
Multifactorial Origin of Neurodevelopmental Disorders: Approaches to Understanding Complex Etiologies



Abstract
:1. Introduction
2. Environmental Chemicals and Neurobehavioral Toxicity: Key Uncertainties and Research Needs
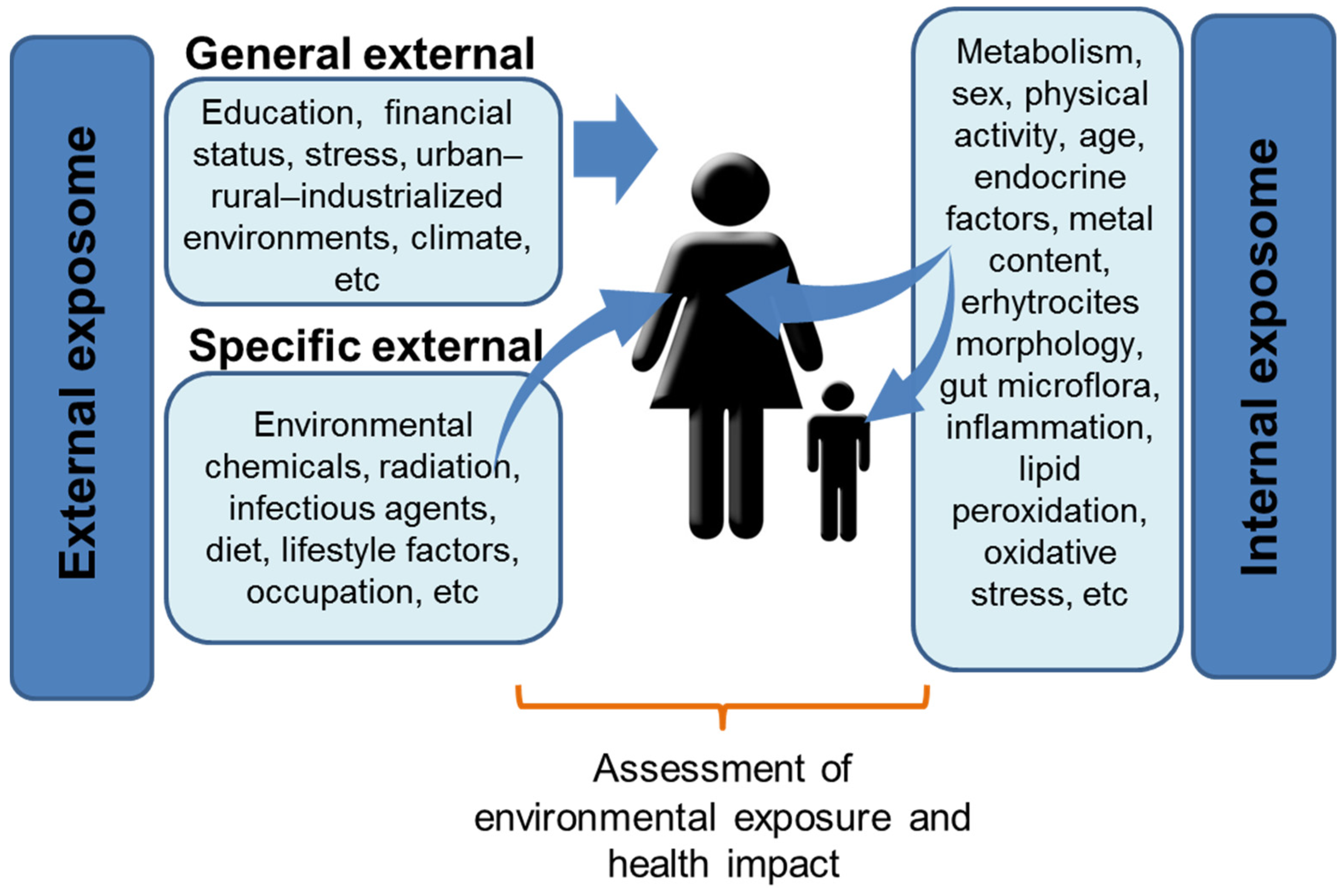
3. Facing Complexity: The Exposome Concept
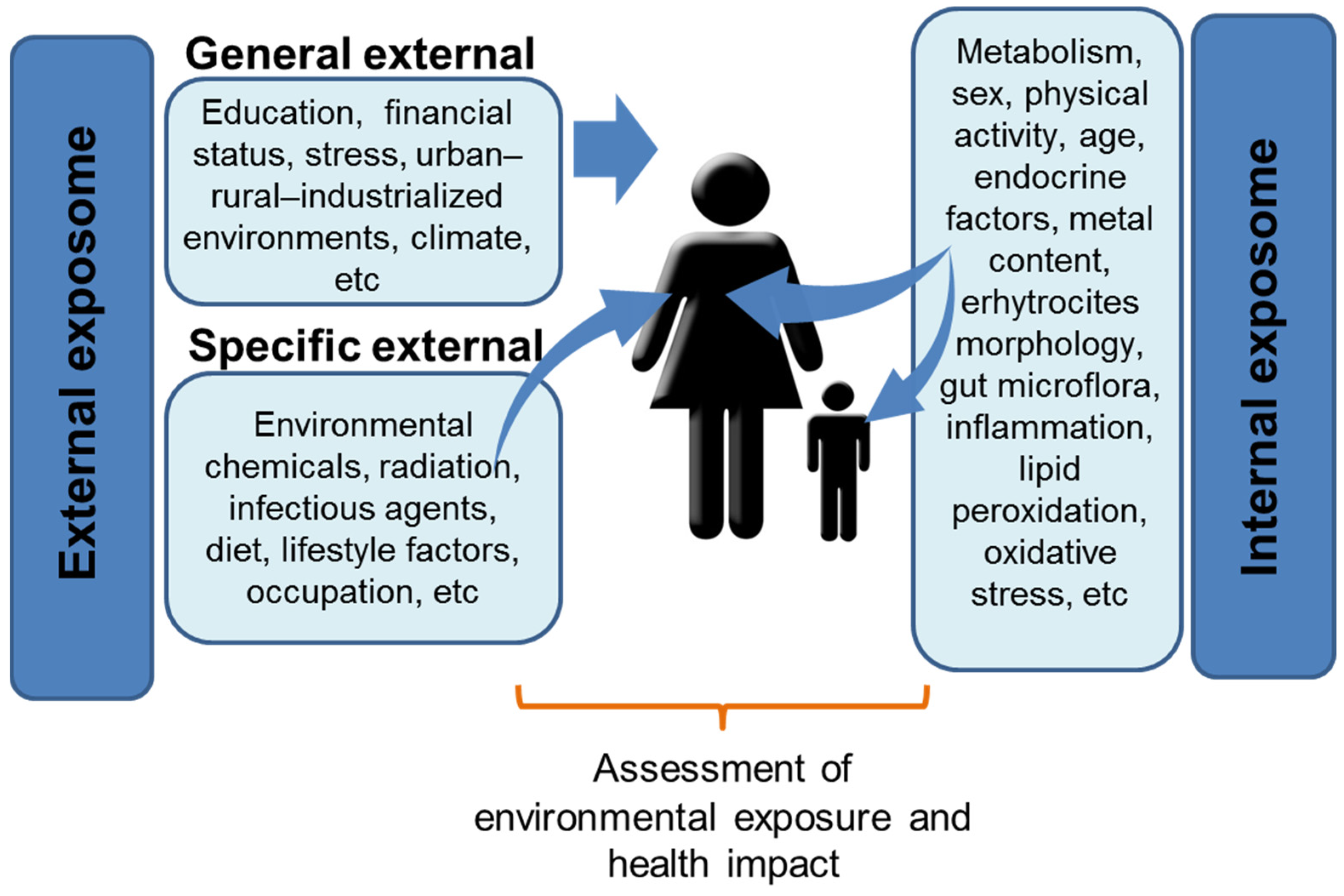
4. Multifactorial Origin of NDDs: The ASD and ADHD Examples
4.1. Etiology of ASD: The Environment Contribution
4.2. Etiology of ADHD: The Environment Contribution
5. Gene Polymorphisms as a Source of Vulnerability
5.1. Gene Polymorphisms Increase Susceptibility to Mercury
5.2. Vulnerability to Organophosphate Pesticides and Paraoxonase 1

{kind=link}
| Single nucleotid polymorphism (SNP) | Hg source | Size of the study (age) | β regression coefficient | Ref |
|---|---|---|---|---|
| ATP Binding Cassette (ABC) transporter genes | Environmental exposure (fish diet) | 1651 (birth cohort) | Interaction Log2 fish intake*genotype on log2 cord blood MeHg | [143] |
| rs2032582: TT vs GG β = −0.49 (CI = −0.71 to −0.26) | ||||
| rs11075290: TT vs CC β = −0.28 (CI = −0.51 to −0.06) | ||||
| rs2273697: GA+AA vs GG β = 0.16 (CI = 0.01 to 0.32) | ||||
| Trasferrin (TF), Brain Derived Neurotrophic Factor (BDNF) | Not known (Avon longitudinal study of parents and children) | 1135 (0–8y) | Log10 cord blood MeHg on WISC-III Performance IQ in selected genotypes | [45] |
| TF rs3811647: AA β = −22.7 (CI = −44.0 to −1.5) | ||||
| BDNF rs2049046: AA β = −13.7 (CI = −26.9 to −0.4) | ||||
| Metallothionein (MT1M, MT2A) | Dental amalgalm tooth filling | 120 boys, 118 girls (8–12y) | Loge urinary Hg on RAVLT8 in selected genotypes | [139] |
| MT1M rs2270837: GA+AA β = −2.11 (SE = 0.74) | ||||
| MT2A rs10636: GC+CC β = −1.96 (SE = 0.86) | ||||
| Loge urinary Hg on Visual Spatial—Digit Symbol in selected genotypes | ||||
| MT2A rs10636: GC+CC β = −12.9 (SE = 4.77) | ||||
| Glutathione related genes: Glutamyl-cysteine-ligases modifier subunit (GCLM) Glutathione-S-transferase μ1 (GSTM1) | Environmental exposure (fish diet) | 400 (adults) | Genotype on Loge blood Hg | [156] |
| GCLM-588: TT β = −0.32 (p = 0.017) | ||||
| GSTM1*0: Homozygous β = 0.20 (p = 0.017) | ||||
| Genotype on Loge hair Hg | ||||
| GCLM-588: TT β = −0.33 (p = 0.009) | ||||
| GSTM1*0: Homozygous β = 0.20 (p = 0.013) | ||||
| Apolipoprotein E (APOE) | Environmental exposure (fish diet) | 180 (0–2y) | Loge cord blood MeHg on CDIIT cognition score in selected genotypes | [141] |
| APOE: ε4 carrier β = −8.47 (CI = −16.1 to −0.84) | ||||
| Loge cord blood MeHg on CDIIT social score in selected genotypes | ||||
| APOE: ε4 carrier β = −11.02 (CI = −20.85 to −1.19) | ||||
| Loge cord blood MeHg on CDIIT whole test score in selected genotypes | ||||
| APOE: ε4 carrier β = −10.45 (CI = −17.36 to −3.54) | ||||
| Coproporphyrinogen oxidase (CPOX); Metallothionine (MT1M, MT2A); Catechol-O-methyltransferase (COMT); Tryptophan 2,3-dioxygenase (TDO2) | Dental amalgalm tooth filling | 120 boys, 118 girls (8–12 y) | Loge urinary Hg on neurobehavioral tests in selected genotypes (boys) | [144] |
| CPOX rs1131857: AC+CC | ||||
| Attention: β range = −2.45 (SE = 0.97) to −18.1 (SE = 4.89) | ||||
| Visual-Spatial: β = −20.7 (SE = 6.08) | ||||
| Executive function: β = 22.26 (SE = 7.03) | ||||
| Learning & Memory: β range = −1.78 (SE = 0.80) to −2.24 (SE = 0.82) | ||||
| Motor: β range = −3.46 (SE = 1.65) to −7.16 (SE = 2.9) | ||||
| MT1M rs2270837: GA+AA | ||||
| Learning & Memory: β range = −1.56 (SE = 0.53) to −2.11 (SE = 0.74) | ||||
| MT2A rs10636: GC+CC | ||||
| Visual-Spatial: β range = −8.21 (SE = 2.62) to −12.9 (SE = 4.77) | ||||
| Learning & Memory: β range = −1.96 (SE = 0.86) to −6.49 (SE = 2.72) | ||||
| COMT rs4680: GA+AA | ||||
| Attention: β = −5.05 (SE = 1.95) | ||||
| Visual-Spatial: β range = −16.49 (SE = 5.52) to −32.46 (SE = 11.28) | ||||
| COMT rs4633: CT+TT | ||||
| Attention: β range = −4.58 (SE = 1.84) to −13.79 (SE = 6.23) | ||||
| Visual-Spatial: β range = −16.19 (SE = 5.53) to −35.71 (SE = 10.87) | ||||
| COMT rs6269: AG+GG | ||||
| Visual-Spatial: β range = −8.48 (SE = 3.83) to −17.92 (SE = 6.8) | ||||
| TDO2 rs3755907: GA+AA | ||||
| Attention: β = −14.66 (SE = 7.12) | ||||
| Visual-Spatial: β = −0.16 (SE = 0.06) | ||||
| Motor: β = −9.36 (SE = 3.90) |
6. Susceptibility to Environmental Chemicals: A Key Role for Epigenetics?
7. Not in Our Genes: Socioeconomic Factors Modulate Environmental Chemicals’ Toxicity
8. Unraveling Complexity: The Need for Experimental Models
8.1. Gene Polymorphisms and Environmental Contaminants: Mouse Studies
8.2. Stress and Environmental Contaminants in Animal Models
9. Designing in Vivo Experiments to Model Complexity
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Andersen, S.L. Trajectories of brain development: Point of vulnerability or window of opportunity? Neurosci. Biobehav. Rev. 2003, 27, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Ladher, R.K.; Wright, T.J.; Moon, A.M.; Mansour, S.L.; Schoenwolf, G.C. FGF8 initiates inner ear induction in chick and mouse. Genes Dev. 2005, 19, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Rakic, P.; Cameron, R.S.; Komuro, H. Recognition, adhesion, transmembrane signaling and cell motility in guided neuronal migration. Curr. Opin. Neurobiol. 1994, 4, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Bystron, I.; Blakemore, C.; Rakic, P. Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 2008, 9, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Levitt, P. Structural and functional maturation of the developing primate brain. J. Pediatr. 2003, 143, S35–S45. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Hasan, K.M.; Trivedi, R.; Pradhan, M.; Das, V.; Parikh, N.A.; Narayana, P.A. Diffusion tensor imaging of the developing human cerebrum. J. Neurosci. Res. 2005, 81, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Tau, G.Z.; Peterson, B.S. Normal development of brain circuits. Neuropsychopharmacology 2010, 35, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Shonkoff, J.P. From Neurons to Neighborhoods: The Science of Early Childhood Development; National Academy Press: Washington, DC, USA, 2000. [Google Scholar]
- Hubel, D.H.; Wiesel, T.N. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J. Physiol. 1970, 206, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Markham, J.A.; Greenough, W.T. Experience-driven brain plasticity: Beyond the synapse. Neuron Glia Biol. 2004, 1, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Baghurst, P.A.; Robertson, E.F.; McMichael, A.J.; Vimpani, G.V.; Wigg, N.R.; Roberts, R.R. The Port Pirie Cohort Study: Lead effects on pregnancy outcome and early childhood development. Neurotoxicology 1987, 8, 395–401. [Google Scholar] [PubMed]
- Dietrich, K.N.; Krafft, K.M.; Bornschein, R.L.; Hammond, P.B.; Berger, O.; Succop, P.A.; Bier, M. Low-level fetal lead exposure effect on neurobehavioral development in early infancy. Pediatrics 1987, 80, 721–730. [Google Scholar] [PubMed]
- Landrigan, P.J.; Whitworth, R.H.; Baloh, R.W.; Staehling, N.W.; Barthel, W.F.; Rosenblum, B.F. Neuropsychological dysfunction in children with chronic low-level lead absorption. Lancet 1975, 1, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Needleman, H.L.; Leviton, A. Lead and neurobehavioural deficit in children. Lancet 1979, 314, 104. [Google Scholar] [CrossRef]
- Jusko, T.A.; Henderson, C.R.; Lanphear, B.P.; Cory-Slechta, D.A.; Parsons, P.J.; Canfield, R.L. Blood lead concentrations <10 microg/dL and child intelligence at 6 years of age. Environ. Health Perspect. 2008, 116, 243–248. [Google Scholar]
- Oken, E.; Bellinger, D.C. Fish consumption, methylmercury and child neurodevelopment. Curr. Opin. Pediatr. 2008, 20, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, B.; Marks, A.R.; Bradman, A.; Harley, K.; Barr, D.B.; Johnson, C.; Morga, N.; Jewell, N.P. Organophosphate pesticide exposure and neurodevelopment in young Mexican-American children. Environ. Health Perspect. 2007, 115, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, B.; Rosas, L.G.; Marks, A.R.; Bradman, A.; Harley, K.; Holland, N.; Johnson, C.; Fenster, L.; Barr, D.B. Pesticide toxicity and the developing brain. Basic Clin. Pharmacol. Toxicol. 2008, 102, 228–236. [Google Scholar] [CrossRef] [PubMed]
- London, L.; Beseler, C.; Bouchard, M.F.; Bellinger, D.C.; Colosio, C.; Grandjean, P.; Harari, R.; Kootbodien, T.; Kromhout, H.; Little, F.; et al. Neurobehavioral and neurodevelopmental effects of pesticide exposures. Neurotoxicology 2012, 33, 887–896. [Google Scholar] [CrossRef]
- Perera, F.P.; Li, Z.; Whyatt, R.; Hoepner, L.; Wang, S.; Camann, D.; Rauh, V. Prenatal airborne polycyclic aromatic hydrocarbon exposure and child IQ at age 5 years. Pediatrics 2009, 124, e195–e202. [Google Scholar] [CrossRef] [PubMed]
- Winneke, G. Developmental aspects of environmental neurotoxicology: Lessons from lead and polychlorinated biphenyls. J. Neurol. Sci. 2011, 308, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Herbstman, J.B.; Sjodin, A.; Kurzon, M.; Lederman, S.A.; Jones, R.S.; Rauh, V.; Needham, L.L.; Tang, D.; Niedzwiecki, M.; Wang, R.Y.; et al. Prenatal exposure to PBDEs and neurodevelopment. Environ. Health Perspect. 2010, 118, 712–719. [Google Scholar] [CrossRef]
- Whyatt, R.M.; Liu, X.; Rauh, V.A.; Calafat, A.M.; Just, A.C.; Hoepner, L.; Diaz, D.; Quinn, J.; Adibi, J.; Perera, F.P.; et al. Maternal prenatal urinary phthalate metabolite concentrations and child mental, psychomotor, and behavioral development at 3 years of age. Environ. Health Perspect. 2012, 120, 290–295. [Google Scholar] [CrossRef]
- Rice, D.S.; Curran, T. Role of the reelin signaling pathway in central nervous system development. Annu. Rev. Neurosci. 2001, 24, 1005–1039. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Landrigan, P.J. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Landrigan, P.J. Developmental neurotoxicity of industrial chemicals. Lancet 2006, 368, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.F.; Bellinger, D.C.; Wright, R.O.; Weisskopf, M.G. Attention-deficit/hyperactivity disorder and urinary metabolites of organophosphate pesticides. Pediatrics 2010, 125, e1270–e1277. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.L.; Sun, G.; Zhang, Y.; Grandjean, P. Developmental fluoride neurotoxicity: A systematic review and meta-analysis. Environ. Health Perspect. 2012, 120, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.M.; Wetmur, J.; Chen, J.; Zhu, C.; Barr, D.B.; Canfield, R.L.; Wolff, M.S. Prenatal exposure to organophosphates, paraoxonase 1, and cognitive development in childhood. Environ. Health Perspect. 2011, 119, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Miodovnik, A.; Engel, S.M.; Zhu, C.; Ye, X.; Soorya, L.V.; Silva, M.J.; Calafat, A.M.; Wolff, M.S. Endocrine disruptors and childhood social impairment. Neurotoxicology 2011, 32, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Rauh, V.; Arunajadai, S.; Horton, M.; Perera, F.; Hoepner, L.; Barr, D.B.; Whyatt, R. Seven-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide. Environ. Health Perspect. 2011, 119, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garciduenas, L.; Mora-Tiscareno, A.; Ontiveros, E.; Gomez-Garza, G.; Barragan-Mejia, G.; Broadway, J.; Chapman, S.; Valencia-Salazar, G.; Jewells, V.; Maronpot, R.R.; et al. Air pollution, cognitive deficits and brain abnormalities: A pilot study with children and dogs. Brain Cogn. 2008, 68, 117–127. [Google Scholar] [CrossRef]
- Volk, H.E.; Lurmann, F.; Penfold, B.; Hertz-Picciotto, I.; McConnell, R. Traffic-related air pollution, particulate matter, and autism. JAMA Psychiatry 2013, 70, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, T.E.; Anixt, J.S.; Loe, I.M.; Chirdkiatgumchai, V.; Kuan, L.; Gilman, R.C. Update on environmental risk factors for attention-deficit/hyperactivity disorder. Curr Psychiatr. Rep. 2011, 13, 333–344. [Google Scholar] [CrossRef]
- Bellinger, D.C. Mercury and pregnancy. Birth Defects Res. A Clin. Mol. Teratol. 2014, 100, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.C. Interpreting epidemiologic studies of developmental neurotoxicity: Conceptual and analytic issues. Neurotoxicol. Teratol. 2009, 31, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Lyall, K.; Schmidt, R.J.; Hertz-Picciotto, I. Maternal lifestyle and environmental risk factors for autism spectrum disorders. Int. J. Epidemiol. 2014, 43, 443–464. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W. Effects of alcohol on the generation and migration of cerebral cortical neurons. Science 1986, 233, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Worley, P.F.; Guilarte, T.R. Lead exposure during synaptogenesis alters NMDA receptor targeting via NMDA receptor inhibition. Neurotoxicology 2011, 32, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Roy, T.S.; Seidler, F.J.; Slotkin, T.A. Morphologic effects of subtoxic neonatal chlorpyrifos exposure in developing rat brain: Regionally selective alterations in neurons and glia. Brain Res. Dev. Brain Res. 2004, 148, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Seidler, F.J. Oxidative and excitatory mechanisms of developmental neurotoxicity: Transcriptional profiles for chlorpyrifos, diazinon, dieldrin, and divalent nickel in PC12 cells. Environ. Health Perspect. 2009, 117, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.S. Bisphenol A: An endocrine disruptor with widespread exposure and multiple effects. J. Steroid Biochem. Mol. Biol. 2011, 127, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; et al. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Crofton, K.M.; Sachana, M.; Shafer, T.J.; Behl, M.; Forsby, A.; Hargreaves, A.; Landesmann, B.; Lein, P.J.; Louisse, J.; et al. Putative adverse outcome pathways relevant to neurotoxicity. Crit. Rev. Toxicol. 2015, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Julvez, J.; Smith, G.D.; Golding, J.; Ring, S.; Pourcain, B.S.; Gonzalez, J.R.; Grandjean, P. Prenatal methylmercury exposure and genetic predisposition to cognitive deficit at age 8 years. Epidemiology 2013, 24, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Hackman, D.A.; Farah, M.J. Socioeconomic status and the developing brain. Trends Cogn. Sci. 2009, 13, 65–73. [Google Scholar] [CrossRef] [PubMed]
- King, M.D.; Bearman, P.S. Socioeconomic status and the increased prevalence of autism in California. Am. Sociol. Rev. 2011, 76, 320–346. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.; Lewis, G.; Lundberg, M.; Araya, R.; Svensson, A.; Dalman, C.; Carpenter, P.; Magnusson, C. Parental socioeconomic status and risk of offspring autism spectrum disorders in a Swedish population-based study. J. Am. Acad. Child. Adolesc. Psychiatr. 2012, 51, 467–476. [Google Scholar] [CrossRef]
- Jurewicz, J.; Polanska, K.; Hanke, W. Chemical exposure early in life and the neurodevelopment of children—An overview of current epidemiological evidence. Ann. Agric. Environ. Med. 2013, 20, 465–486. [Google Scholar] [PubMed]
- Bellinger, D.C. A strategy for comparing the contributions of environmental chemicals and other risk factors to neurodevelopment of children. Environ. Health Perspect. 2012, 120, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Grosse, S.D.; Matte, T.D.; Schwartz, J.; Jackson, R.J. Economic gains resulting from the reduction in childrenʼs exposure to lead in the United States. Environ. Health Perspect. 2002, 110, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef]
- Wild, C.P. The exposome: From concept to utility. Int. J. Epidemiol. 2012, 41, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Vrijheid, M. The exposome: A new paradigm to study the impact of environment on health. Thorax 2014, 69, 876–878. [Google Scholar] [CrossRef] [PubMed]
- EXPOsOMICS. Available online: http://www.exposomicsproject.eu/node/8941 (accessed on 25 September 2014).
- HEALS. Available online: http://www.heals-eu.eu/ (accessed on 25 September 2014).
- HELIX. Available online: http://www.projecthelix.eu/ (accessed on 25 Septmber 2014).
- Sakurai, T.; Dorr, N.P.; Takahashi, N.; McInnes, L.A.; Elder, G.A.; Buxbaum, J.D. Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res. 2011, 4, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Faustman, E.M.; Silbernagel, S.M.; Fenske, R.A.; Burbacher, T.M.; Ponce, R.A. Mechanisms underlying Childrenʼs susceptibility to environmental toxicants. Environ. Health Perspect. 2000, 108, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Hof, P.R. The emerging neuroscience of autism spectrum disorders. Brain Res. 2011, 1380, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; Senman, L.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, T.V.; Sanders, S.J.; Yurkiewicz, I.R.; Ercan-Sencicek, A.G.; Kim, Y.S.; Fishman, D.O.; Raubeson, M.J.; Song, Y.; Yasuno, K.; Ho, W.S.; et al. Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol. Psychiatr. 2012, 71, 392–402. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatr. 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of Autism Spectrum Disorder in a UK Population-Based Twin Sample. JAMA Psychiatry 2015. [Google Scholar] [CrossRef]
- Bohm, H.V.; Stewart, M.G.; Healy, A.M. On the Autistic Spectrum Disorder concordance rates of twins and non-twin siblings. Med. Hypotheses 2013, 81, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Chess, S.; Fernandez, P.; Korn, S. Behavioral consequences of congenital rubella. J. Pediatr. 1978, 93, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Chess, S. Autism in children with congenital rubella. J. Autism Child. Schizophr. 1971, 1, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Christianson, A.L.; Chesler, N.; Kromberg, J.G. Fetal valproate syndrome: Clinical and neuro-developmental features in two sibling pairs. Dev. Med. Child. Neurol. 1994, 36, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, E.G.; Ash, E.; Williams, C.P. Autism and congenital cytomegalovirus. J. Autism Dev. Disord. 1984, 14, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Autism and Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2010. In MMWR Surveillance Summaries; 2014; 63, SS02, pp. 1–21. Available online: http://www.cdc.gov/ncbddd/autism/states/comm_report_autism_2014.pdf (accessed on 19 March 2015). [Google Scholar]
- Elsabbagh, M.; Divan, G.; Koh, Y.J.; Kim, Y.S.; Kauchali, S.; Marcin, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, K. The prevalence puzzle: Autism counts. Nature 2011, 479, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.F.; Tancredi, D.J.; Hertz-Picciotto, I. Independent and dependent contributions of advanced maternal and paternal ages to autism risk. Autism Res. 2010, 3, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Frans, E.M.; Sandin, S.; Reichenberg, A.; Langstrom, N.; Lichtenstein, P.; McGrath, J.J.; Hultman, C.M. Autism risk across generations: A population-based study of advancing grandpaternal and paternal age. JAMA Psychiatr. 2013, 70, 516–521. [Google Scholar] [CrossRef]
- Cheslack-Postava, K.; Liu, K.; Bearman, P.S. Closely spaced pregnancies are associated with increased odds of autism in California sibling births. Pediatrics 2011, 127, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.E.; Croen, L.A.; Braunschweig, D.; Yoshida, C.K.; Grether, J.; Hansen, R.; Kharrazi, M.; Ashwood, P.; van de Water, J. Increased midgestational IFN-gamma, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol. Autism 2011, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Kalkbrenner, A.E.; Daniels, J.L.; Chen, J.C.; Poole, C.; Emch, M.; Morrissey, J. Perinatal exposure to hazardous air pollutants and autism spectrum disorders at age 8. Epidemiology 2010, 21, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatr. 2014, 4, e360. [Google Scholar] [CrossRef]
- Hertz-Picciotto, I.; Croen, L.A.; Hansen, R.; Jones, C.R.; van de Water, J.; Pessah, I.N. The CHARGE study: An epidemiologic investigation of genetic and environmental factors contributing to autism. Environ. Health Perspect. 2006, 114, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Volk, H.E.; Hertz-Picciotto, I.; Delwiche, L.; Lurmann, F.; McConnell, R. Residential proximity to freeways and autism in the CHARGE study. Environ. Health Perspect. 2011, 119, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.F.; Geraghty, E.M.; Tancredi, D.J.; Delwiche, L.D.; Schmidt, R.J.; Ritz, B.; Hansen, R.L.; Hertz-Picciotto, I. Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: The CHARGE study. Environ. Health Perspect. 2014, 122, 1103–1109. [Google Scholar] [PubMed]
- Zerbo, O.; Qian, Y.; Yoshida, C.; Grether, J.K.; van de Water, J.; Croen, L.A. Maternal infection during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 2013. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Hansen, R.L.; Hartiala, J.; Allayee, H.; Schmidt, L.C.; Tancredi, D.J.; Tassone, F.; Hertz-Picciotto, I. Prenatal vitamins, one-carbon metabolism gene variants, and risk for autism. Epidemiology 2011, 22, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.J.; Tancredi, D.J.; Ozonoff, S.; Hansen, R.L.; Hartiala, J.; Allayee, H.; Schmidt, L.C.; Tassone, F.; Hertz-Picciotto, I. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. Am. J. Clin. Nutr. 2012, 96, 80–89. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association (APA). Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; APA: Washington, DC, USA, 2000. [Google Scholar]
- Akinbami, L.J.; Xiang, L.; Pastor, P.N.; Reuben, C.A. Attention Deficit Hyperactivity Disorder Among Children Aged 5–17 Years in the United States, 1998–2009. NCHS data brief, no 70; National Center for Health Statistics: Hyattsville, MD, USA, 2011. Available online: http://www.cdc.gov/nchs/data/databriefs/db70.pdf (accessed on 25 September 2014). [Google Scholar]
- Faraone, S.V.; Mick, E. Molecular genetics of attention deficit hyperactivity disorder. Psychiatr. Clin. N. Am. 2010, 33, 159–180. [Google Scholar] [CrossRef]
- Smith, A.K.; Mick, E.; Faraone, S.V. Advances in genetic studies of attention-deficit/hyperactivity disorder. Curr. Psychiatr. Rep. 2009, 11, 143–148. [Google Scholar] [CrossRef]
- Mick, E.; Todorov, A.; Smalley, S.; Hu, X.; Loo, S.; Todd, R.D.; Biederman, J.; Byrne, D.; Dechairo, B.; Guiney, A.; et al. Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J. Am. Acad. Child. Adolesc. Psychiatr. 2010, 49, 898–905. [Google Scholar] [CrossRef]
- Neale, B.M.; Lasky-Su, J.; Anney, R.; Franke, B.; Zhou, K.; Maller, J.B.; Vasquez, A.A.; Asherson, P.; Chen, W.; Banaschewski, T.; et al. Genome-wide association scan of attention deficit hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147B, 1337–1344. [Google Scholar] [CrossRef]
- DiMaio, S.; Grizenko, N.; Joober, R. Dopamine genes and attention-deficit hyperactivity disorder: A review. J. Psychiatr. Neurosci. 2003, 28, 27–38. [Google Scholar]
- Paloyelis, Y.; Asherson, P.; Mehta, M.A.; Faraone, S.V.; Kuntsi, J. DAT1 and COMT effects on delay discounting and trait impulsivity in male adolescents with attention deficit/hyperactivity disorder and healthy controls. Neuropsychopharmacology 2010, 35, 2414–2426. [Google Scholar] [CrossRef]
- Norton, N.; Owen, M.J. HTR2A: Association and expression studies in neuropsychiatric genetics. Ann. Med. 2005, 37, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Linnet, K.M.; Dalsgaard, S.; Obel, C.; Wisborg, K.; Henriksen, T.B.; Rodriguez, A.; Kotimaa, A.; Moilanen, I.; Thomsen, P.H.; Olsen, J.; et al. Maternal lifestyle factors in pregnancy risk of attention deficit hyperactivity disorder and associated behaviors: Review of the current evidence. Am. J. Psychiatry 2003, 160, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Mick, E.; Biederman, J.; Faraone, S.V.; Sayer, J.; Kleinman, S. Case-control study of attention-deficit hyperactivity disorder and maternal smoking, alcohol use, and drug use during pregnancy. J. Am. Acad. Child. Adolesc. Psychiatr. 2002, 41, 378–385. [Google Scholar] [CrossRef]
- Accornero, V.H.; Amado, A.J.; Morrow, C.E.; Xue, L.; Anthony, J.C.; Bandstra, E.S. Impact of prenatal cocaine exposure on attention and response inhibition as assessed by continuous performance tests. J. Dev. Behav Pediatr 2007, 28, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Bandstra, E.S.; Morrow, C.E.; Anthony, J.C.; Accornero, V.H.; Fried, P.A. Longitudinal investigation of task persistence and sustained attention in children with prenatal cocaine exposure. Neurotoxicol. Teratol. 2001, 23, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.L.; Jacobson, S.W. Prenatal exposure to polychlorinated biphenyls and attention at school age. J. Pediatr. 2003, 143, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.W.; Reihman, J.; Lonky, E.I.; Darvill, T.J.; Pagano, J. Cognitive development in preschool children prenatally exposed to PCBs and MeHg. Neurotoxicol. Teratol. 2003, 25, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M.; Kahn, R.S.; Froehlich, T.; Auinger, P.; Lanphear, B.P. Exposures to environmental toxicants and attention deficit hyperactivity disorder in U.S. children. Environ. Health Perspect. 2006, 114, 1904–1909. [Google Scholar] [PubMed]
- Nigg, J.T.; Nikolas, M.; Mark Knottnerus, G.; Cavanagh, K.; Friderici, K. Confirmation and extension of association of blood lead with attention-deficit/hyperactivity disorder (ADHD) and ADHD symptom domains at population-typical exposure levels. J. Child. Psychol. Psychiatr. 2010, 51, 58–65. [Google Scholar] [CrossRef]
- Wang, H.L.; Chen, X.T.; Yang, B.; Ma, F.L.; Wang, S.; Tang, M.L.; Hao, M.G.; Ruan, D.Y. Case-control study of blood lead levels and attention deficit hyperactivity disorder in Chinese children. Environ. Health Perspect. 2008, 116, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Boucher, O.; Jacobson, S.W.; Plusquellec, P.; Dewailly, E.; Ayotte, P.; Forget-Dubois, N.; Jacobson, J.L.; Muckle, G. Prenatal methylmercury, postnatal lead exposure, and evidence of attention deficit/hyperactivity disorder among Inuit children in Arctic Quebec. Environ. Health Perspect. 2012, 120, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, S.K.; Thurston, S.W.; Bellinger, D.C.; Amarasiriwardena, C.; Korrick, S.A. Prenatal exposure to mercury and fish consumption during pregnancy and attention-deficit/hyperactivity disorder-related behavior in children. Arch. Pediatr. Adolesc. Med. 2012, 166, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Rauh, V.A.; Garfinkel, R.; Perera, F.P.; Andrews, H.F.; Hoepner, L.; Barr, D.B.; Whitehead, R.; Tang, D.; Whyatt, R.W. Impact of prenatal chlorpyrifos exposure on neurodevelopment in the first 3 years of life among inner-city children. Pediatrics 2006, 118, e1845–e1859. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.G.; Heron, J.; Golding, J.; Beveridge, M.; Glover, V. Maternal antenatal anxiety and children's behavioural/emotional problems at 4 years. Report from the Avon Longitudinal Study of Parents and Children. Br. J. Psychiatr. 2002, 180, 502–508. [Google Scholar] [CrossRef]
- Van den Bergh, B.R.; Mulder, E.J.; Mennes, M.; Glover, V. Antenatal maternal anxiety and stress and the neurobehavioural development of the fetus and child: Links and possible mechanisms. A review. Neurosci. Biobehav. Rev. 2005, 29, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Bhutta, A.T.; Cleves, M.A.; Casey, P.H.; Cradock, M.M.; Anand, K.J. Cognitive and behavioral outcomes of school-aged children who were born preterm: A meta-analysis. JAMA 2002, 288, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Hultman, C.M.; Torrang, A.; Tuvblad, C.; Cnattingius, S.; Larsson, J.O.; Lichtenstein, P. Birth weight and attention-deficit/hyperactivity symptoms in childhood and early adolescence: A prospective Swedish twin study. J. Am. Acad. Child. Adolesc. Psychiatr. 2007, 46, 370–377. [Google Scholar] [CrossRef]
- Polanska, K.; Jurewicz, J.; Hanke, W. Exposure to environmental and lifestyle factors and attention-deficit/hyperactivity disorder in children—A review of epidemiological studies. Int. J. Occup. Med. Environ. Health 2012, 25, 330–355. [Google Scholar] [CrossRef] [PubMed]
- Connors, S.L.; Levitt, P.; Matthews, S.G.; Slotkin, T.A.; Johnston, M.V.; Kinney, H.C.; Johnson, W.G.; Dailey, R.M.; Zimmerman, A.W. Fetal mechanisms in neurodevelopmental disorders. Pediatr. Neurol. 2008, 38, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Murray, J.C. What genome-wide association studies can do for medicine. N. Engl. J. Med. 2007, 356, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Livingston, R.J.; von Niederhausern, A.; Jegga, A.G.; Crawford, D.C.; Carlson, C.S.; Rieder, M.J.; Gowrisankar, S.; Aronow, B.J.; Weiss, R.B.; Nickerson, D.A. Pattern of sequence variation across 213 environmental response genes. Genome Res. 2004, 14, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.R.; Russo, J.P.; Yang, S.; Roohi, J.; Blaxill, M.; Kahler, S.G.; Cremer, L.; Hatchwell, E. Autism and environmental genomics. Neurotoxicology 2006, 27, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Parks, J.M.; Johs, A.; Podar, M.; Bridou, R.; Hurt, R.A., Jr.; Smith, S.D.; Tomanicek, S.J.; Qian, Y.; Brown, S.D.; Brandt, C.C.; et al. The genetic basis for bacterial mercury methylation. Science 2013, 339, 1332–1335. [Google Scholar] [CrossRef] [PubMed]
- Joint WHO/Convention Task Force on the Health Aspects of Air Pollution. Health Risks of Heavy Metals from Long-Range Transboundary Air Pollution. Available online: http://www.euro.who.int/__data/assets/pdf_file/0007/78649/E91044.pdf (accessed on 25 September 2014).
- Balshaw, S.; Edwards, J.; Daughtry, B.; Ross, K. Mercury in seafood: Mechanisms of accumulation and consequences for consumer health. Rev. Environ. Health 2007, 22, 91–113. [Google Scholar] [PubMed]
- Mahaffey, K.R.; Clickner, R.P.; Bodurow, C.C. Blood organic mercury and dietary mercury intake: National Health and Nutrition Examination Survey, 1999 and 2000. Environ. Health Perspect. 2004, 112, 562–570. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Children’s exposure to mercury compounds. Available online: http://whqlibdoc.who.int/publications/2010/9789241500456_eng.pdf?ua=1 (accessed on 25 September 2014).
- Debes, F.; Budtz-Jorgensen, E.; Weihe, P.; White, R.F.; Grandjean, P. Impact of prenatal methylmercury exposure on neurobehavioral function at age 14 years. Neurotoxicol. Teratol. 2006, 28, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P. Mercurial uncertainties in environmental health. Ann. N. Y. Acad. Sci. 1997, 837, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Davidson, P.W.; Leste, A.; Benstrong, E.; Burns, C.M.; Valentin, J.; Sloane-Reeves, J.; Huang, L.S.; Miller, W.A.; Gunzler, D.; van Wijngaarden, E.; et al. Fish consumption, mercury exposure, and their associations with scholastic achievement in the Seychelles Child Development Study. Neurotoxicology 2010, 31, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Davidson, P.W.; Myers, G.J.; Weiss, B.; Shamlaye, C.F.; Cox, C. Prenatal methyl mercury exposure from fish consumption and child development: A review of evidence and perspectives from the Seychelles Child Development Study. Neurotoxicology 2006, 27, 1106–1109. [Google Scholar] [CrossRef] [PubMed]
- Myers, G.J.; Davidson, P.W.; Cox, C.; Shamlaye, C.F.; Palumbo, D.; Cernichiari, E.; Sloane-Reeves, J.; Wilding, G.E.; Kost, J.; Huang, L.S.; et al. Prenatal methylmercury exposure from ocean fish consumption in the Seychelles child development study. Lancet 2003, 361, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.; Ramos, R.; Lopez-Espinosa, M.J.; Diez, S.; Vioque, J.; Ballester, F.; Fernandez, M.F. Hair mercury levels, fish consumption, and cognitive development in preschool children from Granada, Spain. Environ. Res. 2010, 110, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Valent, F.; Horvat, M.; Sofianou-Katsoulis, A.; Spiric, Z.; Mazej, D.; Little, D.; Prasouli, A.; Mariuz, M.; Tamburlini, G.; Nakou, S.; et al. Neurodevelopmental effects of low-level prenatal mercury exposure from maternal fish consumption in a Mediterranean cohort: Study rationale and design. J. Epidemiol. 2013, 23, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, K.S.; Wennberg, M.; Stromberg, U.; Bergdahl, I.A.; Hallmans, G.; Jansson, J.H.; Lundh, T.; Norberg, M.; Rentschler, G.; Vessby, B.; et al. Evaluation of the impact of genetic polymorphisms in glutathione-related genes on the association between methylmercury or n-3 polyunsaturated long chain fatty acids and risk of myocardial infarction: Acase-control study. Environ. Health 2011, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Gundacker, C.; Gencik, M.; Hengstschlager, M. The relevance of the individual genetic background for the toxicokinetics of two significant neurodevelopmental toxicants: Mercury and lead. Mutat. Res. 2010, 705, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Oken, E.; Radesky, J.S.; Wright, R.O.; Bellinger, D.C.; Amarasiriwardena, C.J.; Kleinman, K.P.; Hu, H.; Gillman, M.W. Maternal fish intake during pregnancy, blood mercury levels, and child cognition at age 3 years in a US cohort. Am. J. Epidemiol. 2008, 167, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Strain, J.J.; Davidson, P.W.; Thurston, S.W.; Harrington, D.; Mulhern, M.S.; McAfee, A.J.; van Wijngaarden, E.; Shamlaye, C.F.; Henderson, J.; Watson, G.E.; et al. Maternal PUFA status but not prenatal methylmercury exposure is associated with childrenʼs language functions at age five years in the Seychelles. J. Nutr. 2012, 142, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, D.; Woods, J.S.; Heyer, N.J.; Rohlman, D.; Farin, F.M.; Li, T.; Garabedian, C.E. The association between a genetic polymorphism of coproporphyrinogen oxidase, dental mercury exposure and neurobehavioral response in humans. Neurotoxicol. Teratol. 2006, 28, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.S.; Heyer, N.J.; Echeverria, D.; Russo, J.E.; Martin, M.D.; Bernardo, M.F.; Luis, H.S.; Vaz, L.; Farin, F.M. Modification of neurobehavioral effects of mercury by a genetic polymorphism of coproporphyrinogen oxidase in children. Neurotoxicol. Teratol. 2012, 34, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Woods, J.S. Cloning, expression, and biochemical properties of CPOX4, a genetic variant of coproporphyrinogen oxidase that affects susceptibility to mercury toxicity in humans. Toxicol. Sci. 2009, 109, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Syversen, T.; Souza, D.O.; Rocha, J.B. Metallothioneins: Mercury species-specific induction and their potential role in attenuating neurotoxicity. Exp. Biol. Med. (Maywood) 2006, 231, 1468–1473. [Google Scholar]
- Wang, Y.; Goodrich, J.M.; Gillespie, B.; Werner, R.; Basu, N.; Franzblau, A. An investigation of modifying effects of metallothionein single-nucleotide polymorphisms on the association between mercury exposure and biomarker levels. Environ. Health Perspect. 2012, 120, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.S.; Heyer, N.J.; Russo, J.E.; Martin, M.D.; Pillai, P.B.; Farin, F.M. Modification of neurobehavioral effects of mercury by genetic polymorphisms of metallothionein in children. Neurotoxicol. Teratol. 2013, 39, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Eddins, D.; Petro, A.; Pollard, N.; Freedman, J.H.; Levin, E.D. Mercury-induced cognitive impairment in metallothionein-1/2 null mice. Neurotoxicol. Teratol. 2008, 30, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Lin, C.C.; Hwang, Y.H.; Hsieh, W.S.; Liao, H.F.; Chen, P.C. Mercury, APOE, and children’s neurodevelopment. Neurotoxicology 2013, 37, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Stewart, W.F.; Schwartz, B.S.; Simon, D.; Kelsey, K.; Todd, A.C. ApoE genotype, past adult lead exposure, and neurobehavioral function. Environ. Health Perspect. 2002, 110, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Llop, S.; Engstrom, K.; Ballester, F.; Franforte, E.; Alhamdow, A.; Pisa, F.; Tratnik, J.S.; Mazej, D.; Murcia, M.; Rebagliato, M.; et al. Polymorphisms in ABC transporter genes and concentrations of mercury in newborns—Evidence from two Mediterranean birth cohorts. PLoS One 2014, 9, e97172. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.S.; Heyer, N.J.; Russo, J.E.; Martin, M.D.; Farin, F.M. Genetic polymorphisms affecting susceptibility to mercury neurotoxicity in children: Summary findings from the Casa Pia Childrenʼs Amalgam Clinical Trial. Neurotoxicology 2014, 44C, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.C. Overview of modifiers of methylmercury neurotoxicity: Chemicals, nutrients, and the social environment. Neurotoxicology 2008, 29, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.C. Interpretation of small effect sizes in occupational and environmental neurotoxicology: Individual versus population risk. Neurotoxicology 2007, 28, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Sultatos, L.G. Mammalian toxicology of organophosphorus pesticides. J. Toxicol. Environ. Health 1994, 43, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.J.; McIntosh, L.J.; Mink, P.J.; Jurek, A.M.; Li, A.A. Pesticide exposure and neurodevelopmental outcomes: Review of the epidemiologic and animal studies. J. Toxicol. Environ. Health B Crit. Rev. 2013, 16, 127–283. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.L.; Daroff, R.B.; Autrup, H.; Bridges, J.; Buffler, P.; Costa, L.G.; Coyle, J.; McKhann, G.; Mobley, W.C.; Nadel, L.; et al. Review of the toxicology of chlorpyrifos with an emphasis on human exposure and neurodevelopment. Crit. Rev. Toxicol. 2008, 38, 1–125. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Meyer, A.; Seidler, F.J.; Slotkin, T.A. Alterations in central nervous system serotonergic and dopaminergic synaptic activity in adulthood after prenatal or neonatal chlorpyrifos exposure. Environ. Health Perspect. 2005, 113, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Seidler, F.J.; Tate, C.A.; Cousins, M.M.; Slotkin, T.A. Fetal chlorpyrifos exposure: Adverse effects on brain cell development and cholinergic biomarkers emerge postnatally and continue into adolescence and adulthood. Environ. Health Perspect. 2003, 111, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Ricceri, L.; Venerosi, A.; Capone, F.; Cometa, M.F.; Lorenzini, P.; Fortuna, S.; Calamandrei, G. Developmental neurotoxicity of organophosphorous pesticides: Fetal and neonatal exposure to chlorpyrifos alters sex-specific behaviors at adulthood in mice. Toxicol. Sci. 2006, 93, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.M.; Berkowitz, G.S.; Barr, D.B.; Teitelbaum, S.L.; Siskind, J.; Meisel, S.J.; Wetmur, J.G.; Wolff, M.S. Prenatal organophosphate metabolite and organochlorine levels and performance on the Brazelton Neonatal Behavioral Assessment Scale in a multiethnic pregnancy cohort. Am. J. Epidemiol. 2007, 165, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, G.S.; Wetmur, J.G.; Birman-Deych, E.; Obel, J.; Lapinski, R.H.; Godbold, J.H.; Holzman, I.R.; Wolff, M.S. In utero pesticide exposure, maternal paraoxonase activity, and head circumference. Environ. Health Perspect. 2004, 112, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.F.; Chevrier, J.; Harley, K.G.; Kogut, K.; Vedar, M.; Calderon, N.; Trujillo, C.; Johnson, C.; Bradman, A.; Barr, D.B.; et al. Prenatal exposure to organophosphate pesticides and IQ in 7-year-old children. Environ. Health Perspect. 2011, 119, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Barcelos, G.R.; Grotto, D.; de Marco, K.C.; Valentini, J.; Lengert, A.; de Oliveira, A.Á.; Garcia, S.C.; Braga, G.Ú.L.; Engström, K.S.; de Syllos Cólus, I.M.; et al. Polymorphisms in glutathione-related genes modify mercury concentrations and antioxidant status in subjects environmentally exposed to methylmercury. Sci. Total Environ. 2013, 463–464, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Li, W.F.; Richter, R.J.; Shih, D.M.; Lusis, A.; Furlong, C.E. The role of paraoxonase (PON1) in the detoxication of organophosphates and its human polymorphism. Chem. Biol. Interact. 1999, 119–120, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, B.; Huen, K.; Marks, A.; Harley, K.G.; Bradman, A.; Barr, D.B.; Holland, N. PON1 and neurodevelopment in children from the CHAMACOS study exposed to organophosphate pesticides in utero. Environ. Health Perspect. 2010, 118, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Cole, T.B.; Vitalone, A.; Furlong, C.E. Measurement of paraoxonase (PON1) status as a potential biomarker of susceptibility to organophosphate toxicity. Clin. Chim. Acta 2005, 352, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Giordano, G.; Cole, T.B.; Marsillach, J.; Furlong, C.E. Paraoxonase 1 (PON1) as a genetic determinant of susceptibility to organophosphate toxicity. Toxicology 2013, 307, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Vitalone, A.; Cole, T.B.; Furlong, C.E. Modulation of paraoxonase (PON1) activity. Biochem. Pharmacol. 2005, 69, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Cole, T.B.; Furlong, C.E. Polymorphisms of paraoxonase (PON1) and their significance in clinical toxicology of organophosphates. J. Toxicol. Clin. Toxicol. 2003, 41, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Humbert, R.; Adler, D.A.; Disteche, C.M.; Hassett, C.; Omiecinski, C.J.; Furlong, C.E. The molecular basis of the human serum paraoxonase activity polymorphism. Nat. Genet. 1993, 3, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kumar, M.; Chan, W.; Berkowitz, G.; Wetmur, J.G. Increased influence of genetic variation on PON1 activity in neonates. Environ. Health Perspect. 2003, 111, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Holland, N.; Furlong, C.; Bastaki, M.; Richter, R.; Bradman, A.; Huen, K.; Beckman, K.; Eskenazi, B. Paraoxonase polymorphisms, haplotypes, and enzyme activity in Latino mothers and newborns. Environ. Health Perspect. 2006, 114, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Huen, K.; Harley, K.; Brooks, J.; Hubbard, A.; Bradman, A.; Eskenazi, B.; Holland, N. Developmental changes in PON1 enzyme activity in young children and effects of PON1 polymorphisms. Environ. Health Perspect. 2009, 117, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Huen, K.; Barcellos, L.; Beckman, K.; Rose, S.; Eskenazi, B.; Holland, N. Effects of PON polymorphisms and haplotypes on molecular phenotype in Mexican-American mothers and children. Environ. Mol. Mutagen. 2011, 52, 105–116. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, M.; Ricci, I.; Sacco, R.; Liu, X.; D’Agruma, L.; Muscarella, L.A.; Guarnieri, V.; Militerni, R.; Bravaccio, C.; Elia, M.; et al. Paraoxonase gene variants are associated with autism in North America, but not in Italy: Possible regional specificity in gene-environment interactions. Mol. Psychiatr. 2005, 10, 1006–1016. [Google Scholar] [CrossRef]
- Eskenazi, B.; Kogut, K.; Huen, K.; Harley, K.G.; Bouchard, M.; Bradman, A.; Boyd-Barr, D.; Johnson, C.; Holland, N. Organophosphate pesticide exposure, PON1, and neurodevelopment in school-age children from the CHAMACOS study. Environ. Res. 2014, 134C, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Kucukali, C.I.; Aydin, M.; Ozkok, E.; Orhan, N.; Cakir, U.; Kilic, G.; Ozbek, Z.; Ince, N.; Kara, I. Paraoxonase-1 55/192 genotypes in schizophrenic patients and their relatives in Turkish population. Psychiatr. Genet. 2008, 18, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Day, I.N.; Gaunt, T.R.; Hinks, L.J.; Timpson, N.; Ebrahim, S.; Davey Smith, G. The association of the paraoxonase (PON1) Q192R polymorphism with depression in older women: Findings from the British Womenʼs Heart and Health Study. J. Epidemiol. Commun. Health 2007, 61, 85–87. [Google Scholar] [CrossRef]
- Pasca, S.P.; Dronca, E.; Nemes, B.; Kaucsar, T.; Endreffy, E.; Iftene, F.; Benga, I.; Cornean, R.; Dronca, M. Paraoxonase 1 activities and polymorphisms in autism spectrum disorders. J. Cell Mol. Med. 2010, 14, 600–607. [Google Scholar] [PubMed]
- Pasca, S.P.; Nemes, B.; Vlase, L.; Gagyi, C.E.; Dronca, E.; Miu, A.C.; Dronca, M. High levels of homocysteine and low serum paraoxonase 1 arylesterase activity in children with autism. Life Sci. 2006, 78, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Kofink, D.; Boks, M.P.; Timmers, H.T.; Kas, M.J. Epigenetic dynamics in psychiatric disorders: Environmental programming of neurodevelopmental processes. Neurosci. Biobehav. Rev. 2013, 37, 831–845. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.O.; Szyf, M. The epigenetics of social adversity in early life: Implications for mental health outcomes. Neurobiol. Dis. 2010, 39, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Rutten, B.P.; Mill, J. Epigenetic mediation of environmental influences in major psychotic disorders. Schizophr. Bull. 2009, 35, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Somogyi, E.; Coulon, N.; Kermarrec, S.; Cohen, D.; Bronsard, G.; Bonnot, O.; Weismann-Arcache, C.; Botbol, M.; Lauth, B.; et al. Gene x Environment interactions in autism spectrum disorders: Role of epigenetic mechanisms. Front. Psychiatr. 2014, 5, 53. [Google Scholar] [CrossRef]
- Chuang, J.C.; Jones, P.A. Epigenetics and microRNAs. Pediatr. Res. 2007, 61, 24R–29R. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.; Renthal, W.; Kumar, A.; Nestler, E.J. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Bollati, V. Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 2009, 21, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, M.L.; Adams, S.; Dunaway, K.W.; LaSalle, J.M. Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol. Cell Biol. 2012, 32, 2894–2903. [Google Scholar] [CrossRef] [PubMed]
- Murgatroyd, C.; Spengler, D. Epigenetics of early child development. Front. Psychiatr. 2011, 2, 16. [Google Scholar] [CrossRef]
- Gonzales, M.L.; LaSalle, J.M. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatr. Rep. 2010, 12, 127–134. [Google Scholar] [CrossRef]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M.; Yasui, D.H. Evolving role of MeCP2 in Rett syndrome and autism. Epigenomics 2009, 1, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Nardone, S.; Sams, D.S.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatr. 2014, 4, e433. [Google Scholar] [CrossRef]
- Bromley, R.L.; Mawer, G.; Clayton-Smith, J.; Baker, G.A.; Liverpool and Manchester Neurodevelopment Group. Autism spectrum disorders following in utero exposure to antiepileptic drugs. Neurology 2008, 71, 1923–1924. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, M.; Nii, T.; Ishimaru, N.; Minamino, A.; Hara, D.; Takasaki, I.; Tabuchi, A.; Tsuda, M. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci. Res. 2009, 65, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Mohammed, S.A.; Leavell, J.; Collins, C. Race, socioeconomic status, and health: complexities, ongoing challenges, and research opportunities. Ann. N. Y. Acad. Sci. 2010, 1186, 69–101. [Google Scholar] [CrossRef] [PubMed]
- Young, G.S.; Fox, M.A.; Trush, M.; Kanarek, N.; Glass, T.A.; Curriero, F.C. Differential exposure to hazardous air pollution in the United States: A multilevel analysis of urbanization and neighborhood socioeconomic deprivation. Int. J. Environ. Res. Public Health 2012, 9, 2204–2225. [Google Scholar] [CrossRef] [PubMed]
- Environmental Protection Agency. Science and Decisions: Advancing Risk Assessment; National Academies Press: Washington, DC, USA, 2009. [Google Scholar]
- Committee on the Health Risks of Phthalates, National Research Council. Phthalates and Cumulative Risk Assessment: The Task Ahead. Available online: http://www.nap.edu/catalog/12528/phthalates-and-cumulative-risk-assessment-the-task-ahead (accessed on 15 January 2015).
- Sexton, K.; Linder, S.H. Cumulative risk assessment for combined health effects from chemical and nonchemical stressors. Am. J. Public Health 2011, 101, S81–S88. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Tucker, P. Critical biological pathways for chronic psychosocial stress and research opportunities to advance the consideration of stress in chemical risk assessment. Am. J. Public Health 2011, 101, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Morello-Frosch, R.; Shenassa, E.D. The environmental “riskscape” and social inequality: Implications for explaining maternal and child health disparities. Environ. Health Perspect. 2006, 114, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; Rice, G.E.; Teuschler, L.K.; Wright, J.M. Joint exposure to chemical and nonchemical neurodevelopmental stressors in U.S. women of reproductive age in NHANES. Int. J. Environ. Res. Public Health 2014, 11, 4384–4401. [Google Scholar] [CrossRef] [PubMed]
- Cory-Slechta, D.A. Studying toxicants as single chemicals: Does this strategy adequately identify neurotoxic risk? Neurotoxicology 2005, 26, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Kobrosly, R.W.; Parlett, L.E.; Stahlhut, R.W.; Barrett, E.S.; Swan, S.H. Socioeconomic factors and phthalate metabolite concentrations among United States women of reproductive age. Environ. Res. 2012, 115, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Stangle, D.E.; Strawderman, M.S.; Smith, D.; Kuypers, M.; Strupp, B.J. Reductions in blood lead overestimate reductions in brain lead following repeated succimer regimens in a rodent model of childhood lead exposure. Environ. Health Perspect. 2004, 112, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.E.; Ashwood, P. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicol. Teratol. 2013, 36, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Umansky, S.R. Circulating cell-free microRNA as biomarkers for screening, diagnosis and monitoring of neurodegenerative diseases and other neurologic pathologies. Front. Cell Neurosci. 2013, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, M.; Xia, W.; Hofmann, O.; Gregas, M.; Ho Sui, S.; Hide, W.; Yang, T.; Needleman, H.L.; Bellinger, D.C. Prenatal lead levels, plasma amyloid beta levels, and gene expression in young adulthood. Environ. Health Perspect. 2012, 120, 702–707. [Google Scholar] [CrossRef] [PubMed]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabri, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, L.; Greco, A.; Zanardo, V.; Suppiej, A. Early-life sex-dependent vulnerability to oxidative stress: The natural twining model. J. Matern. Fetal Neonatal Med. 2013, 26, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, R.A. Handbook of Behavioral Teratology; Plenum Press: New York, NY, USA, 1986. [Google Scholar]
- Crawley, J.N. Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin. Neurosci. 2012, 14, 293–305. [Google Scholar] [PubMed]
- Laviola, G.; Adriani, W.; Gaudino, C.; Marino, R.; Keller, F. Paradoxical effects of prenatal acetylcholinesterase blockade on neuro-behavioral development and drug-induced stereotypies in reeler mutant mice. Psychopharmacology (Berl) 2006, 187, 331–344. [Google Scholar] [CrossRef]
- Mullen, B.R.; Khialeeva, E.; Hoffman, D.B.; Ghiani, C.A.; Carpenter, E.M. Decreased reelin expression and organophosphate pesticide exposure alters mouse behaviour and brain morphology. ASN Neuro 2013, 5, e00106. [Google Scholar]
- Abrahams, B.S.; Geschwind, D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Folsom, T.D.; Fatemi, S.H. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 2013, 68, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Michetti, C.; Caruso, A.; Laviola, G.; Scattoni, M.L. Characterization of neonatal vocal and motor repertoire of reelin mutant mice. PLoS One 2013, 8, e64407. [Google Scholar] [CrossRef] [PubMed]
- Abazyan, B.; Dziedzic, J.; Hua, K.; Abazyan, S.; Yang, C.; Mori, S.; Pletnikov, M.V.; Guilarte, T.R. Chronic exposure of mutant DISC1 mice to lead produces sex-dependent abnormalities consistent with schizophrenia and related mental disorders: A gene-environment interaction study. Schizophr Bull. 2014, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Venerosi, A.; Ricceri, L.; Tait, S.; Calamandrei, G. Sex dimorphic behaviors as markers of neuroendocrine disruption by environmental chemicals: The case of chlorpyrifos. Neurotoxicology 2012, 33, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- Furlong, C.E. Genetic variability in the cytochrome P450-paraoxonase 1 (PON1) pathway for detoxication of organophosphorus compounds. J. Biochem. Mol. Toxicol. 2007, 21, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.B.; Fisher, J.C.; Burbacher, T.M.; Costa, L.G.; Furlong, C.E. Neurobehavioral assessment of mice following repeated postnatal exposure to chlorpyrifos-oxon. Neurotoxicol. Teratol. 2012, 34, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Honda, M.; Watanabe, C.; Satoh, M.; Yasutake, A. Neurobehavioral changes and alteration of gene expression in the brains of metallothionein-I/II null mice exposed to low levels of mercury vapor during postnatal development. J. Toxicol. Sci. 2011, 36, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Giordano, G.; Kavanagh, T.J.; Costa, L.G. Neurotoxicity of a polybrominated diphenyl ether mixture (DE-71) in mouse neurons and astrocytes is modulated by intracellular glutathione levels. Toxicol. Appl. Pharmacol. 2008, 232, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.P.; Chen, Y.; Schneider, S.N.; Nebert, D.W.; Shertzer, H.G. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic. Biol. Med. 2004, 37, 1511–1526. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Kugiyama, K.; Sugiyama, S.; Miyamoto, S.; Koide, S.; Fukushima, H.; Honda, O.; Yoshimura, M.; Ogawa, H. Polymorphism in the 5'-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation 2002, 105, 2968–2973. [Google Scholar] [CrossRef] [PubMed]
- Yochum, C.L.; Bhattacharya, P.; Patti, L.; Mirochnitchenko, O.; Wagner, G.C. Animal model of autism using GSTM1 knockout mice and early post-natal sodium valproate treatment. Behav. Brain Res. 2010, 210, 202–210. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, H.G.; Kusek, G.K.; Yang, M.; Phoenix, J.L.; Bolivar, V.J.; Crawley, J.N. Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav. 2008, 7, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Malkova, N.V.; Yu, C.Z.; Hsiao, E.Y.; Moore, M.J.; Patterson, P.H. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 2012, 26, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Schwartzer, J.J.; Careaga, M.; Onore, C.E.; Rushakoff, J.A.; Berman, R.F.; Ashwood, P. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl. Psychiatr. 2013, 3, e240. [Google Scholar] [CrossRef]
- De Felice, A.; Scattoni, M.L.; Ricceri, L.; Calamandrei, G. Prenatal exposure to a common organophosphate insecticide delays motor development in a mouse model of idiopathic autism. PLoS One 2015, in press. [Google Scholar]
- Hougaard, K.S.; Hansen, A.M. Enhancement of developmental toxicity effects of chemicals by gestational stress. A review. Neurotoxicol. Teratol. 2007, 29, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Weston, H.I.; Sobolewski, M.E.; Allen, J.L.; Weston, D.; Conrad, K.; Pelkowski, S.; Watson, G.E.; Zareba, G.; Cory-Slechta, D.A. Sex-dependent and non-monotonic enhancement and unmasking of methylmercury neurotoxicity by prenatal stress. Neurotoxicology 2014, 41, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Colomina, M.T.; Albina, M.L.; Domingo, J.L.; Corbella, J. Effects of maternal stress on methylmercury-induced developmental toxicity in mice. Physiol. Behav. 1995, 58, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Colomina, M.T.; Albina, M.L.; Domingo, J.L.; Corbella, J. Influence of maternal stress on the effects of prenatal exposure to methylmercury and arsenic on postnatal development and behavior in mice: A preliminary evaluation. Physiol. Behav. 1997, 61, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Rasco, J.F.; Hood, R.D. Effects of maternal restraint stress and sodium arsenate in mice. Reprod. Toxicol. 1994, 8, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Rossi-George, A.; Virgolini, M.B.; Weston, D.; Cory-Slechta, D.A. Alterations in glucocorticoid negative feedback following maternal Pb, prenatal stress and the combination: A potential biological unifying mechanism for their corresponding disease profiles. Toxicol. Appl. Pharmacol. 2009, 234, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Cory-Slechta, D.A.; Virgolini, M.B.; Thiruchelvam, M.; Weston, D.D.; Bauter, M.R. Maternal stress modulates the effects of developmental lead exposure. Environ. Health Perspect. 2004, 112, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Cory-Slechta, D.A.; Weston, D.; Liu, S.; Allen, J.L. Brain hemispheric differences in the neurochemical effects of lead, prenatal stress, and the combination and their amelioration by behavioral experience. Toxicol. Sci. 2013, 132, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Virgolini, M.B.; Bauter, M.R.; Weston, D.D.; Cory-Slechta, D.A. Permanent alterations in stress responsivity in female offspring subjected to combined maternal lead exposure and/or stress. Neurotoxicology 2006, 27, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.L.; Grace, C.E.; Braun, A.A.; Schaefer, T.L.; Skelton, M.R.; Tang, P.H.; Vorhees, C.V.; Williams, M.T. Effects of developmental stress and lead (Pb) on corticosterone after chronic and acute stress, brain monoamines, and blood Pb levels in rats. Int. J. Dev. Neurosci. 2011, 29, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Huff, N.C.; Smith, S.H.; Mason, S.N.; Foster, W.M.; Auten, R.L.; Bilbo, S.D. Maternal stress and effects of prenatal air pollution on offspring mental health outcomes in mice. Environ. Health Perspect. 2013, 121, 1075–1082. [Google Scholar] [PubMed]
- Levin, E.D.; Cauley, M.; Johnson, J.E.; Cooper, E.M.; Stapleton, H.M.; Ferguson, P.L.; Seidler, F.J.; Slotkin, T.A. Prenatal dexamethasone augments the neurobehavioral teratology of chlorpyrifos: Significance for maternal stress and preterm labor. Neurotoxicol. Teratol. 2014, 41, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Welshons, W.V.; Vom Saal, F.S.; Toutain, P.L.; Myers, J.P. Should oral gavage be abandoned in toxicity testing of endocrine disruptors? Environ. Health 2014, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Seidler, F.J.; Aldridge, J.E.; Tate, C.A.; Cousins, M.M.; Slotkin, T.A. Critical periods for chlorpyrifos-induced developmental neurotoxicity: Alterations in adenylyl cyclase signaling in adult rat brain regions after gestational or neonatal exposure. Environ. Health Perspect. 2004, 112, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.A.; Clemens, L.G.; Nunez, A.A. Mother counts: How effects of environmental contaminants on maternal care could affect the offspring and future generations. Front. Neuroendocrinol. 2010, 31, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Nephew, B.; Murgatroyd, C. The role of maternal care in shaping CNS function. Neuropeptides 2013, 47, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Chiarotti, F.; Alleva, E.; Bignami, G. Problems of test choice and data analysis in behavioral teratology: The case of prenatal benzodiazepines. Neurotoxicol. Teratol. 1987, 9, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Zorrilla, E.P. Multiparous species present problems (and possibilities) to developmentalists. Dev. Psychobiol. 1997, 30, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Lazic, S.E.; Essioux, L. Improving basic and translational science by accounting for litter-to-litter variation in animal models. BMC Neurosci. 2013, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.K.; Hogberg, H.T.; Buzanska, L.; Lenas, P.; van Vliet, E.; Hartung, T. In vitro developmental neurotoxicity (DNT) testing: Relevant models and endpoints. Neurotoxicology 2010, 31, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Hines, R.N.; Sargent, D.; Autrup, H.; Birnbaum, L.S.; Brent, R.L.; Doerrer, N.G.; Cohen Hubal, E.A.; Juberg, D.R.; Laurent, C.; Luebke, R.; et al. Approaches for assessing risks to sensitive populations: Lessons learned from evaluating risks in the pediatric population. Toxicol. Sci. 2010, 113, 4–26. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Felice, A.; Ricceri, L.; Venerosi, A.; Chiarotti, F.; Calamandrei, G. Multifactorial Origin of Neurodevelopmental Disorders: Approaches to Understanding Complex Etiologies. Toxics 2015, 3, 89-129. https://doi.org/10.3390/toxics3010089
De Felice A, Ricceri L, Venerosi A, Chiarotti F, Calamandrei G. Multifactorial Origin of Neurodevelopmental Disorders: Approaches to Understanding Complex Etiologies. Toxics. 2015; 3(1):89-129. https://doi.org/10.3390/toxics3010089
Chicago/Turabian StyleDe Felice, Alessia, Laura Ricceri, Aldina Venerosi, Flavia Chiarotti, and Gemma Calamandrei. 2015. "Multifactorial Origin of Neurodevelopmental Disorders: Approaches to Understanding Complex Etiologies" Toxics 3, no. 1: 89-129. https://doi.org/10.3390/toxics3010089
APA StyleDe Felice, A., Ricceri, L., Venerosi, A., Chiarotti, F., & Calamandrei, G. (2015). Multifactorial Origin of Neurodevelopmental Disorders: Approaches to Understanding Complex Etiologies. Toxics, 3(1), 89-129. https://doi.org/10.3390/toxics3010089