Antiproliferative Activity of (-)-Rabdosiin Isolated from Ocimum sanctum L.


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. General Experimental Procedures
2.3. Extraction and Isolation
2.4. Cytotoxic Effects against Cancer Cell Lines
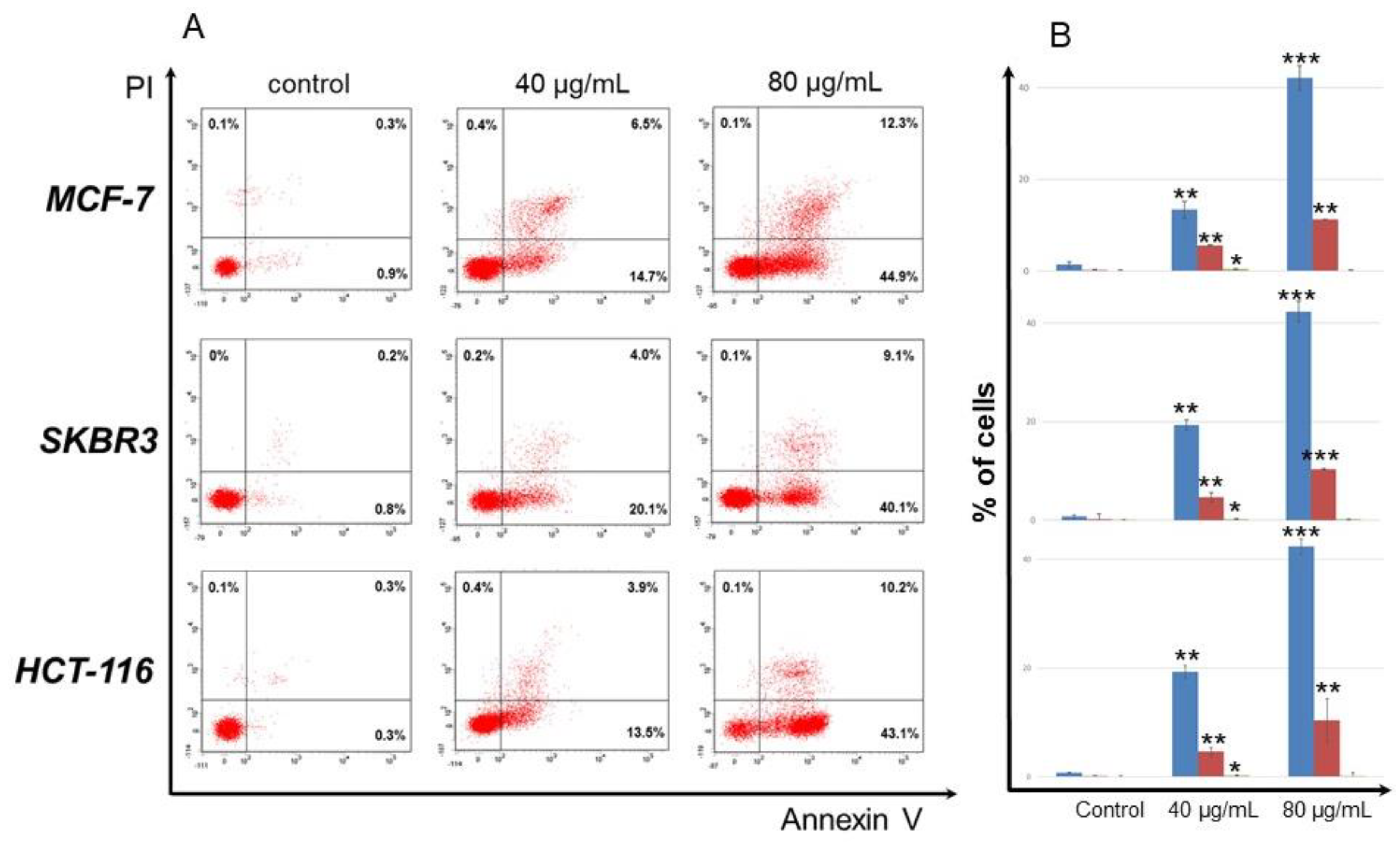
2.5. Flow Cytometry Analysis
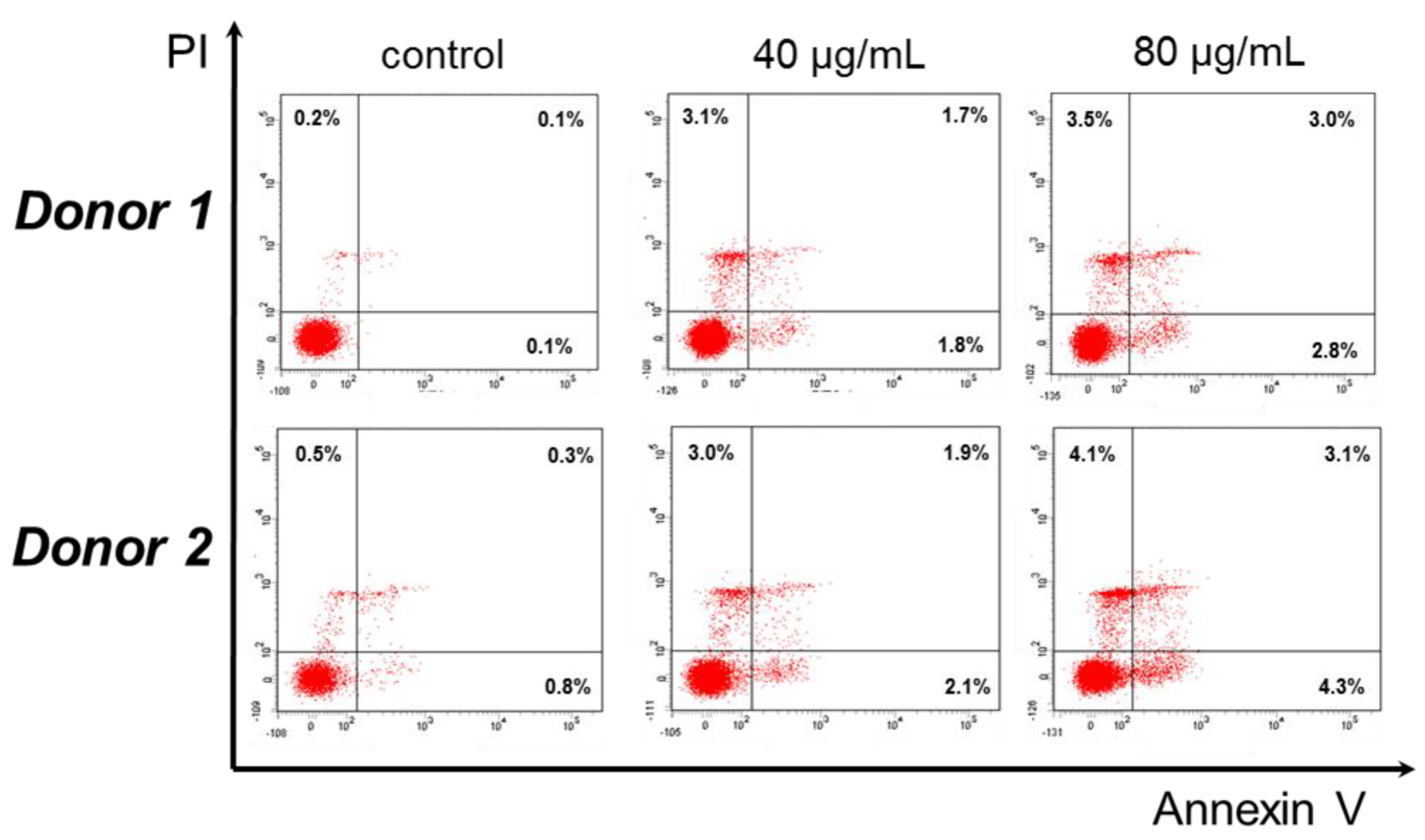
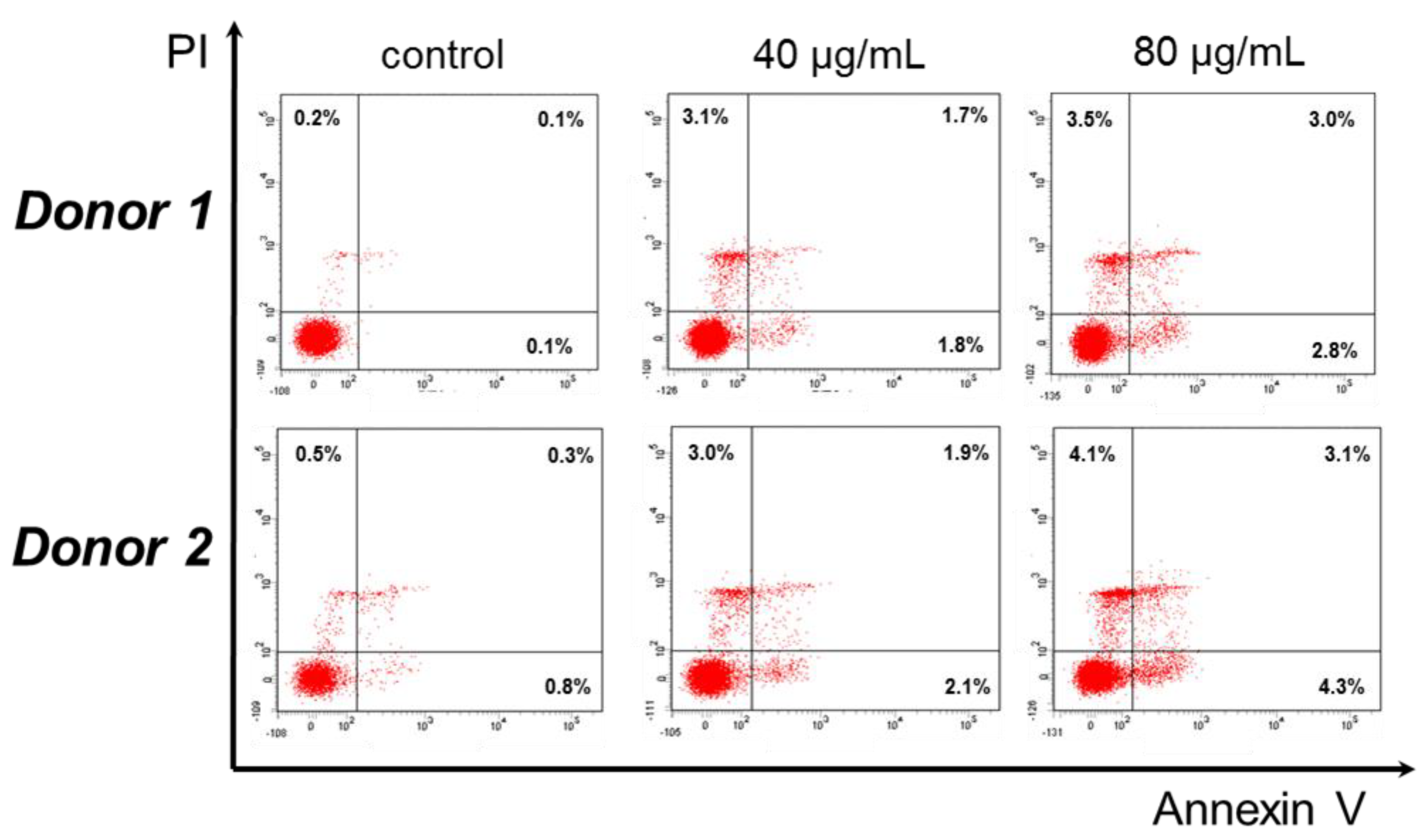
2.6. Cytotoxic Effect against Human Peripheral Blood Mononuclear Cells
3. Results and Discussion
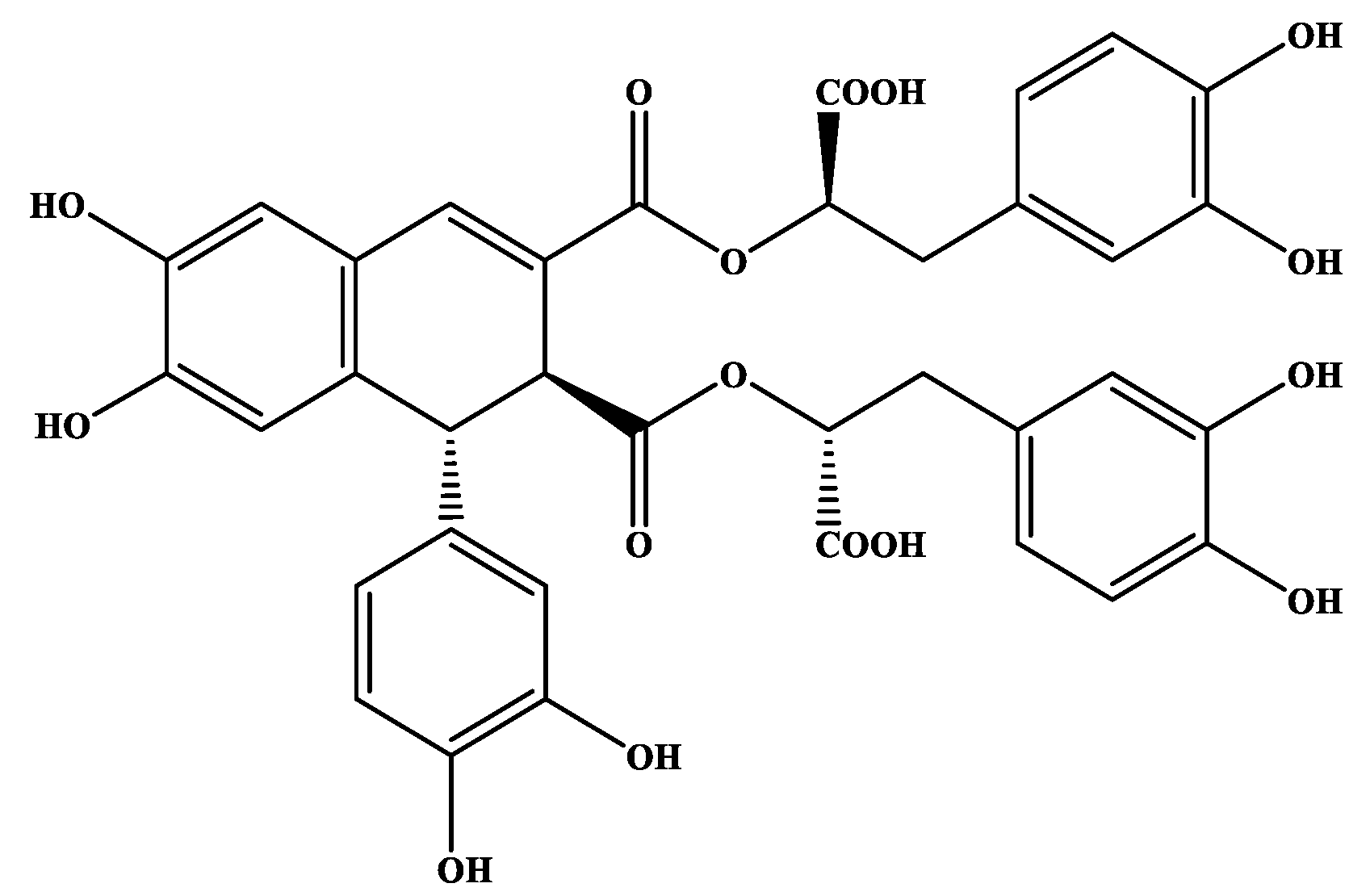
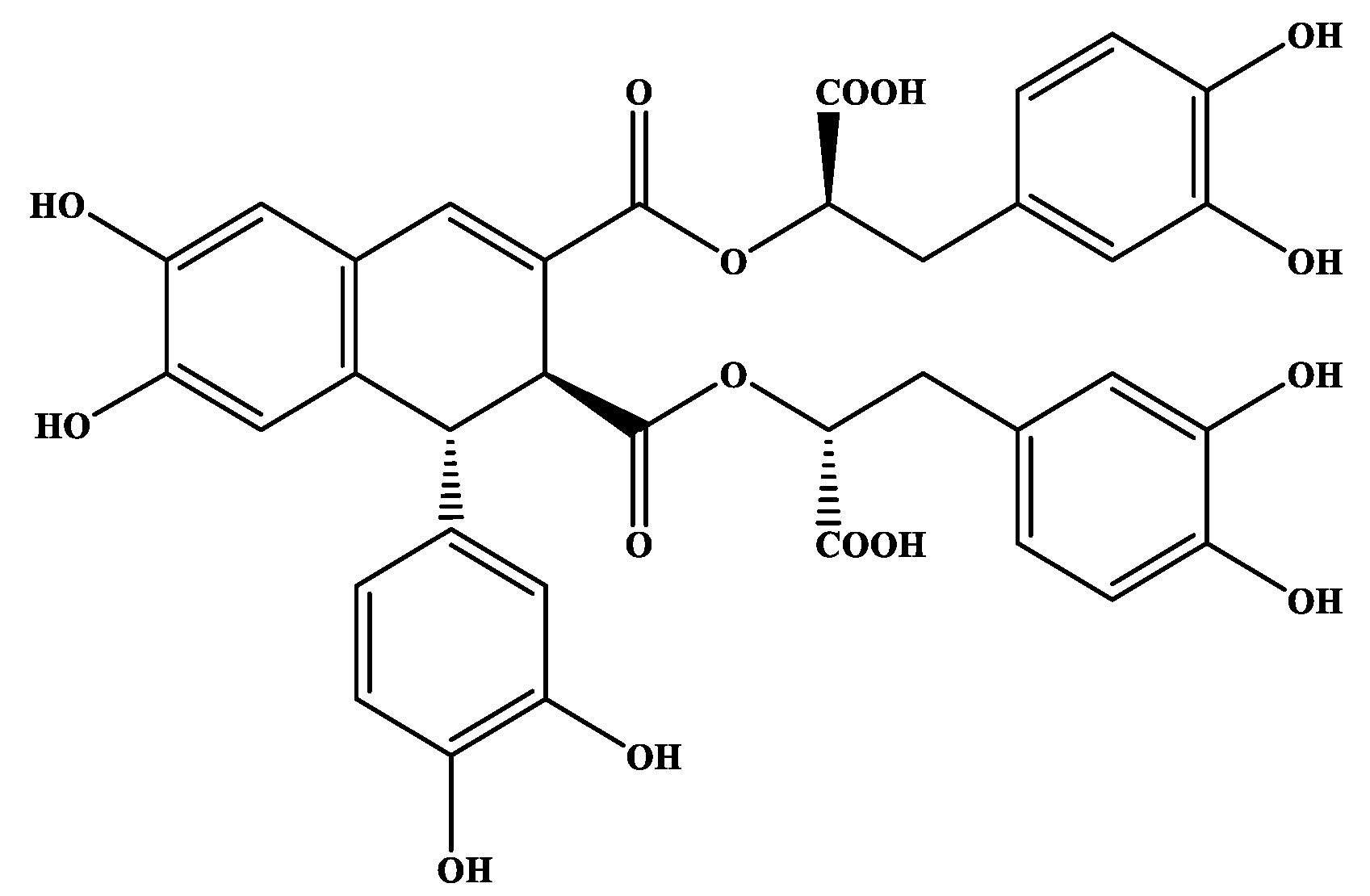
3.1. Secondary Metabolites Isolated from O. sanctum
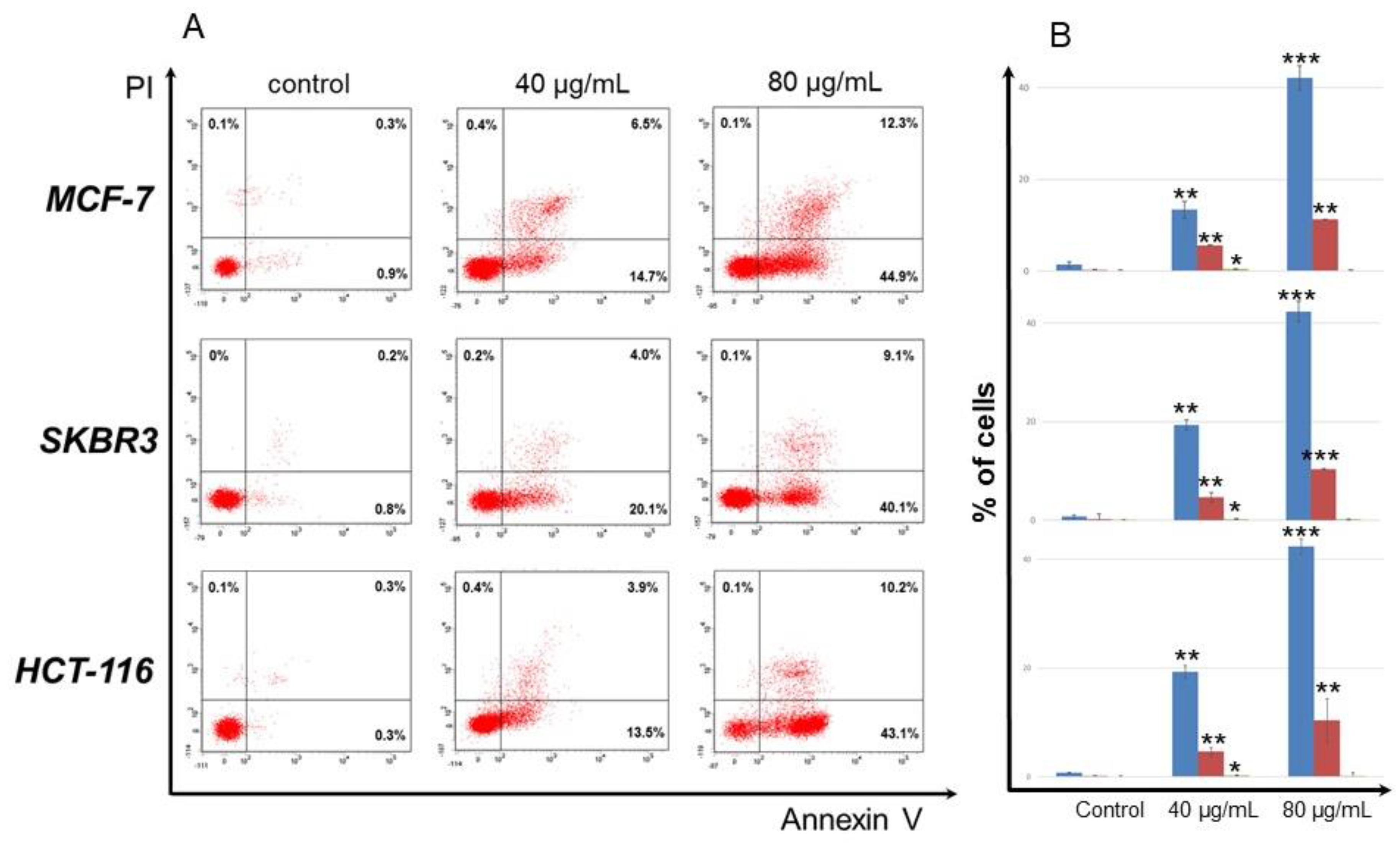
3.2. Antiproliferative Activityod of Secondary Metabolites of O. sanctum
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Prajapati, N.D.; Purohit, S.S.; Sharma, A.K.; Kumar, T.A. Hand Book of Medicinal Plant, 1st ed.; Agrobios: Jodhpur, India, 2003; p. 367. [Google Scholar]
- Cohen, M.M. Tulsi-Ocimum sanctum: A herb for all reasons. J. Ayurveda Integr. Med. 2014, 5, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Pandey, B. Anita. In Economic Botany; Chand and Company Ltd.: New Delhi, India, 1990; p. 294. [Google Scholar]
- Rajeshwari, S. Ocimum sanctum: The Indian home remedy. In Current Medical Scene; Cipla Ltd.: Bombay, India, 1992. [Google Scholar]
- Gupta, S.K.; Prakash, J.; Srivastava, S. Validation of traditional claim of Tulsi, Ocimum sanctum Linn. as a medicinal plant. Indian J. Exp. Biol. 2002, 40, 765–773. [Google Scholar]
- Das, S.K.; Vasudevan, D.M. Tulsi: The Indian holy power plant. Nat. Prod. Radiance 2006, 5, 279–283. [Google Scholar]
- Mondal, S.; Mirdha, B.R.; Mahapatra, S.C. The Science behind sacredness of Tulsi (Ocimum sanctum L.). Indian J. Physiol. Pharmacol. 2009, 53, 291–306. [Google Scholar] [PubMed]
- Pandey, G.; Madhuri, S. Pharmacological activities of Ocimum sanctum (Tulsi): A review. JPSR 2010, 5, 61–66. [Google Scholar]
- Mohan, L.; Amberkar, M.V.; Kumari, M. Ocimum sanctum linn (TULSI)—An overview. JPSR 2011, 7, 51–53. [Google Scholar]
- Pattanayak, Ρ.; Pritishova, B.; Debajyoti, D.; Panda, S.K. Ocimum sanctum Linn. A reservoir plant for therapeutic applications: An overview. Pharmacogn. Rev. 2010, 4, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.; Bhatia, J. Antinociceptive action of Ocimum sanctum (Tulsi) in mice: Possible mechanisms involved. J. Ethnopharmacol. 2003, 88, 293–296. [Google Scholar] [CrossRef]
- Babu, K.; Uma Maheswari, K.C. In vivo studies on the effect of Ocimum sanctum L. leaf extract in mordifying the genotoxicity induced by chromium and mercury in Allium root meristems. J. Environ. Biol. 2006, 27, 93–95. [Google Scholar]
- Narendhirakannan, R.T.; Subramanian, S.; Kandaswamy, M. Biochemical evaluation of antidiabetogenic properties of some commonly used Indian plants on streptozotocin-induced diabetes in experimental rats. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1150–1157. [Google Scholar] [CrossRef]
- Hannan, J.M.; Marenah, L.; Ali, L.; Rokeya, B.; Flatt, P.R.; Abdel-Wahab, Y.H. Ocimum sanctum leaf extracts stimulate insulin secretion from perfusd pancreas, isolated islets and clonal pancreatic beta-cells. J. Endocrinol. 2006, 189, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Grovel, J.K.; Vats, V.; Yadav, S.S. Pterocarpus marsupium extract (Vijayasar) prevented the alteration in metabolic patterns induced in the normal rat by feeding an adequate diet containing fructose as sole carbohydrate. Diabetes Obes. Metab. 2005, 7, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Shirota, O.; Mori, K.; Sekita, S.; Fuchino, H.; Takano, A.; Kuroyanagi, M. Leishmanicidal active constituents from Nepalese medicinal plant Tulsi (Ocimum sanctum L.). Chem. Pharm. Bull. 2009, 57, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, J.; Cohen, M.M. The Clinical Efficacy and Safety of Tulsi in Humans: A Systematic Review of the Literature. Evid. Based Complement. Altern. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.himalayawellness.com/herbfinder/ocimum-tenuiflorum.htm (accessed on 31 January 2019).
- Skaltsa, H.; Tzakou, O.; Singh, M. Polyphenols of Οcimum sanctum L. from Suriname. Pharm. Biol. 1999, 37, 92–94. [Google Scholar] [CrossRef]
- Skaltsa, H.; Couladi, M.; Philianos, S.; Singh, M. Phytochemical Study of the leaves of Ocimum sanctum L. Fitoterapia 1987, 4, 286. [Google Scholar]
- Skaltsa, H.; Tzakou, O.; Loukis, A.; Argyriadou, N. Analyse de l’huile essentielle d’Ocimum sanctum L. Plant. Méd. Phytoth. 1990, 2, 79–81. [Google Scholar]
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013. [Google Scholar] [CrossRef]
- Desai, S.; Desai, D.G.; Kaur, H. Saponins and their biological activities. Pharma Times 2009, 41, 13–16. [Google Scholar]
- Bezerra, D.P.; Militão, G.C.G.; Castro de Morais, M.; Pergentino de Sousa, D. The Dual Antioxidant/Prooxidant Effect of Eugenol and Its Action in Cancer Development and Treatment. Nutrients 2017, 9, 1367. [Google Scholar] [CrossRef]
- Manaharan, T.; Thirugnanasampandan, R.; Jayakumar, R.; Kanthimathi, M.S.; Ramya, G.; Gogul Ramnath, M. Purified essential oil from Ocimum sanctum Linn. triggers the apoptotic mechanism in human breast cancer cells. Pharmacogn. Mag. 2016, 12, S327–S331. [Google Scholar]
- Mabry, T.G.; Markham, K.R.; Thomas, M.B. The Systematic Identification of Flavonoids; Springer Science & Business Media: Berlin/Heidelberg, Germany; New York, NY, USA, 1970. [Google Scholar]
- Coll, J.C.; Bowden, B.F. The application of Vacuum Liquid Chromatography to the separation of terpene mixtures. J. Nat. Prod. 1986, 49, 934–936. [Google Scholar] [CrossRef]
- Neu, R. Chelate von Diarylborsäuren mit aliphatischen oxyalkylaminen als reagenzien für den nachweis von oxyphenyl-benzo-γ-pyronen. Die Naturwissenchaften 1957, 44, 181. [Google Scholar] [CrossRef]
- Mosman, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ioannou, K.; Derhovanessian, E.; Tsakiri, E.; Samara, P.; Kalbacher, H.; Voelter, W.; Trougakos, I.P.; Pawelec, G.; Tsitsilonis, O.E. Prothymosin α and a prothymosin α-derived peptide enhance TH1-type immune responses against defined HER-2/neu epitopes. BMC Immunol. 2013, 14, 43–55. [Google Scholar] [CrossRef]
- Liu, J. Pharmacology of oleanolic acid and ursolic acid. J. Ethnopharmacol. 1995, 49, 57–58. [Google Scholar]
- Μoghaddam, G.M.; Ahmad, F.; Samzadeh-Kermani, A. Biological Activity of Betulinic Acid: A Review. Pharmacol. Pharm. 2012, 3, 119–123. [Google Scholar] [CrossRef]
- Batta, A.K.; Xu, G.; Honda, A.; Miyazaki, T.; Salen, G. Stigmasterol reduces plasma cholesterol levels and inhibits hepatic synthesis and intestinal absorption in the rat. Metabolism 2006, 55, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Ghelardini, C.; Galeotti, N.; Di Cesare, L.; Mazzanti, G.; Bartolini, A. Local anaesthetic activity of β-caryophyllene. Farmaco 2001, 56, 5–7. [Google Scholar] [CrossRef]
- Agata, I.; Hatanp, T.; Okudaq, T.A. Tetrameric derivative of caffeic acid from Rabdosia japonica. Phytochemistry 1989, 28, 2447–2450. [Google Scholar] [CrossRef]
- Inyushkina, V.Y.; Bulgakov, P.V.; Veselova, V.M.; Bryukhanov, M.V.; Zverev, F.Y.; Lampatov, V.V.; Azarova, V.O.; Tchernoded, K.G.; Fedoreyev, A.S.; Zhuravlev, N.Y. High Rabdosiin and Rosmarinic acid production in Eritrichium sericeum callus cultures and the effect of the calli on Masugi-Nephritis in rats. Biosci. Biotechnol. Biochem. 2007, 71, 1286–1293. [Google Scholar] [CrossRef]
- Murata, T.; Miyase, T.; Yoshizaki, F. Hyalurodinase inhibitors from Keiskea japonica. Chem. Pharm. Bull. 2008, 60, 121–128. [Google Scholar] [CrossRef]
- López-Lázaro, M. Distribution and biological activities of the flavonoid luteolin. Mini Rev. Med. Chem. 2009, 9, 31–59. [Google Scholar] [CrossRef] [PubMed]
- Grayer, R.J.; Kite, G.C.; Veitch, N.C.; Eckert, M.; Marin, P.D.; Senanayake, P.; Paton, A.J. Leaf flavonoid glycosides as chemosystematic characters in Ocimum. Biochem. Syst. Ecol. 2002, 30, 327–342. [Google Scholar] [CrossRef]
- Markham, R.K.; Porter, J.L.; Mues, R.; Zinsmeister, D.H.; Brehmm, G.B. Flavonoid variation in the liverwort Conocephalum conicum: Evidence for geographic races. Phytochemistry 1976, 15, 147–150. [Google Scholar] [CrossRef]
- Lu, Y.; Foo, L.Y. Flavonoid and phenolic glycosides from Salvia officinalis. Phytochemistry 2000, 55, 263–267. [Google Scholar] [CrossRef]
- Agrawal, P.K.; Bansal, M.C. Flavonoid glycosides. In Carbon-13 NMR of Flavonoids; Agrawal, P.K., Ed.; Elsevier: Amsterdam, The Netherlands, 1989; pp. 283–364. [Google Scholar]
- Markham, K.R.; Geiger, H. 1H NMR spectroscopy of flavonoids and their glycosides in DMSO-d6. In The Flavonoids, Advances in Research Since 1986; Harborne, J.B., Ed.; Chapman and Hall: London, UK, 1994; pp. 441–497. [Google Scholar]
- Foo, L.Y.; Molan, A.L.; Woodfield, D.R.; McNabb, W.C. The phenols and prodelphinidins of white cover flowers. Phytochemistry 2000, 54, 539–548. [Google Scholar] [CrossRef]
- Yahara, S.; Satoshiro, M.; Nishioka, I.; Nagasawa, T.; Oura, H. Isolation and Characterization of Phenolic Compounds from Coptidis Rhizoma. Chem. Pharm. Bull. 1985, 33, 527. [Google Scholar] [CrossRef]
- Norr, H.; Wagner, H. New constituents from Ocimum sanctum. Planta Med. 1992, 58, 574. [Google Scholar] [CrossRef]
- Βogucki, D.; Charlton, J. A non-enzymatic synthesis of (S)-(-)-rosmarinic acid and a study of a biomimetic route to (+)-rabdosiin. Can. J. Chem. 1997, 75, 1783–1794. [Google Scholar] [CrossRef]
- Agata, I.; Hatano, T.; Nishibe, S.; Okuda, T. Rabdosiin, a new rosmarinic acid dimer with a lignan skeleton, from Rabdosia japonica. Chem. Pharm. Bull. 1988, 36, 3223–3225. [Google Scholar] [CrossRef]
- Nishizawa, M.; Tsuda, M.; Hayashi, K. Two caffeic acid tetramers having enantiomeric phenyldihydronaphthalene moieties from Macrotomia euchroma. Phytochemistry 1990, 29, 2645–2649. [Google Scholar] [CrossRef]
- Yamamoto, H.; Inoue, K.; Yazaki, K. Cafeic acid oligomers in Lithospermum erythrorhizon cell suspension cultures. Phytochemistry 2000, 53, 651–657. [Google Scholar] [CrossRef]
- Ito, H.; Miyazaki, T.; Ono, M.; Sakurai, H. Antiallergic activities of rabdosiin and its related compounds: Chemical and biochemical evaluations. Bioorg. Med. Chem. 1988, 6, 1051–1056. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Nishizawa, M.; Yamagishi, T.; Tanaka, T.; Nonaka, G.; Cosentino, L.M.; Snoder, J.V.; Lee, K. Anti-AIDS agents, 18. Sodium and potassium salts of caffeic acid tetramers from Arnebia euchromaas anti-HIV agents. J. Nat. Prod. 1995, 58, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Dhandayuthapani, S.; Azad, H.; Rathinavelu, A. Apoptosis Induction by Ocimum sanctum extract in LNCaP prostate cancer cells. J. Med. Food 2015, 18, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, P.; Bishayee, A. Ocimum sanctum Linn. (Tulsi): An ethnomedicinal plant for the prevention and treatment of cancer. Anticancer Drugs 2013, 24, 659–666. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 ± SD (in μg/mL) a | IC50 ± SD (in μΜ) | |||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | 6 | 7 | 9 | 11 | 12 | 13 | Extract * | Doxorubicin |
| MCF-7 | 75 ± 2.12 a | 142 ± 3.54 | 141 ± 1.41 | 139 ± 7.78 | 140 ± 12.02 | 140 ± 4.95 | 45 ± 2.12 | 0.092 ± 0.007 |
| SKBR3 | 83 ± 3.54 | ΝΤ | ΝΤ | ΝΤ | ΝΤ | ΝΤ | 46 ± 5.66 | 0.095 ± 0.008 |
| HCT-116 | 84 ± 7.78 | ΝΤ | ΝΤ | ΝΤ | ΝΤ | ΝΤ | 57 ± 14.14 | 0.192 ± 0.029 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flegkas, A.; Milosević Ifantis, T.; Barda, C.; Samara, P.; Tsitsilonis, O.; Skaltsa, H. Antiproliferative Activity of (-)-Rabdosiin Isolated from Ocimum sanctum L. Medicines 2019, 6, 37. https://doi.org/10.3390/medicines6010037
Flegkas A, Milosević Ifantis T, Barda C, Samara P, Tsitsilonis O, Skaltsa H. Antiproliferative Activity of (-)-Rabdosiin Isolated from Ocimum sanctum L. Medicines. 2019; 6(1):37. https://doi.org/10.3390/medicines6010037
Chicago/Turabian StyleFlegkas, Alexandros, Tanja Milosević Ifantis, Christina Barda, Pinelopi Samara, Ourania Tsitsilonis, and Helen Skaltsa. 2019. "Antiproliferative Activity of (-)-Rabdosiin Isolated from Ocimum sanctum L." Medicines 6, no. 1: 37. https://doi.org/10.3390/medicines6010037
APA StyleFlegkas, A., Milosević Ifantis, T., Barda, C., Samara, P., Tsitsilonis, O., & Skaltsa, H. (2019). Antiproliferative Activity of (-)-Rabdosiin Isolated from Ocimum sanctum L. Medicines, 6(1), 37. https://doi.org/10.3390/medicines6010037