
A Comparative Study of Tumor-Specificity and Neurotoxicity between 3-Styrylchromones and Anti-Cancer Drugs


 , ,
, ,  , ,
, ,
Abstract
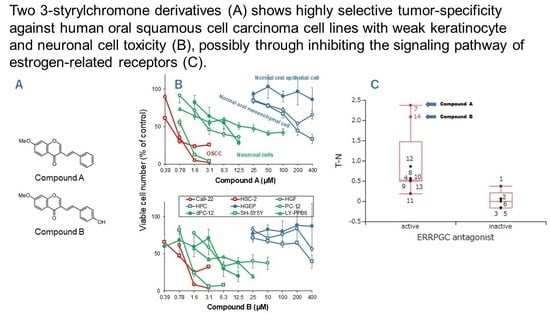
1. Introduction
2. Materials and Methods
2.1. Experimental Materials and Reagents
2.2. Synthesis of Novel Chromone Derivatives
2.3. Cells
2.4. Cytotoxicity Assay
2.5. Tumor-Specificity and Neurotoxicity
2.6. Calculation of Chemical Descriptors
2.7. Statistical Processing
3. Results
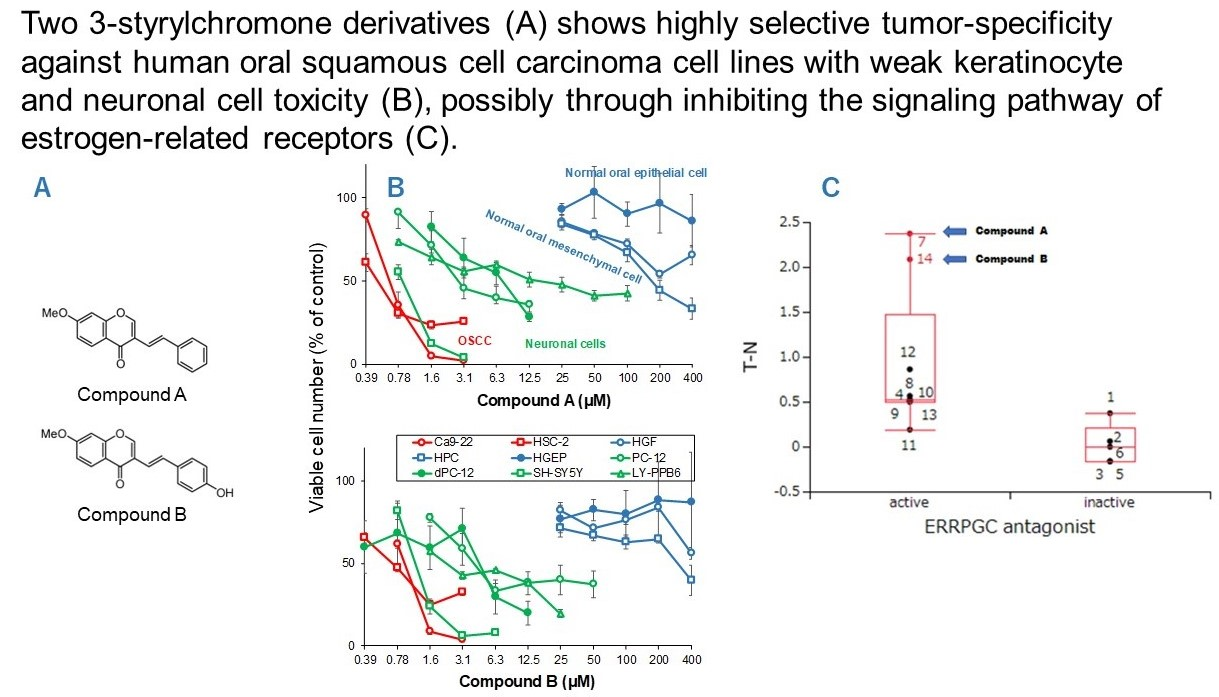
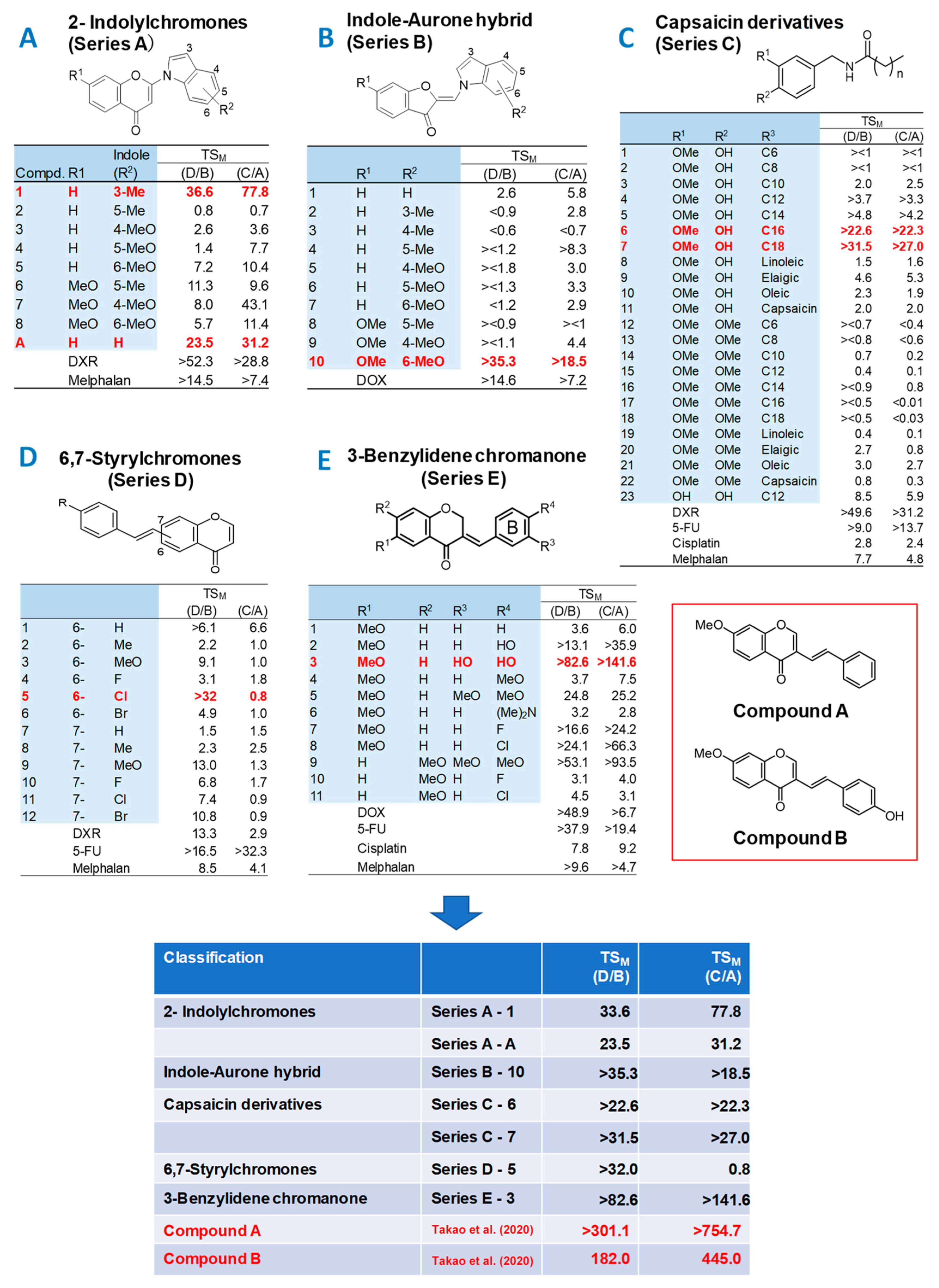
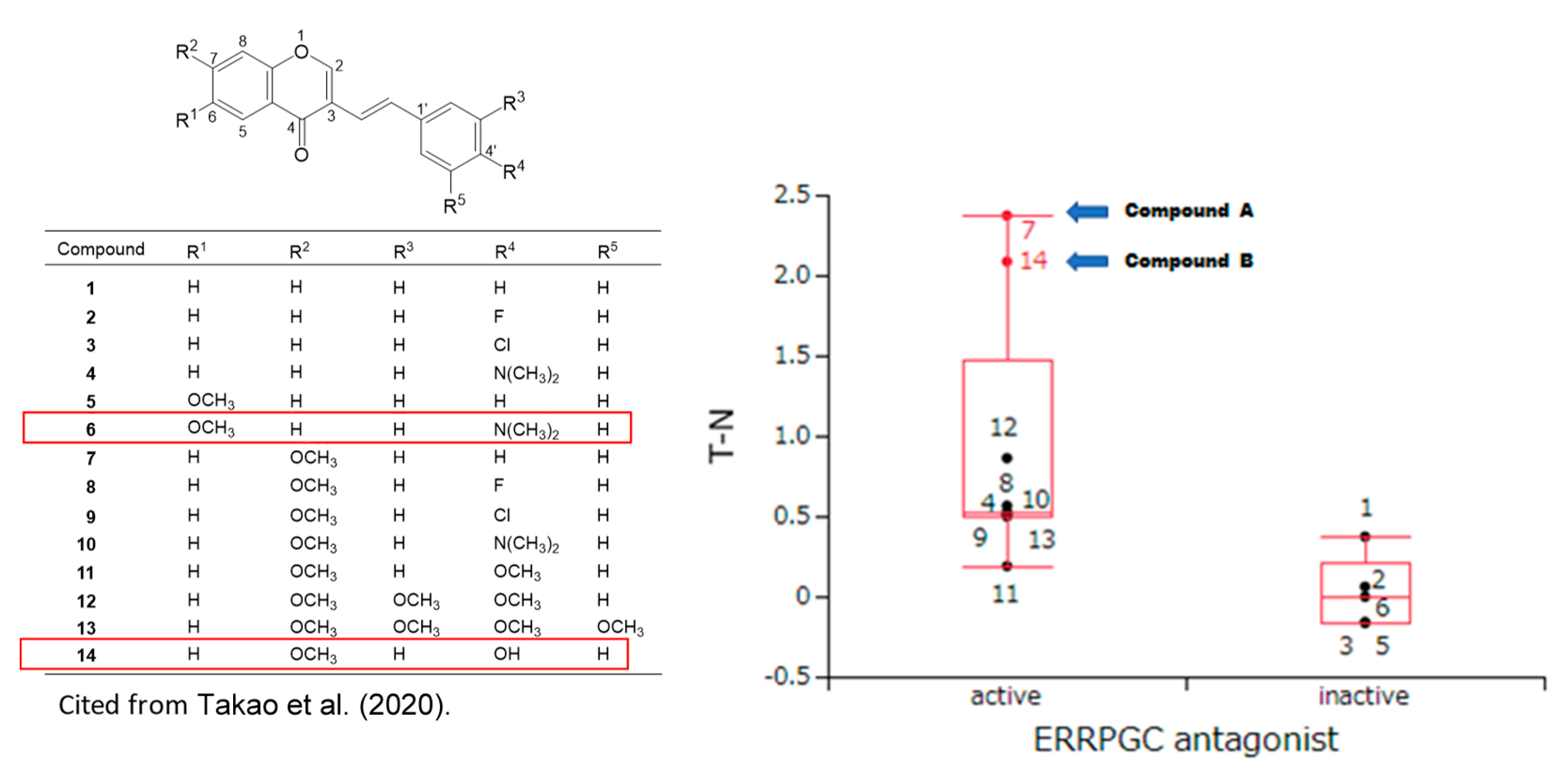
3.1. Continuous Search for New Chromone Derivatives with Higher TSM—Confirmation of Prominent Tumor-Specificity of Compounds A and B
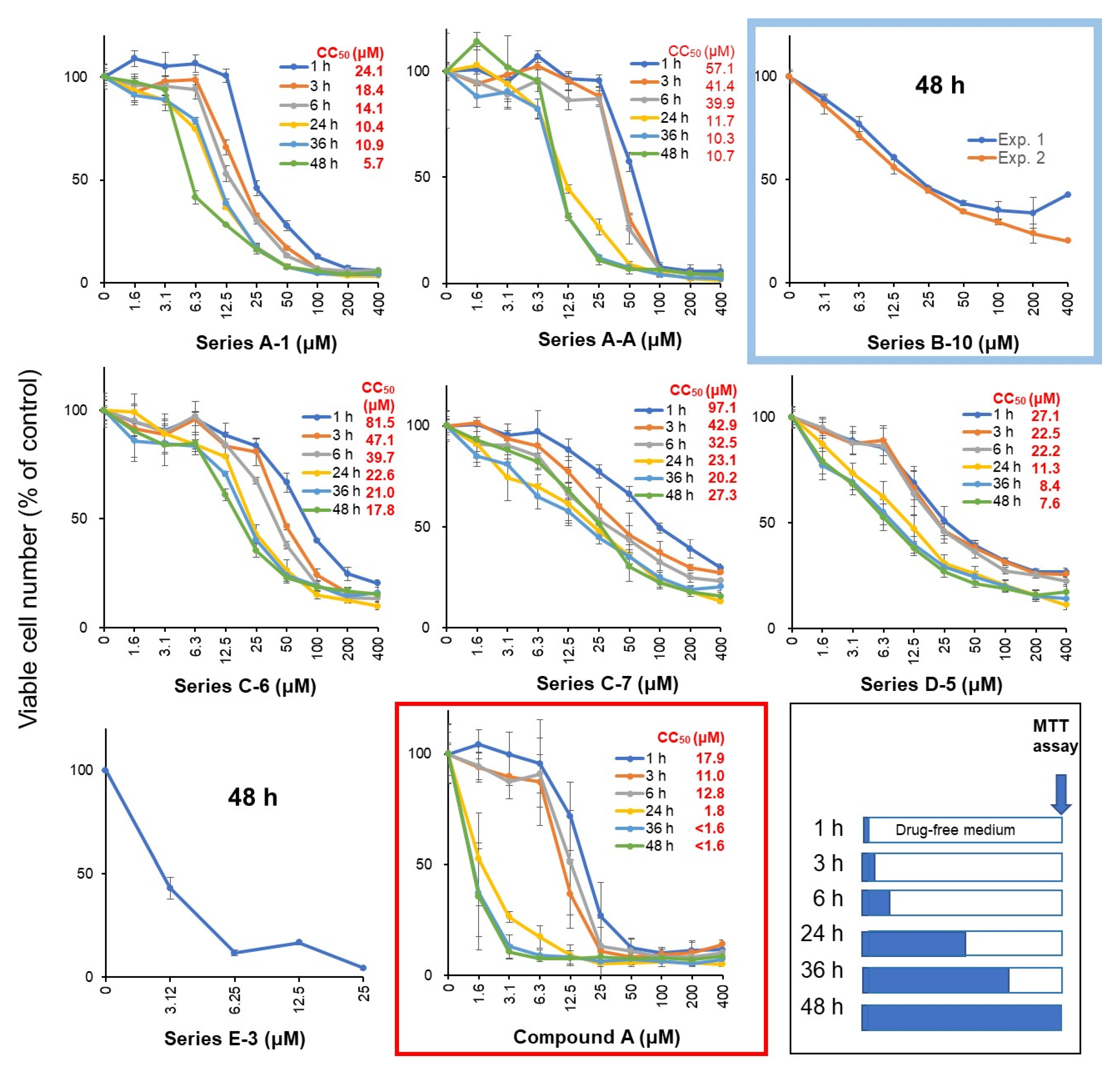
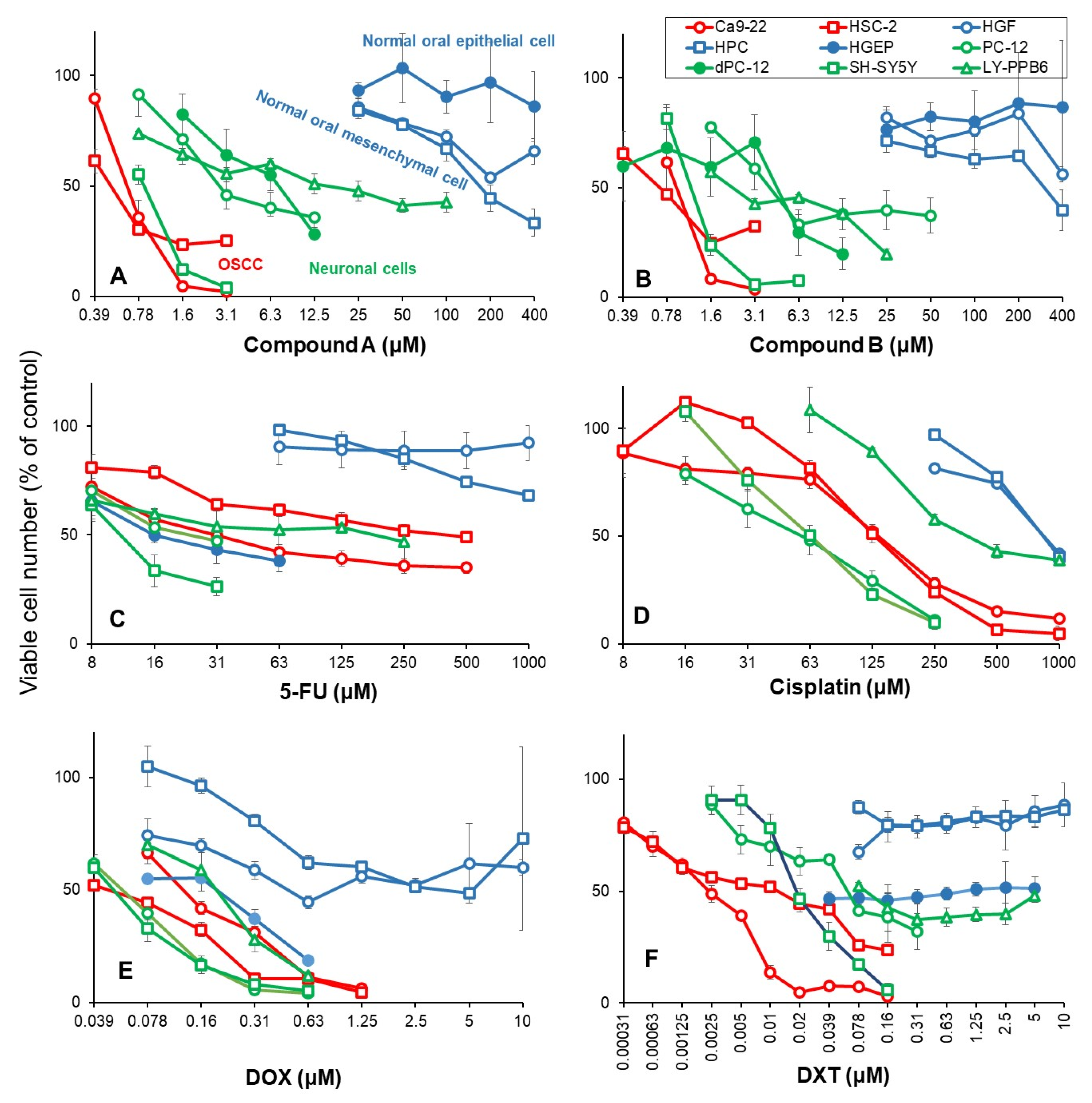
3.2. Rapid Decay of Cell Growth by Chromone Derivatives
3.3. Higher Tumor-Specificities and Less Neurotoxicities of Compounds A and B than Those of Popular Anti-Cancer Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|
| Compound A | Compound B | 5-FU | Cisplatin | DOX | DTX | ||
| Human oral squamous cell carcinoma cells | |||||||
| Ca9-22 | 0.68 | 0.95 | 31.0 | 137.4 | 0.13 | 0.002 | |
| HSC-2 | 0.53 | 0.72 | 411.3 | 130.1 | 0.05 | 0.013 | |
| mean | (A) | 0.60 | 0.83 | 221.1 | 133.7 | 0.09 | 0.008 |
| Human normal oral mesenchymal cells | |||||||
| HGF | >400 | >400 | >1000 | 876.1 | >10 | >10 | |
| HPC | 174.9 | 317.3 | >1000 | 871.3 | 3.7 | >10 | |
| mean | (B) | >287.4 | >358.6 | >1000 | 873.7 | >6.8 | >10 |
| Human normal oral epithelial cells | |||||||
| HGEP | (C) | >400 | >400 | 15.5 | N.D. 1 | 0.21 | 0.039 |
| Undifferentiated neuronal cells | |||||||
| PC-12 | 2.9 | 4.2 | 24.0 | 58.2 | 0.060 | 0.063 | |
| SH-SY5Y | 0.9 | 1.2 | 11.3 | 63.7 | 0.053 | 0.019 | |
| LY-PPB6 | 16.3 | 2.4 | 190.4 | 381.0 | 0.21 | 0.10 | |
| mean | (D) | 6.7 | 2.6 | 75.2 | 167.6 | 0.11 | 0.060 |
| Differentiated PC12 cells | |||||||
| dPC-12 | (E) | 7.5 | 4.7 | ||||
| Tumor-specificity | |||||||
| TSM | (B/A) | >475.6 | >429.9 | >4.5 | 6.5 | >75.9 | >1316.9 |
| TSE | (C/A) | >661.8 | >479.4 | 0.070 | N.D. 1 | 2.3 | 5.1 |
| TSN | (D/A) | 11.1 | 3.1 | 0.34 | 1.3 | 1.2 | 7.9 |
| TSDN | (E/A) | 12.4 | 5.6 | ||||
4. Discussion
4.1. Rational of Using Human Normal Oral Cells as a First Stage of Screening of High TSM Cells
4.2. Failure of 65 Newly Synthesized Chromones Derivatives to Exceed the TSM of Chromones A and B
4.3. Comparison of Anti-Tumor Potentials and Adverse Effects of Chromones A and B with Four Popular Anti-Cancer Drugs
4.3.1. Tumor-Specificity
4.3.2. Keratinocyte Toxicity
4.3.3. Neurotoxicity
4.4. Search for Target Molecules
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elad, S.; Yarom, N.; Zadik, Y.; Kuten-Shorrer, M.; Sonis, S.T. The broadening scope of oral mucositis and oral ulcerative mucosal toxicities of anticancer therapies. CA Cancer J. Clin. 2022, 72, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Murdock, J.L.; Reeves, D.J. Chemotherapy-induced oral mucositis management: A retrospective analysis of MuGard, Caphosol, and standard supportive care measures. J. Oncol. Pharm. Prac. 2020, 26, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Ueno, T.; Yoshida, N.; Akutsu, Y.; Takeuchi, H.; Baba, H.; Matsubara, H.; Kitagawa, Y.; Yoshida, K. Is Oral Mucositis Occurring During Chemotherapy for Esophageal Cancer Patients Correctly Judged? EPOC Observational Cohort Study. Anticancer Res. 2019, 39, 4441–4448. [Google Scholar] [CrossRef]
- Brinkmann, V.; Fritz, G. Prevention of anticancer therapy-induced neurotoxicity: Putting DNA damage in perspective. Neurotoxicology 2022, 91, 1–10. [Google Scholar] [CrossRef]
- Li, T.; Mizrahi, D.; Goldstein, D.; Kiernan, M.C.; Park, S.B. Chemotherapy and peripheral neuropathy. Neurol. Sci. 2021, 42, 4109–4121. [Google Scholar] [CrossRef]
- Fumagalli, G.; Monza, L.; Cavaletti, G.; Rigolio, R.; Meregalli, C. Neuroinflammatory Process Involved in Different Preclinical Models of Chemotherapy-Induced Peripheral Neuropathy. Front. Immunol. 2020, 11, 626687. [Google Scholar] [CrossRef]
- Ehmke, N. Chemotherapy Extravasation: Incidence of and Factors Associated with Events in a Community Cancer Center. Clin. J. Oncol. Nurs. 2021, 25, 680–686. [Google Scholar] [CrossRef]
- Boulanger, J.; Ducharme, A.; Dufour, A.; Fortier, S.; Almanric, K. Management of the extravasation of anti-neoplastic agents. Support. Care Cancer 2015, 23, 1459–1471. [Google Scholar] [CrossRef]
- Bahrami, M.; Karimi, T.; Yadegarfar, G.; Norouzi, A. Assessing the Quality of Existing Clinical Practice Guidelines for Chemotherapy Drug Extravasation by Appraisal of Guidelines for Research and Evaluation II. Iran J. Nurs. Midwifery Res. 2019, 24, 410–416. [Google Scholar] [PubMed]
- Sakagami, H.; Okudaira, N.; Masuda, Y.; Amano, O.; Yokose, S.; Kanda, Y.; Suguro, M.; Natori, T.; Oizumi, H.; Oizumi, T. Induction of Apoptosis in Human Oral Keratinocyte by Doxorubicin. Anticancer Res. 2017, 37, 1023–1029. [Google Scholar]
- Sakagami, H.; Chowdhury, S.A.; Suzuki, F.; Hashimoto, K.; Hatano, H.; Takekawa, H.; Ishihara, M.; Kikuchi, H.; Nishikawa, H.; Taniguchi, S.; et al. Tumor-specific cytotoxic activity of polyphenols, terpenoids, ketones and other synthetic compounds. In Functional Polyphenols and Carotenes with Antioxidative Action; Motohashi, Ed.; Research Signpost: Kerala, India, 2005; pp. 133–176. [Google Scholar]
- Sakagami, H. Biological activities and possible dental application of three major groups of polyphenols. J. Pharmacol. Sci. 2014, 126, 92–106. [Google Scholar] [CrossRef]
- Sugita, Y.; Takao, K.; Uesawa, Y.; Nagai, J.; Iijima, Y.; Sano, M.; Sakagami, H. Development of Newly Synthesized Chromone Derivatives with High Tumor Specificity against Human Oral Squamous Cell Carcinoma. Medicines 2020, 7, 50. [Google Scholar] [CrossRef]
- Takao, K.; Hoshi, K.; Sakagami, H.; Shi, H.; Bandow, K.; Nagai, J.; Uesawa, Y.; Tomomura, A.; Tomomura, M.; Sugita, Y. Further Quantitative Structure-Cytotoxicity Relationship Analysis of 3-Styrylchromones. Anticancer Res. 2020, 40, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, H.; Okudaira, N.; Uesawa, Y.; Takao, K.; Kagaya, H.; Sugita, Y. Quantitative Structure-Cytotoxicity Relationship of 2-Azolylchromones. Anticancer Res. 2018, 38, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.; Noguchi, K.; Hashimoto, Y.; Shirahata, A.; Sugita, Y. Synthesis and evaluation of fatty acid amides on the N-oleoylethanolamide-like activation of peroxisome proliferator activated receptor α. Chem. Pharm. Bull. 2015, 63, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Takao, K.; Shiori, U.; Kamauchi, H.; Sugita, Y. Design, synthesis and evaluation of 2-(indolylmethylidene)-2,3-dihydro-1-benzofuran-3-one and 2-(indolyl)-4H-chromen-4-one derivatives as novel monoamine oxidases inhibitors. Bioorg. Chem. 2019, 87, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, H.; Uesawa, Y.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Sugita, Y. Quantitative Structure-Cytotoxicity Relationship of Oleoylamides. Anticancer Res. 2015, 35, 5341–5351. [Google Scholar] [PubMed]
- Patonay, T.; Vasas, A.; Kiss-Szikszai, A.; Silva, A.M.S.; Cavaleiro, J.A.S. Efficient Synthesis of Chromones with Alkenyl Functionalities by the Heck Reaction. Aust. J. Chem. 2010, 63, 1582–1593. [Google Scholar] [CrossRef]
- Uesawa, Y.; Sakagami, H.; Kagaya, H.; Yamashita, M.; Takao, K.; Sugita, Y. Quantitative Structure-cytotoxicity Relationship of 3-Benzylidenechromanones. Anticancer Res. 2016, 36, 5803–5812. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.Y.; Yue, X.H.; Dong, M.J.; Li, J.; Zhang, C.P. Assessment of neoadjuvant chemotherapy with docetaxel, cisplatin, and fluorouracil in patients with oral cavity cancer. Cancer Med. 2023, 12, 2417–2426. [Google Scholar] [CrossRef]
- Takao, K.; Takemura, Y.; Nagai, J.; Kamauchi, H.; Hoshi, K.; Mabashi, R.; Uesawa, Y.; Sugita, Y. Synthesis and biological evaluation of 3-styrylchromone derivatives as selective monoamine oxidase B inhibitors. Bioorg. Med. Chem. 2021, 42, 116255. [Google Scholar] [CrossRef]
- Kantoh, K.; Ono, M.; Nakamura, Y.; Nakamura, Y.; Hashimoto, K.; Sakagami, H.; Wakabayashi, H. Hormetic and anti-radiation effects of tropolone-related compounds. Vivo 2010, 24, 843–851. [Google Scholar]
- Sakagami, H.; Shi, H.; Bandow, K.; Tomomura, M.; Tomomura, A.; Horiuchi, M.; Fujisawa, T.; Oizumi, T. Search of Neuroprotective Polyphenols Using the “Overlay” Isolation Method. Molecules 2018, 23, 1840. [Google Scholar] [CrossRef] [PubMed]
- Tagai, N.; Tanaka, A.; Sato, A.; Uchiumi, F.; Tanuma, S.I. Low Levels of Brain-Derived Neurotrophic Factor Trigger Self-aggregated Amyloid β-Induced Neuronal Cell Death in an Alzheimer’s Cell Model. Biol. Pharm. Bull. 2020, 43, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Yamate, J.; Sakamori, M.; Kuwamura, M.; Kotani, T. Neoplastic and non-neoplastic cell lines from a malignant peripheral nerve sheath tumour of the cervix of a rat. J. Comp. Pathol. 2007, 137, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, K.; Wu, R.; Uesawa, Y. A Toxicity Prediction Tool for Potential Agonist/Antagonist Activities in Molecular Initiating Events Based on Chemical Structures. Int. J. Mol. Sci. 2020, 21, 7853. [Google Scholar] [CrossRef] [PubMed]
- Iijima, Y.; Bandow, K.; Sano, M.; Hino, S.; Kaneko, T.; Horie, N.; Sakagami, H. In Vitro Assessment of Antitumor Potential and Combination Effect of Classical and Molecular-targeted Anticancer Drugs. Anticancer Res. 2019, 39, 6673–6684. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Lee, J.H.; Kim, S.H.; Um, J.W. Oxaliplatin-based adjuvant chemotherapy rather than fluorouracil-based chemotherapy in rectal cancer is more efficient to decrease distant metastasis and increase survival after preoperative chemoradiotherapy and surgery: A meta-analysis. Int. J. Color. Dis. 2022, 37, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, H.; Shimada, C.; Kanda, Y.; Amano, O.; Sugimoto, M.; Ota, S.; Soga, T.; Tomita, M.; Sato, A.; Tanuma, S.-I.; et al. Effects of 3-styrylchromones on metabolic profiles and cell death in oral squamous cell carcinoma cells. Toxicol. Rep. 2015, 2, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Sakagami, H.; Shi, H.; Abe, T.; Tamura, N.; Takeshima, H.; Horie, N.; Kaneko, T.; Shiratsuchi, H.; Kaneko, T. Partial Protection of Paclitaxel-induced Neurotoxicity by Antioxidants. Vivo 2018, 32, 745–752. [Google Scholar] [CrossRef]
- Okudan, N.; Belviranlı, M.; Sezer, T. Potential Protective Effect of Coenzyme Q10 on Doxorubicin-Induced Neurotoxicity and Behavioral Disturbances in Rats. Neurochem. Res. 2022, 47, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Majhi, S.; Singh, L.; Yasir, M. Evaluation of Ameliorative Effect of Quercetin and Candesartan in Doxorubicin-Induced Cardiotoxicity. Vasc. Health Risk Manag. 2022, 18, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhao, C.; Xu, B.; Huang, Y.; Liu, C. Effect of docetaxel on mechanical properties of ovarian cancer cells. Exp. Cell Res. 2021, 408, 112853. [Google Scholar] [CrossRef]
- Yu, B.; Liu, Y.; Luo, H.; Fu, J.; Li, Y.; Shao, C. Androgen receptor splicing variant 7 (ARV7) inhibits docetaxel sensitivity by inactivating the spindle assembly checkpoint. J. Biol. Chem. 2021, 296, 100276. [Google Scholar] [CrossRef]
- Tanuma, S.I.; Oyama, T.; Okazawa, M.; Yamazaki, H.; Takao, K.; Sugita, Y.; Amano, S.; Abe, T.; Sakagami, H. A Dual Anti-Inflammatory and Anti-Proliferative 3-Styrylchromone Derivative Synergistically Enhances the Anti-Cancer Effects of DNA-Damaging Agents on Colon Cancer Cells by Targeting HMGB1-RAGE-ERK1/2 Signaling. Int. J. Mol. Sci. 2022, 23, 3426. [Google Scholar] [CrossRef]
- Saranya, R.; Chandini, R.; Mohideen, K.; Adtani, P.N.; Subramani, V.; Balasubramaniam, M.; Adtani, P. Expression of Sex Hormones in Oral Squamous Cell Carcinoma: A Systematic Review on Immunohistochemical Studies. Cureus 2022, 14, e25384. [Google Scholar] [CrossRef] [PubMed]
- Doll, C.; Bestendonk, C.; Kreutzer, K.; Neumann, K.; Pohrt, A.; Trzpis, I.; Koerdt, S.; Dommerich, S.; Heiland, M.; Raguse, J.D.; et al. Prognostic Significance of Estrogen Receptor Alpha in Oral Squamous Cell Carcinoma. Cancers 2021, 13, 5763. [Google Scholar] [CrossRef] [PubMed]




| Signaling Pathway Investigated | p-Value |
|---|---|
| ERRPGC_ant (estrogen-related receptor with PGC antagonist) | 0.0307 1 |
| CAR_ant (constitutive androstane receptor antagonist) | 0.1385 |
| RAR_ant (retinoic acid receptor antagonist) | 0.1472 |
| ARant_ago (androgen receptor with antagonist agonist) | 0.1567 |
| TSHR_ago (thyroid stimulating hormone receptor agonist) | 0.1619 |
| H2AX_ago (histone variant H2AX agonist) | 0.1825 |
| GR_ant (glucocorticoid receptor antagonist) | 0.2328 |
| TRHR_ago (thyrotropin-releasing hormone receptor agonist) | 0.2913 |
| TR_ant (thyroid receptor antagonist) | 0.3295 |
| PPARg_ant (peroxisome proliferator-activated receptor gamma antagonist) | 0.3348 |
| CaspC_ind (caspase-3/7 in CHO-K1 inducer) | 0.3508 |
| HDAC_ant (histone deacetylase antagonist) | 0.3539 |
| TRHR_ant (thyrotropin-releasing hormone receptor antagonist) | 0.3812 |
| ERb_ant (estrogen receptor beta antagonist) | 0.4774 |
| RXR_ago (retinoid X receptor-alpha agonist) | 0.4933 |
| FXR_ago (farnesoid-X-receptor agonist) | 0.5155 |
| TSHR_ant (thyroid stimulating hormone receptor antagonist) | 0.5234 |
| ERRPGC_ago (estrogen-related receptor with PGC agonist) | 0.5461 |
| TGFb_ant (transforming growth factor beta antagonist) | 0.5573 |
| CaspH_ind (caspase-3/7 in HepG2 inducer) | 0.5673 |
| ERlbd_ago (estrogen receptor alpha lbd agonist) | 0.6411 |
| ROR_ant (retinoid-related orphan receptor gamma antagonist) | 0.6582 |
| PPARd_ant (peroxisome proliferator-activated receptor delta antagonist) | 0.6642 |
| PR_ant (progesterone receptor antagonist) | 0.724 |
| ERsr_ago (endoplasmic reticulum stress response agonist) | 0.8102 |
| ARlbd_ant (androgen receptor lbd antagonist) | 0.8227 |
| ERR_ago (estrogen-related receptor agonist) | 0.8391 |
| MMP_disr (mitochondrial membrane potential disruptor) | 0.8582 |
| ARfull_ant (androgen receptor full antagonist) | 0.8582 |
| GR_ago (glucocorticoid receptor agonist) | 0.8849 |
| HSR_act (heat shock response activator) | 0.8966 |
| CAR_ago (constitutive androstane receptor agonist) | 0.9127 |
| ARfulls_ant (androgen receptor with stimulator antagonist) | 0.9127 |
| ATAD5_ind (ATAD5 genotoxic inducer) | 0.9318 |
| NFkB_ago (NFkB agonist) | 0.9318 |
| AhR_ago (aryl hydrocarbon receptor agonist) | 0.933 |
| AP1_ago (activator protein-1 agonist) | 0.9366 |
| PPARg_ago (peroxisome proliferator-activated receptor gamma agonist) | 0.9472 |
| p53_ago (p53 agonist) | - |
| ARlbd_ago (androgen receptor lbd agonist) | - |
| ERlbd_ant (estrogen receptor alpha lbd antagonist) | - |
| ERfull_ant (estrogen receptor alpha full antagonist) | - |
| Arom_ant (aromatase antagonist) | - |
| ARE_ago (antioxidant response element agonist) | - |
| PPARd_ago (peroxisome proliferator-activated receptor delta agonist) | - |
| FXR_ant (farnesoid-X-receptor antagonist) | - |
| VDR_ago (vitamin D receptor agonist) | - |
| VDR_ant (vitamin D receptor antagonist) | - |
| HIF1_ago (hypoxia agonist) | - |
| ERfulls_ant (estrogen receptor alpha with stimulator antagonist) | - |
| Shh_ago (sonic hedgehog signaling agonist) | - |
| ERaant_ago (estrogen receptor alpha with antagonist–agonist) | - |
| Shh_ant (sonic hedgehog signaling antagonist) | - |
| TSHR_agoant (thyroid stimulating hormone receptor agonist–antagonist) | - |
| ERb_ago (estrogen receptor beta agonist) | - |
| ERR_ant (estrogen-related receptor antagonist) | - |
| PXR_ago (human pregnane X receptor agonist) | - |
| TGFb_ago (transforming growth factor beta agonist) | - |
| PR_ago (progesterone receptor agonist) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, T.; Sakagami, H.; Amano, S.; Uota, S.; Bandow, K.; Uesawa, Y.; U, S.; Shibata, H.; Takemura, Y.; Kimura, Y.; et al. A Comparative Study of Tumor-Specificity and Neurotoxicity between 3-Styrylchromones and Anti-Cancer Drugs. Medicines 2023, 10, 43. https://doi.org/10.3390/medicines10070043
Abe T, Sakagami H, Amano S, Uota S, Bandow K, Uesawa Y, U S, Shibata H, Takemura Y, Kimura Y, et al. A Comparative Study of Tumor-Specificity and Neurotoxicity between 3-Styrylchromones and Anti-Cancer Drugs. Medicines. 2023; 10(7):43. https://doi.org/10.3390/medicines10070043
Chicago/Turabian StyleAbe, Tomoyuki, Hiroshi Sakagami, Shigeru Amano, Shin Uota, Kenjiro Bandow, Yoshihiro Uesawa, Shiori U, Hiroki Shibata, Yuri Takemura, Yu Kimura, and et al. 2023. "A Comparative Study of Tumor-Specificity and Neurotoxicity between 3-Styrylchromones and Anti-Cancer Drugs" Medicines 10, no. 7: 43. https://doi.org/10.3390/medicines10070043
APA StyleAbe, T., Sakagami, H., Amano, S., Uota, S., Bandow, K., Uesawa, Y., U, S., Shibata, H., Takemura, Y., Kimura, Y., Takao, K., Sugita, Y., Sato, A., Tanuma, S.-i., & Takeshima, H. (2023). A Comparative Study of Tumor-Specificity and Neurotoxicity between 3-Styrylchromones and Anti-Cancer Drugs. Medicines, 10(7), 43. https://doi.org/10.3390/medicines10070043