A.; Ohtaka, M.; Nagoya, T.; Mataki, C.; Hamakubo, T.; Kodama, T. A novel cell adhesion inhibitor, K-7174, reduces the endothelial VCAM-1 induction by inflammatory cytokines, acting through the regulation of GATA. Biochem. Biophys. Res. Commun. 2000, 272, 370–374. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Lanz, R.B.; Fiskus, W.; Geng, C.; Yi, P.; Hartig, S.M.; Rajapakshe, K.; Shou, J.; Wei, L.; Shah, S.; et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc. Natl. Acad. Sci. USA 2014, 111, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Wade, M.; Nakjang, S.; Chaytor, L.; Grey, J.; Robson, C.N. FOXA1 regulates androgen receptor variant activity in models of castrate-resistant prostate cancer. Oncotarget 2015, 6, 29782. [Google Scholar] [CrossRef] [PubMed]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Reisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef]
- Welti, J.; Sharp, A.; Yuan, W.; Dolling, D.; Nava Rodrigues, D.; Figueiredo, I.; Gil, V.; Neeb, A.; Clarke, M.; Seed, G.; et al. Targeting bromodomain and extra-terminal (BET) family proteins in castration-resistant prostate cancer (CRPC). Clin. Cancer Res. 2018, 24, 3149–3162. [Google Scholar] [CrossRef]
- Urbanucci, A.; Barfeld, S.J.; Kytola, V.; Itkonen, H.M.; Coleman, L.M.; Vodak, D.; Sjoblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen receptor deregulation drives bromodomain -mediated chromatin alterations in prostate cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef]
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol. Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef]
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Aggarwal, R.; Abida, W.; Schweizer, M.; Pantuck, A.; Nanus, D.; Heath, E.; Lakhotia, S.; Hansen, H.C.; Silverman, H.; Bauman, L.; et al. CTo95: A phase Ib/IIa study of the BET bromodomain inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer (mCRPC). In Proceedings of the AACR Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Zarrinpar, A.; Lee, D.K.; Silva, A.; Datta, N.; Kee, T.; Eriksen, C.; Weigle, K.; Agopian, V.; Kaldas, F.; Farmer, D.; et al. Individualizing liver transplant immunosuppression using a phenotypic personalized medicine platform. Sci. Transl. Med. 2016, 8, 333ra49. [Google Scholar] [CrossRef]
- Pantuck, A.J.; Lee, D.K.; Kee, T.; Wang, P.; Lakhotia, S.; Silverman, M.H.; Mathis, C.; Drakaki, A.; Belldegrun, A.S.; Ho, C.M.; et al. Modulating BET bromodomain inhibitor ZEN-3694 and Enzalutamide combination dosing in a metastatic prostate cancer patient using CURATE.A1, an artificial intelligence platform. Adv. Ther. 2018, 1, 1800104. [Google Scholar] [CrossRef]
- Pawar, A.; Gollavilli, P.N.; Wang, S.; Asangani, I.A. Resistance to BET inhibitor leads to alternative therapeutic vulnerabilities in castration-resistant prostate cancer. Cell. Rep. 2018, 22, 2236–2245. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Scher, H.I.; Rosen, N. HSP90 as a therapeutic target in propstate cancer. Sem. Oncol. 2003, 30, 709–716. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Farah, E.; Sarkar, S.; Ahmad, N.; Gupta, S.; Larner, J.; Liu, X. Cotargeting HSP90 and its client proteins for treatment of prostate cancer. Mol. Cancer Ther. 2016, 15, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Dagar, M.; Singh, J.P.; Dagar, G.; Tyagi, R.K.; Bagchi, G. Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J. Biol. Chem. 2019, 294, 8699–8710. [Google Scholar] [CrossRef] [PubMed]
- Jansson, K.H.; Tucker, J.B.; Stahl, L.E.; Simmons, J.K.; Fuller, C.; Beshiri, M.L.; Agarwal, S.; Fang, L.; Hynes, P.G.; Allin, A.N.; et al. High-throughput screens identifiey HSP90 inhibitors as potent therapeutics that target inter-related growth and survival pathways in advanced prostate cancer. Sci. Rep. 2018, 8, 17239. [Google Scholar] [CrossRef]
- Thakur, M.K.; Hellbrun, L.K.; Sheng, S.; Stein, M.; Liu, G.; Anatonarakis, E.S.; Vaishampayan, U.; Dzinic, S.H.; Li, X.; Freeman, S.; et al. A phase II trial of ganetespib, a heat shock protein 90 (Hsp90) inhibitor, in patients with docetaxel-pretreated metastatic castrate-resistant prostate cancer (CRPC)-a prostate cancer clinical trial consortium (PCCTC) study. Invest. New Drugs 2016, 34, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Ferraldeschi, R.; Welti, J.; Powers, M.V.; Yuan, W.; Smyth, T.; Seed, G.; Riisnaes, R.; Hedayat, S.; Wang, H.; Crespo, M.; et al. Second-generation SHP90 inhibitor Onalespib blocks mRNA spilicing of androgen receptor varian 7 in prostate cancer cells. Cancer Res. 2016, 76, 2731–2742. [Google Scholar] [CrossRef]
- Slovin, S.F.; Hussain, S.; Saad, F.; Garcia, J.A.; Picus, J.; Ferraldeschi, R.; Crespo, M.; Flohr, P.R.; Riisnaes, R.; Lin, C.; et al. Pharmacodynamic and clinical results from a phase 1/2 study of the HSP90 inhibitor Onalespib in combination with Abirateronew acetate in prostate cancer. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Khor, L.Y.; Desilvio, M.; Al-Saleem, T.; Hammond, M.E.; Grignon, D.J.; Sause, W.; Pilepich, M.; Okunieff, P.; Sandler, H.; Pollack, A.; et al. MDM2 as a predictor of prostate carcinoma outcome: An analysis of Radiation Therapy Oncology Group Protocol 8610. Cancer 2005, 104, 962–967. [Google Scholar] [CrossRef]
- Khor, L.Y.; Bae, K.; Paulus, R.; Al-Saleem, T.; Hammond, M.E.; Grignon, D.J.; Che, M.; Venkatesan, V.; Byhardt, R.W.; Rotman, M.; et al. MDM2 and Ki-67 predict for distant metastasis and mortality in men treated with radiotherapy and androgen deprivation for prostate cancer: RTOG 982-02. J. Clin. Oncol. 2009, 27, 3177–3184. [Google Scholar] [CrossRef] [PubMed]
- Giridhar, P.V.; William, K.; Vonttandorf, A.P.; Defrol, P.L.; Kasper, S. Constant degradation of the androgen receptor by MDM2 conserves prostate cancer stem cell integrity. Cancer Res. 2019, 79, 1125–1137. [Google Scholar]
- Feng, F.Y.; Zhang, Y.; Kothari, V.; Evans, J.R.; Jackson, W.C.; Chen, W.; Johson, S.B.; Luczak, C.; Wang, S.; Hamstra, D.A. MDM2 inhibition sensitizes prostate cancer cells ablalation and radiotherapy in a p53-dependent manner. Neoplasia 2016, 18, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Roxburgh, P.; Currie, D.; Morrisonm, P.; Kelly, C.; Thomson, F.; McCormick, C.; Bilsiand, A.; Jones, R.H.; Quinton, A.; McCartney, E.; et al. Abstract CT027: Safety, pharmacokinetics (PK) & pharmacodynamics (PD) of idasanutlin (idasa), combined with abiraterone (abi)/prednisone (pred) or enzalutamide (enza) in castrate resistant prostate cancer (CRPC). In Proceedings of the AACR Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Gavyert, K.; Sailer, V.; Conteduca, V.; Dardenne, E.; Sigouros, M.; Isse, K.; Kearney, M.; Vosoughi, A.; Fernandez, L.; et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci. Transl. Med. 2019, 11, eaav0891. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Shelkh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Mosquerra, J.M.; Beltran, H.; Park, K.; MacDonald, T.Y.; Robinson, B.D.; Tagawa, S.T.; Perner, S.; Bismar, T.A.; Erbersdobler, A.; Dhir, R.; et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013, 15, 1–10. [Google Scholar] [CrossRef]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, B.; Wu, W.; Li, L.; Broom, B.M.; Basourakos, S.P.; Korentzelos, D.; Luan, Y.; Wang, J.; Yang, G.; et al. Targeting the MYCN-PARP-DNA damage response pathway in neuroendocrine prostate cancer. Clin. Cancer Res. 2018, 24, 696–707. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, L.; Chang, Y.; Zhang, T.; Chen, X.; Wang, A.; Groth, J.; Foo, W.C.; Liang, C.; Hu, H.; et al. N-Myc promotes therapeutic resistance development of neuroendocrine development of neuroendocrine prostate cancer by differentially regulating miR-421/ATM pathway. Mol. Cancer 2019, 18, 11. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Azzad, A.A.; Volik, S.V.; Annala, M.; Beja, K.; McConeghy, B.; Haegert, A.; Warner, E.W.; Mo, F.; Brahmbhatt, S. Genomic alterations in cell-free DNA and ERnzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol. 2016, 2, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Alshalalfa, M.; Zhao, S.G.; Liu, Y.; Mahal, B.A.; Quigley, D.A.; Wei, T.; Davicioni, E.; Rebbeck, T.R.; Kanotoff, P.W.; et al. Novel RB1-loss transcriptomic signature is associated with poor clinical outcomes across cancer types. Clin. Cancer Res. 2019, 25, 4290–4299. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Gao, Y.; He, H.H.; Dan, D.; Han, W.; Avery, A.; Macoska, J.A.; Liu, X.; Chen, S.; Ma, F.; et al. Androgen receptor tumor suppressor function is mediated by recruitment of retinoblastoma protein. Cell Rep. 2016, 17, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.D.; Beltran, H. Retinoblastoma loss in cancer: Casting a wider net. Clin. Cancer Res. 2019, 25, 4199–4201. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.M.; Yates, T.J.; Ruiz-Torres, M.; Kim-Kiselak, C.; Gudiel, A.A.; Dershpande, C.; Wang, W.Z.; Cicchini, M.; Stokes, K.L.; Tobias, J.W.; et al. RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature 2019, 569, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jenning, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells lacking the RB1 tumor suppressor gene are hyperdependent in Aurora B kinase for survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Du, J.; Persons, S.H.; Merzough, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Lake, E.W.; Muretta, J.M.; Thompson, A.R.; Rasmussen, D.M.; Majumdar, A.; Faber, E.B.; Ruff, E.F.; Thomas, D.D.; Levinson, N.M. Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for Aurora kinase activation states. Proc. Natl. Acad. Sci. USA 2018, 115, E11894–E11903. [Google Scholar] [CrossRef]
- Bleijs, M.; van de Watering, M.; Clevers, H.; Drost, J. Xenograft and organoid model systems in cancer research. EMBO J. 2019, e101654. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Wyatt, A.W.; Xue, H.; Wang, Y.; Dong, X.; Haegert, A.; Wu, R.; Brahmbhatt, S.; Mo, F.; Joimg, L.; et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014, 74, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Vessella, R.L.; Morissey, C.; Coleman, L.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.M.; Roudier, M.; et al. Lucap prostate cancer patient-derived xenografts reflect the molecular heterogeneity of advanced disease and serve as models for evaluating cancer therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Lin, T.P.; Campbell, M.; Pan, C.C.; Lee, S.H.; Lee, H.C.; Yang, M.H.; Kung, H.J.; Chang, P.C. REST is a crucial regulator for acquiring EMT-like and stemness phenotypes in hormone-refractory prostate cancer. Sci. Rep. 2017, 7, 42795. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, A.; Bermann, T.B.; Lavallee, C.; Batth, T.S.; Lin, D.; Lerdrup, M.; Friis, S.; Bartels, A.; Kristensen, G.; Krzyzanowska, A.; et al. Proteogenomic characterization of patient-derived xenografts highlights the role of REWST in neuroendocrine differentiation of castration-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Lange, T.; Oh-Hohenhorst, S.J.; Joosse, S.A.; Pantel, K.; Hahn, O.; Gosau, T.; Dyshlovov, S.A.; Wellbrock, J.; Feldhaus, S.; Maar, H.; et al. Development and characterization of a spontaneously metastatic patient-derived xenograft model of human prostate cancer. Sci. Rep. 2018, 8, 17535. [Google Scholar] [CrossRef]
- Navone, N.M.; van Weerden, W.M.; Vessella, R.L.; Williams, E.D.; Wang, Y.; Isaacs, J.T.; Nguyen, H.M.; Culig, Z.; van der Pluijm, G.; Rentsch, C.A.; et al. Movember gap1 pdx project: An international collection of serially transplantable cancer patient-derived xenograft (pdx) models. Prostate 2018, 78, 1262–1282. [Google Scholar] [CrossRef] [PubMed]
- Centenera, M.M.; Hickey, T.E.; Jindal, S.; Ryan, N.K.; Ravindranathan, P.; Mohammed, H.; Robinson, J.L.; Schiewer, M.J.; Ma, S.; Kapur, P.; et al. A patient-derived explant (PDE) model of hormone-dependent cancer. Mol. Oncol. 2018, 12, 1608–1622. [Google Scholar] [CrossRef]
- Park, J.; Lee, J.K.; Phillips, J.W.; Huang, P.; Cheng, D.; Huang, J.; Witte, O.N. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc. Natl. Acad. Sci. USA 2016, 113, 4482–4487. [Google Scholar] [CrossRef]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigorous, M.; Donoghue, A.; Kossai, M.; et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.D.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Invest. 2019, 130, 127961. [Google Scholar] [CrossRef]
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin remodeling ATPase BRG1 and PTEN are synthetic lethal in prostate cancer. J. Clin. Invest. 2019, 129, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Wang, Y.; Zhang, X.; Goodrich, D.W. Generation of tumor organoids from genetically engineered mouse models of prostate cancer. J. Vis. Exp. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Beshiri, M.L.; Tice, C.M.; Tran, C.; Nguyen, H.M.; Sowalsky, A.G.; Agarwal, S.; Jansson, K.H.; Yang, Q.; McGowen, K.M.; Yin, J.; et al. A PDX/organoid biobank of advanced prostate cancers captures genomic and phenotypic heterogeneity for disease modeling and therapeutic screening. Clin. Cancer Res. 2018, 24, 4332–4345. [Google Scholar] [CrossRef] [PubMed]
- Richards, Z.; McCray, T.; Marsili, J.; Zenner, M.L.; Maniucu, J.T.; Garcia, J.; Kajdacsy-Balla, A.; Murray, M.; Voisine, C.; Murphy, A.B.; et al. Prostate stroma increases viability and maintains the branching phenotype of human prostate organoids. iScience 2019, 12, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective genome profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis. Oncol. 2017. [Google Scholar] [CrossRef]
- Chung, J.H.; Deawl, N.; Sokol, E.; Mathew, P.; Whitehead, R.; Millis, S.Z.; Frampton, G.M.; Bratslavsky, G.; Pal, S.K.; Lee, R.J.; et al. Prospective comprehensive genomic profiling of primary and metastatic prostate tumors. J. Clin. Oncol. Prec. Oncol. 2019. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Lliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukakla, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leogamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef]
- Bratt, O.; Drevin, L.; Akre, O.; Garmo, H.; Statin, P. Family history and probability of prostate cancer, differentiated by risk category: A nationwide population-based study. J. Natl. Cancer Inst. 2016, 108, djw110. [Google Scholar] [CrossRef] [PubMed]
- Momozawa, Y.; Iwasaki, Y.; Hirata, M.; Liu, X.; Kamatani, Y.; Takahashi, A.; Sugano, K.; Yoshida, T.; Murakami, Y.; Matsuda, K.; et al. Germline pathogenic variants in 7636 Japanese patients with prostate cancere and 12,366 controls. J. Natl. Cancer Inst. 2019, djz124. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Salami, S.S.; Spratt, D.E.; Kaffenberger, S.D.; Jacobs, M.F.; Morgan, T. Bringing prostate cancer germline genetics into clinical practice. J. Urol. 2019, 202, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.I.; Giri, V.N.; Paller, C.J.; Abida, W.; Alumkal, J.J.; Beer, T.M.; Beltran, H.; George, D.J.; Heath, E.I.; Higano, C.S.; et al. Evolving intersection between inherited cancer genetics and therapeutic clinical trials in prostate cancer: A white paper from the germline genetics working group of the prostate cancer clinical trilas consortium. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, D.; Morra, F.; Giannella, R.; Cerrato, A.; Celetti, A. Identification of novel biomarkers of homologous recombination defect in DNA repair to predict sensitivity of prostate cancer cells to PARP-inhibitors. Int. J. Mol. Sci. 2019, 20, E3100. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Gui, B.; Gui, F.; Takai, T.; Feng, C.; Bai, X.; Fazli, L.; Liu, S.; Zhang, X.; Kibel, A.S.; Jia, L. Selective targeting of PARP-2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function. Proc. Natl. Acad. Sci. USA 2019, 116, 14573–14582. [Google Scholar] [CrossRef]
- Chatterjee, P.; Schweizer, M.T.; Lucas, J.M.; Coleman, I.; Nyquist, M.D.; Frank, S.B.; Tharakan, R.; Mostaghel, E.; Luo, J.; Pritchard, C.C.; et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J. Clin. Invest. 2019, 130, 127613. [Google Scholar] [CrossRef]
- Torquato, S.; Pallavajjala, A.; Goldstein, A.; Toro, P.V.; Silberstein, J.L.; Lee, J.; Nakazawa, M.; Waters, I.; CXhu, D.; Shinn, D.; et al. Genetic alterations detected in cell-free DNA are associated with enzalutamide and abiraterone resistance in castration-resistant prostate cancer. JCO Precis. Oncol. 2019. [Google Scholar] [CrossRef]
- Salami, S.; Singhal, U.; Spratt, D.E.; Palapattu, G.S.; Hollenbeck, B.K.; Schonoft, J.D.; Graf, R.; Louw, I.; Iendrisak, A.; Dugan, L.; et al. Circulating tumor cells as a predictor of treatment response in clinically localized prostate cancer. JCO Precis. Oncol. 2019. [Google Scholar] [CrossRef]
- Baumgart, S.J.; Nevedomskaya, E.; Haendler, B. Dysregulated transcriptional control in prostate cancer. Int. J. Med. Sci. 2019, 20, 2883. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yan, Y.; Wang, D.; Ding, D.; Ma, T.; Ye, Z.; Jimenez, R.; Wang, L.; Wu, H.; Huang, H. DUB3 promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol. Cell 2018, 761, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Bosotti, R.; Timpanaro, A.; Vazquez, R.; Merulla, J.; Pandit, S.; Rossi, S.; Albino, D.; Allegrini, S.; Mitra, A.; et al. Epigenetic control of mitochondrial fission enables self-renewal of stem-like tumor cells in human prostate cancer. Cell Metab. 2019, S1550-4131. [Google Scholar] [CrossRef] [PubMed]
- Shafran, J.S.; Andrieu, G.P.; Gyorffy, B.; Denis, G.V. BRD4 regulates metastatic potential of castration-resistant prostate cancer through AHNAK. Mol. Cancer Res. 2019, molcanres.1279.2018. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Welti, J.C.; Lambros, M.B.K.; Dolling, D.; Rodrigues, D.N.; Pope, L.; Aversa, C.; Figueiredo, I.; Fraser, J.; Ahmad, Z.; et al. clinical utility of circulating tumour cell androgen receptor splice variant-7 status in metastatic castration-resistant prostate cancer. Eur. Urol. 2019, in press. [Google Scholar] [CrossRef]
- Stoykova, G.E.; Schlaepfer, I.R. Lipid metabolism and endocrine resistance in prostate cancer, and new opportunities for therapy. Int. J. Mol. Sci. 2019, 20, E2626. [Google Scholar] [CrossRef]
- Frame, F.M.; Maitland, N.J. Epigenetic control of gene expression in the normal and malignant human prostate: A rapid response which promotes therapeutic resistance. Int. J. Mol. Sci. 2019, 20, 2437. [Google Scholar] [CrossRef]
- Parolia, A.; Cieslik, M.; Chu, S.C.; Xiao, L.; Ouchi, T.; Zhang, Y.; Wang, X.; Vats, P.; Cao, X.; Pitchiya, S.; et al. Distinct structural classes of FOXA1 alterations in advancer prostate cancer. Nature 2019, 571, 413–418. [Google Scholar] [CrossRef]
- Adams, E.J.; Karthaus, W.R.; Hoover, E.; Liu, D.; Gruet, A.; Zhang, Z.; Cho, H.; DiLoreto, R.; Chhangawala, S.; Liu, Y.; et al. FOXA1 mutations alter pioneering, differentiation and prostate cancer phenotypes. Nature 2019, 571, 408–412. [Google Scholar] [CrossRef]






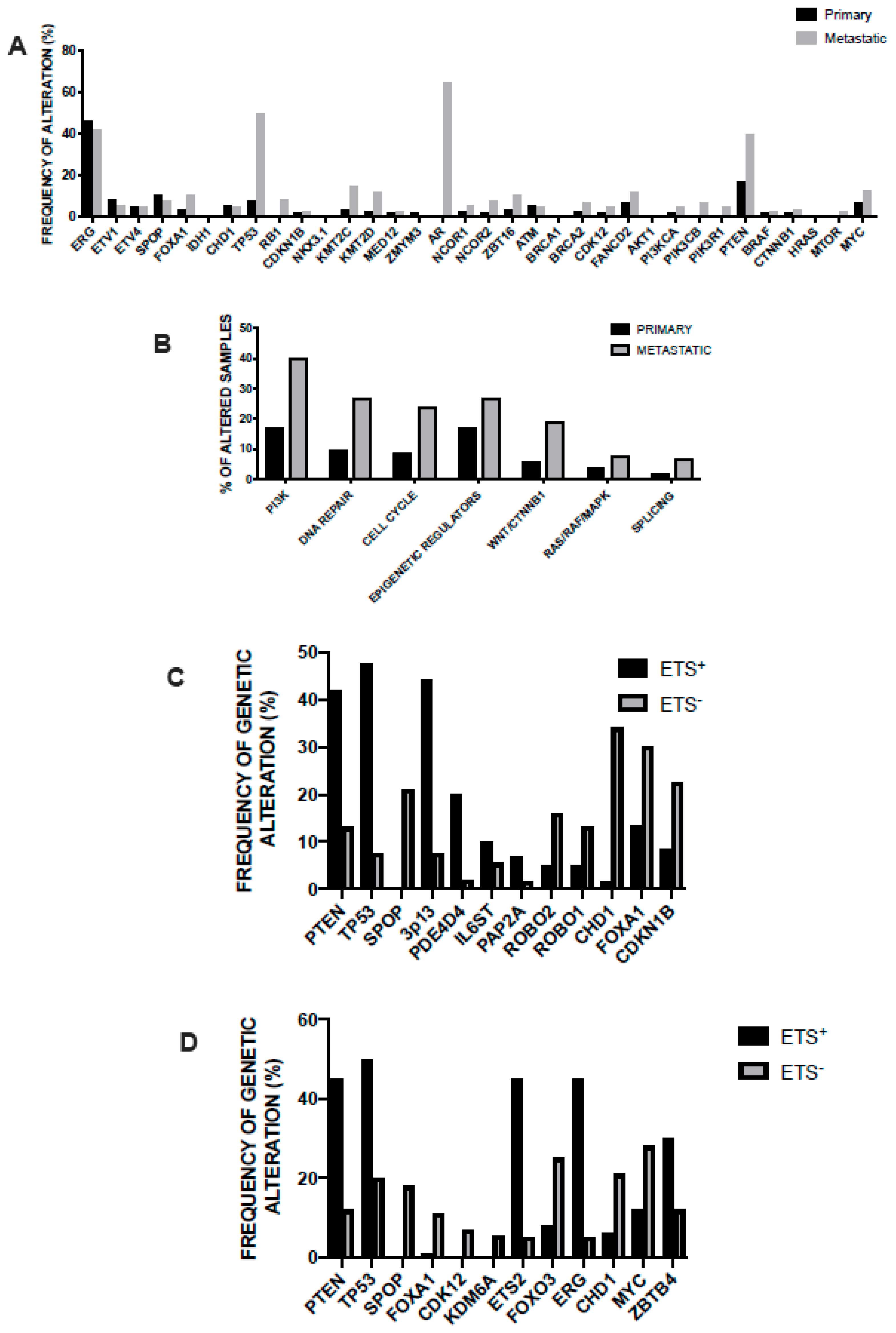{kind=link}
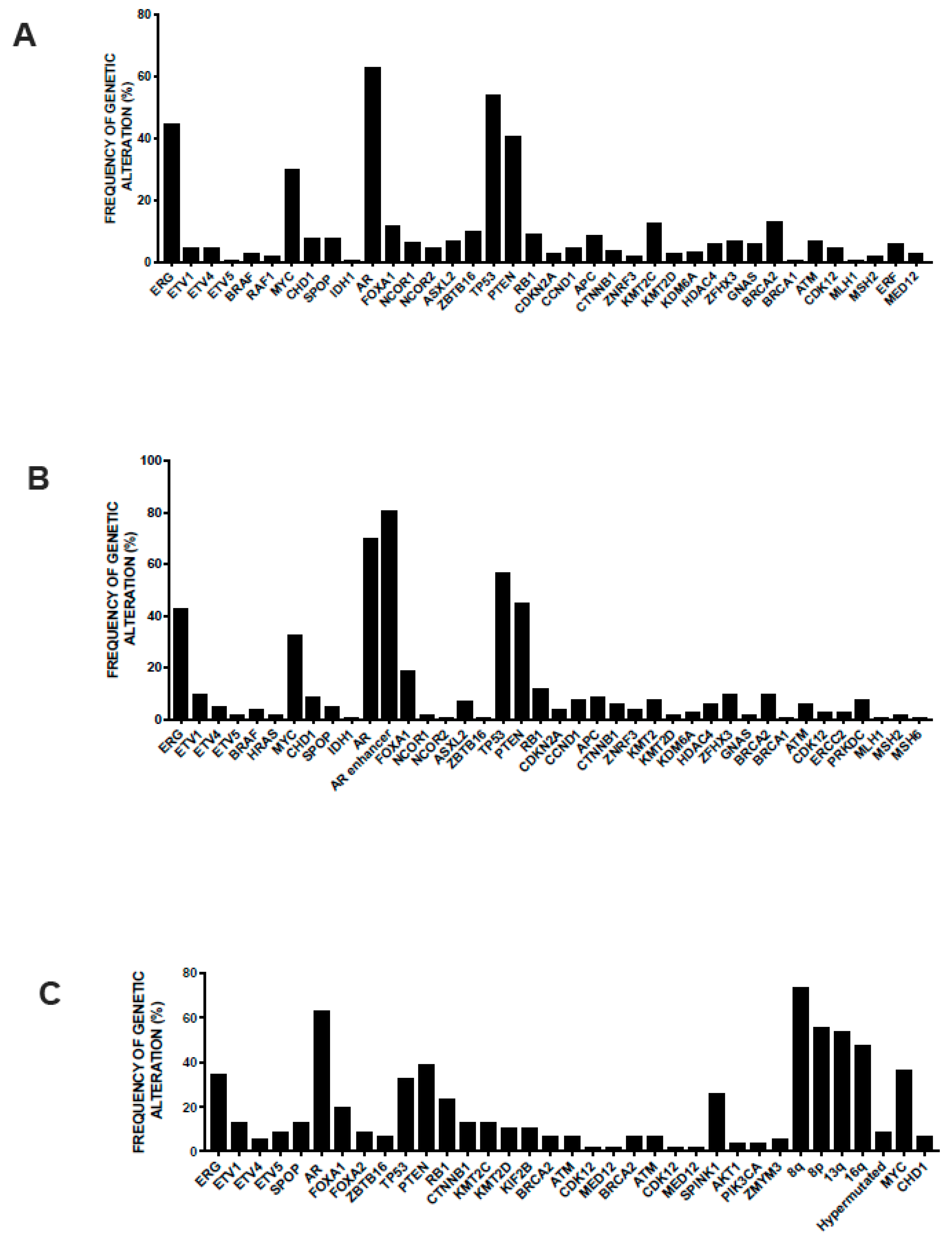{kind=link}
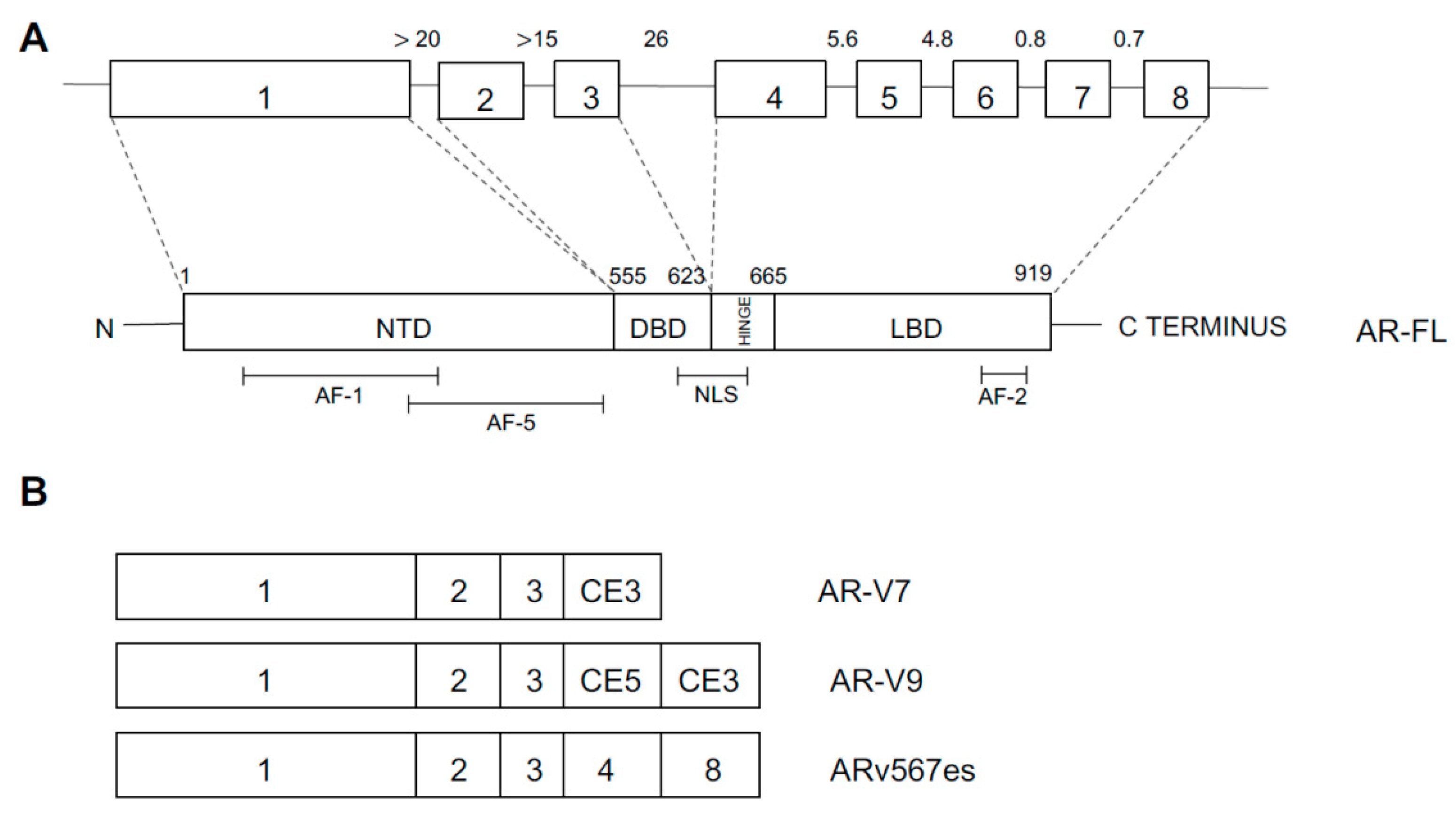{kind=link}
{kind=link}
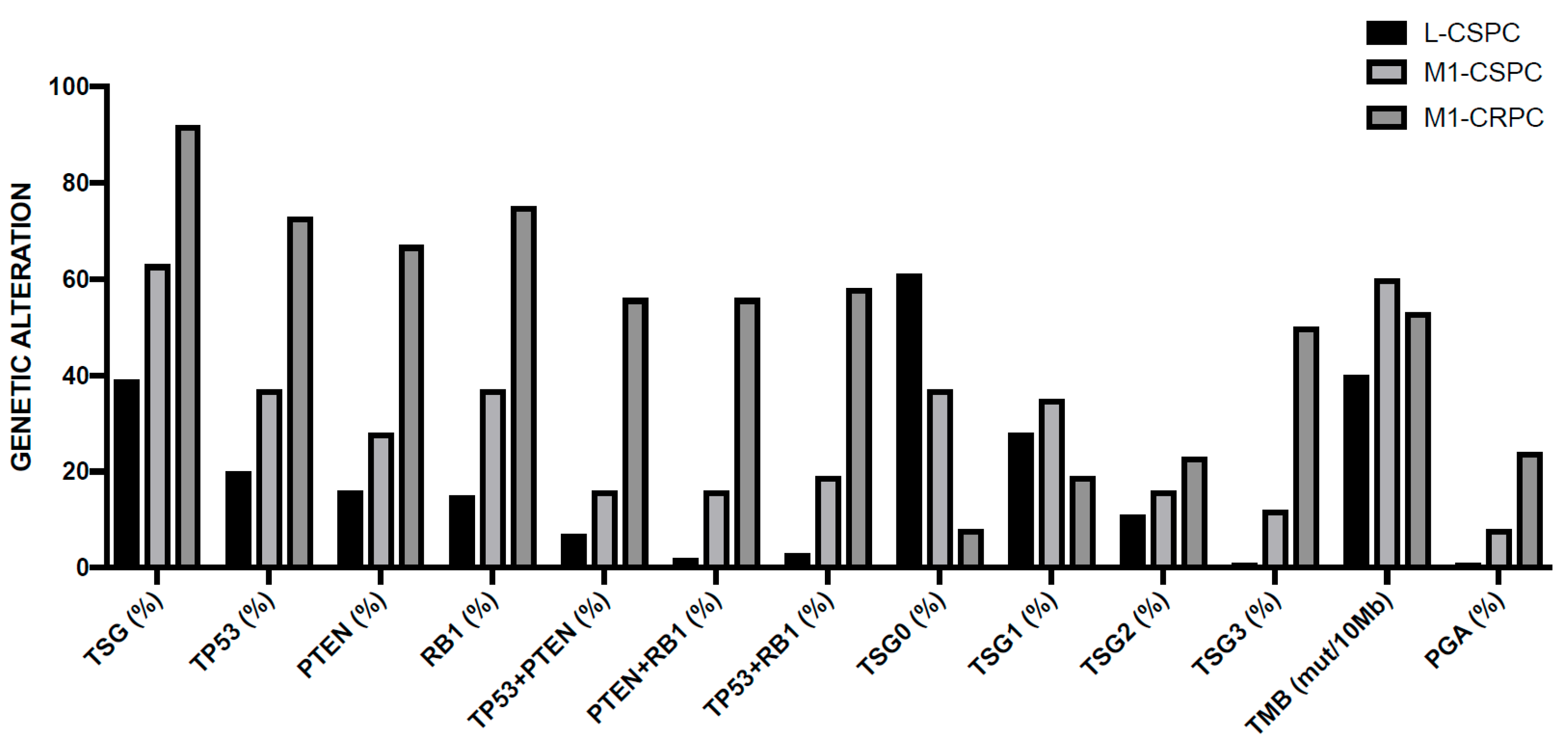{kind=link}
| Chromosome Region | Genetic Event | Genes Involved | Frequency in Primary Tumors (%) | Frequency in Advanced Tumors (%) |
|---|---|---|---|---|
| 2q | Deletion | CXCR4 | 23 | 61 |
| 3p13 | Deletion | FOXP1, RYBP, SQQ1 | 20 | 32 |
| 5q | Deletion | CHD1, APC | 36 | 76 |
| 6q | Deletion | MAP3K7, ZNF292 | 41 | 74 |
| 8p | Deletion | NKX3-1, PPP2B2A | 56 | 90 |
| 10q | Deletion | PTEN | 26 | 83 |
| 12p | Deletion | CDKN1B | 24 | 53 |
| 13q | Deletion | BRCA2, RB1 | 45 | 90 |
| 16q | Deletion | CDH1 | 44 | 90 |
| 17p | Deletion | TP53 | 28 | 78 |
| 17q | Deletion | BRCA1, ETV4 | 17 | 41 |
| 18q | Gain | SMAD4, BCL2 | 25 | 67 |
| 3q | Gain | PI3KCA, ETV5 | 10 | 61 |
| 7 | Gain | ETV1, EGFR, MCM7, BRAF | 14 | 75 |
| 8q | Gain | MYC | 21 | 84 |
| 16p | Gain | - | 18 | 64 |
| 21q | Fusion | ERG, TMPRSS2 | 25 | 48 |
| Drug Category | Compound | Chemical Structure | Mechanism of Action |
|---|---|---|---|
| First-generation antiandrogen | Bicalutamide | Nonsteroidal antiandrogen | It blocks the effects of androgens at AR level |
| First-generation antiandrogen | Flutamide | Nonsteroidal antiandrogen | It blocks the effects of androgens at AR level |
| First-generation antiandrogen | Nilutamide | Nonsteroidal antiandrogen | It blocks the effects of androgens at AR level |
| First-generation antiandrogen | Cyproterone | Derivative of progesterone | It binds to AR and blocks the effects of testosterone and DHT |
| GnRH Agonist | Leuprorelin | Synthetic analog of gonadotropin-releasing hormone (GnRH) | It binds with high affinity to GnRH receptor on anterior pituitary cells, where it acts as an agonist |
| GnRH Agonist | Triptorelin | Synthetic analog of GnRH: more potent than native hormone and more resistant to proteolysis | It binds with high affinity to GnRH receptor on anterior pituitary cells, where it acts as an agonist |
| GnRH Agonist | Goserelin | Synthetic analog of GnRH: more potent than native hormone and more resistant to proteolysis | It binds with high affinity to GnRH receptor on anterior pituitary cells, where it acts as an agonist |
| GnRH Agonist | Degorelix | Synthetic peptide derivate of GnRH | It binds with high affinity to GnRH receptor on anterior pituitary cells, where it acts as an agonist |
| GnRH Agonist | Relugolix | Synthetic nonpeptide analog of GnRH | It binds with high affinity to GnRH receptor on anterior pituitary cells, inhibits the secretion of FSH and LH, preventing the release of testosterone by Leidig cells |
| Second-generation androgen inhibitors | Abiraterone | Steroidal compound inhibitor of androgen synthesis | It blocks the enzyme cytochrome P450 1 alpha-hydroxylase (CYP17), an enzyme required for testosterone synthesis |
| Second-generation androgen inhibitors | Enzalutamide | Synthetic AR signaling inhibitor | It blocks AR signaling at three key stages: it blocks the binding of androgens to AR, it inhibits nuclear translocation of activated AR and impairs binding of activated AR with DNA |
| Second-generation androgen inhibitors | Apalutamide | Small molecule synthetic AR antagonist | It selectively binds to the ligand-binding domain of AR and blocks nuclear translocation and binding to androgen response elements |
| Second-generation androgen inhibitors | Darolutamide | Nonsteroidal AR antagonist | It selectively binds to the ligand-binding domain of AR and blocks nuclear translocation and binding to androgen response elements |
| Second-generation androgen inhibitors | Galeterone | Steroidal compound inhibitor of androgen synthesis | It blocks the enzyme CY17, acts as an AR antagonist, promotes AR degradation |
| Second-generation androgen inhibitors | EPI-506 | Nonsteroidal small molecule AR antagonist | It selectively binds to the NTD of AR and blocks AR signaling |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. https://doi.org/10.3390/medicines6030082
Testa U, Castelli G, Pelosi E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines. 2019; 6(3):82. https://doi.org/10.3390/medicines6030082
Chicago/Turabian StyleTesta, Ugo, Germana Castelli, and Elvira Pelosi. 2019. "Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications" Medicines 6, no. 3: 82. https://doi.org/10.3390/medicines6030082
APA StyleTesta, U., Castelli, G., & Pelosi, E. (2019). Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines, 6(3), 82. https://doi.org/10.3390/medicines6030082