
Contamination Characteristics of Antibiotic Resistance Genes in Multi-Vector Environment in Typical Regional Fattening House

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Ethics
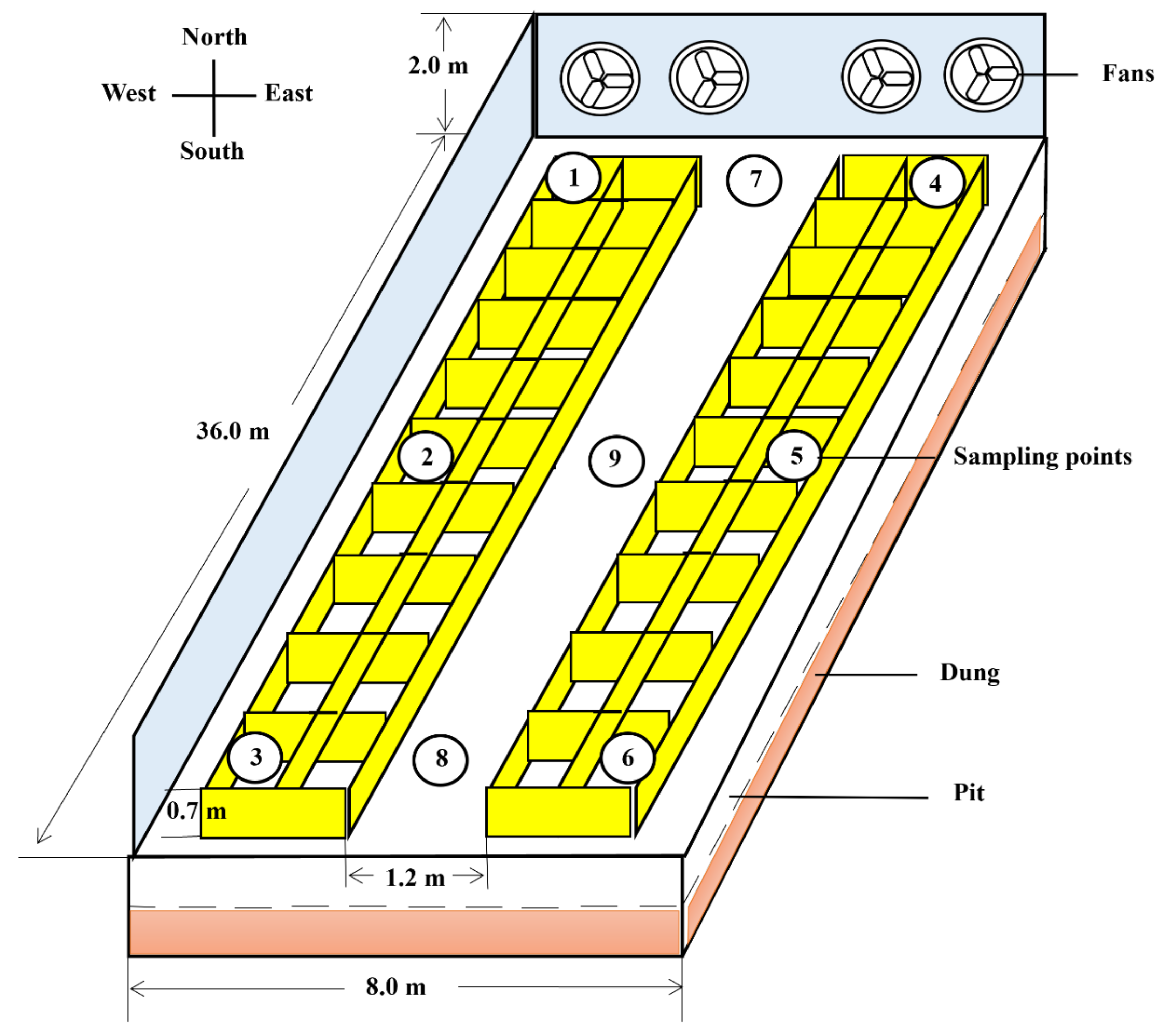
2.2. Fattening House Description
2.3. Sample Collection
2.4. Metagenome Analysis
2.5. PCR Assay
2.6. Statistical Analysis
3. Results
3.1. Microbial Gene Number and Microbial Components in Different Vectors
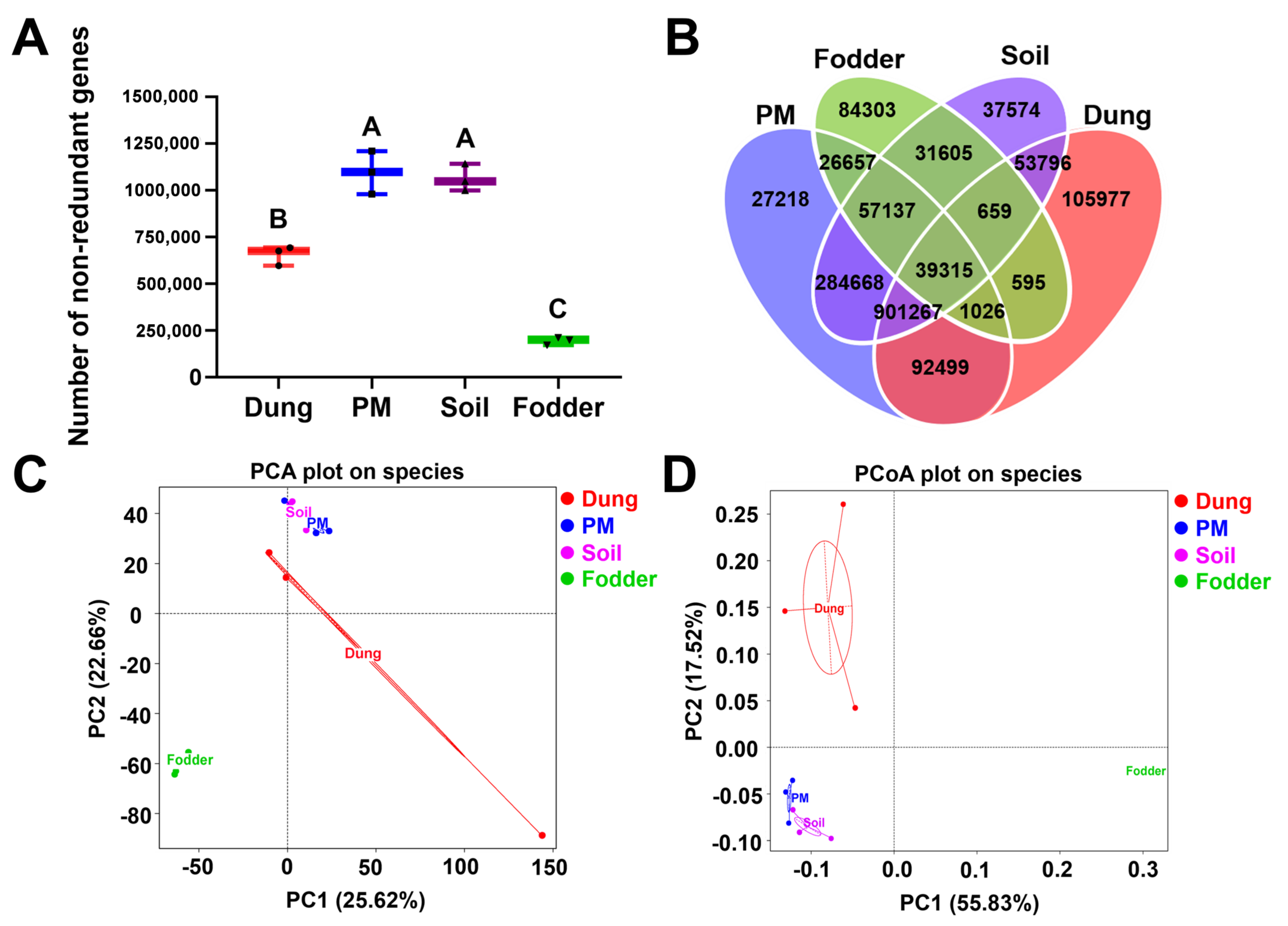
3.2. Dominant Microbiota in Different Vectors
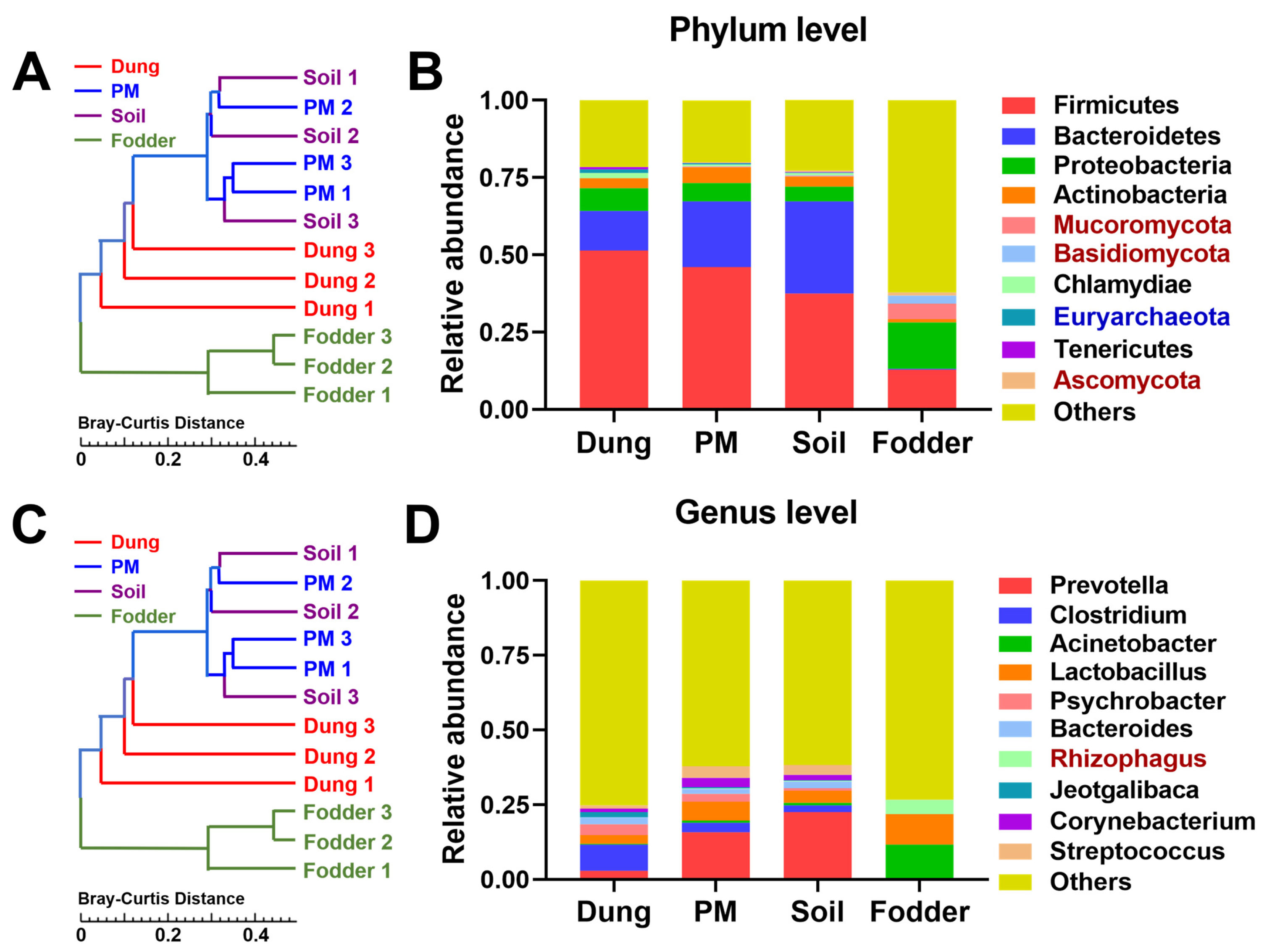
3.3. Differential Microbiota in Different Vectors
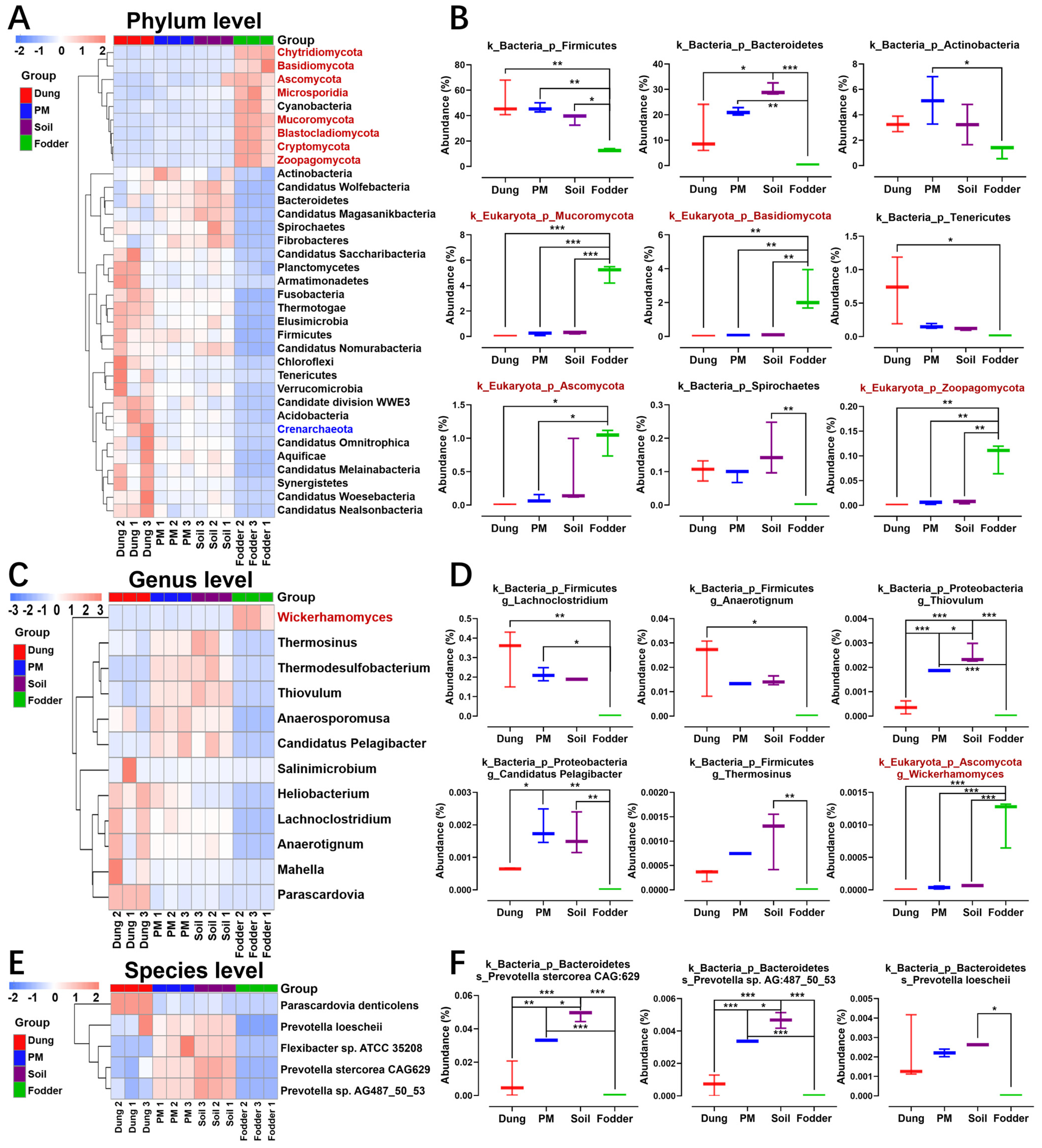
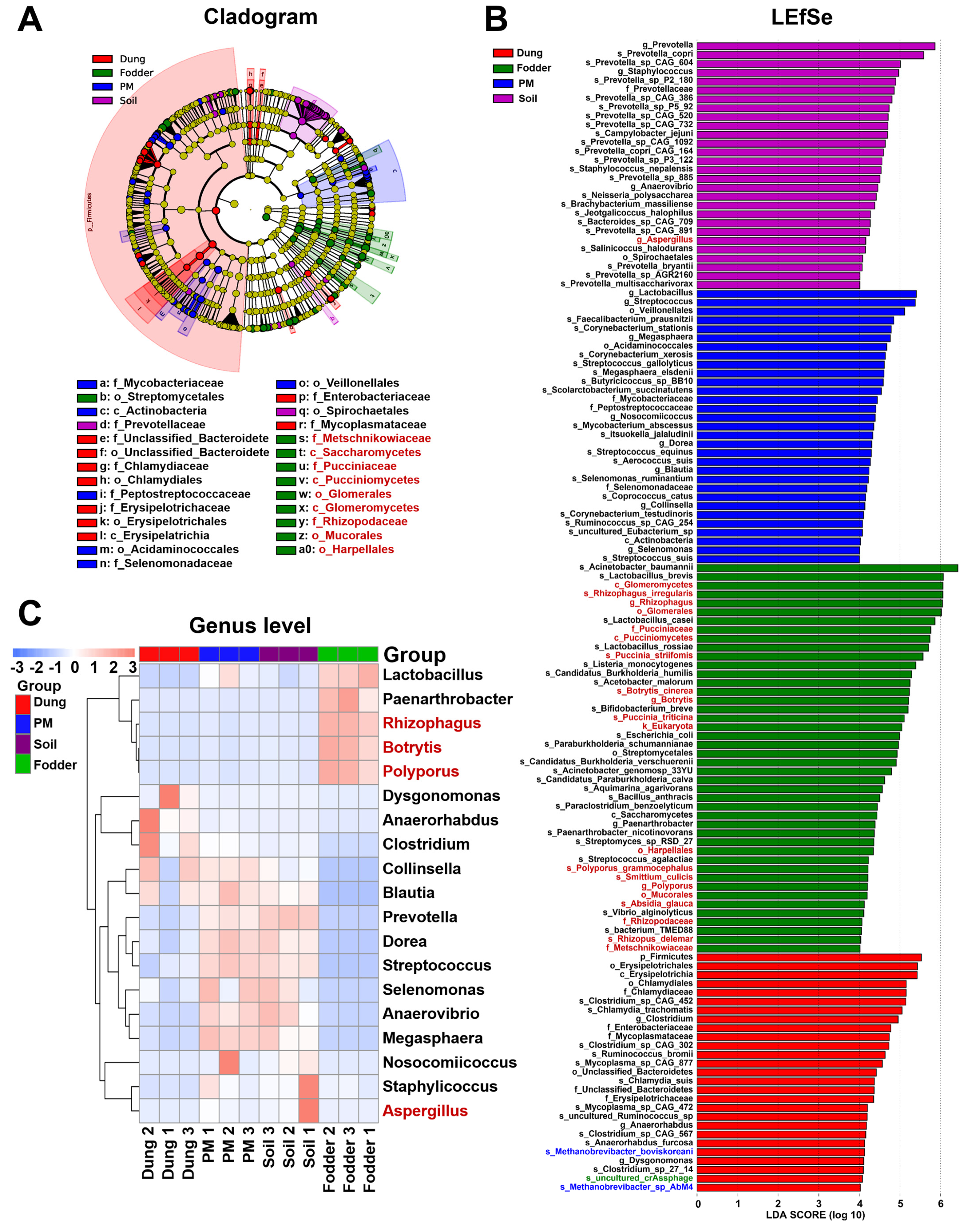
3.4. Featured Microbial Taxa in Different Vectors
3.5. Microbial Functional Prediction in Different Vectors
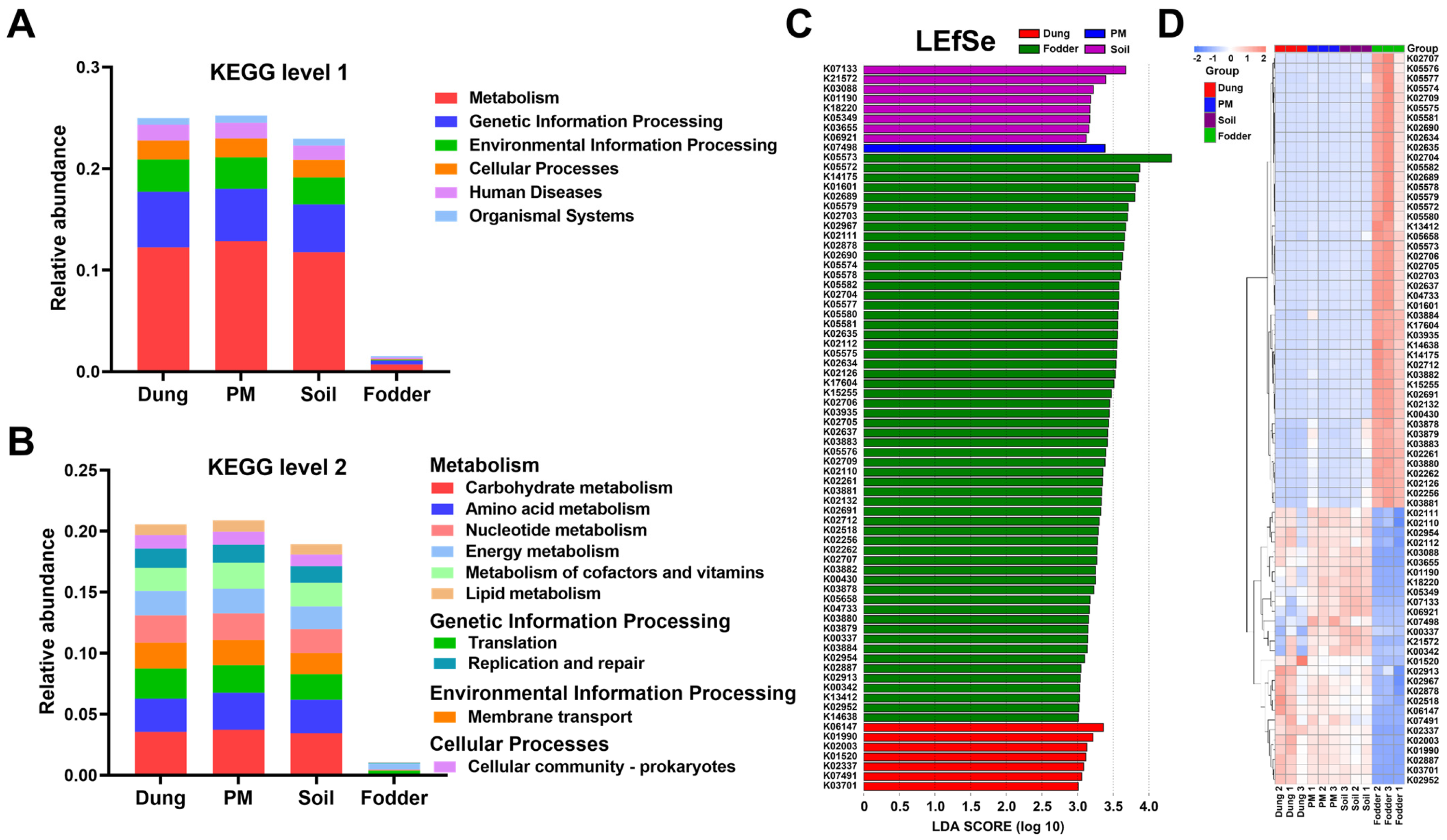
3.6. ARG Number and ARO Components in Different Vectors
3.7. Featured ARGs in Different Vectors
3.8. ARGs and Microbiota Attribution
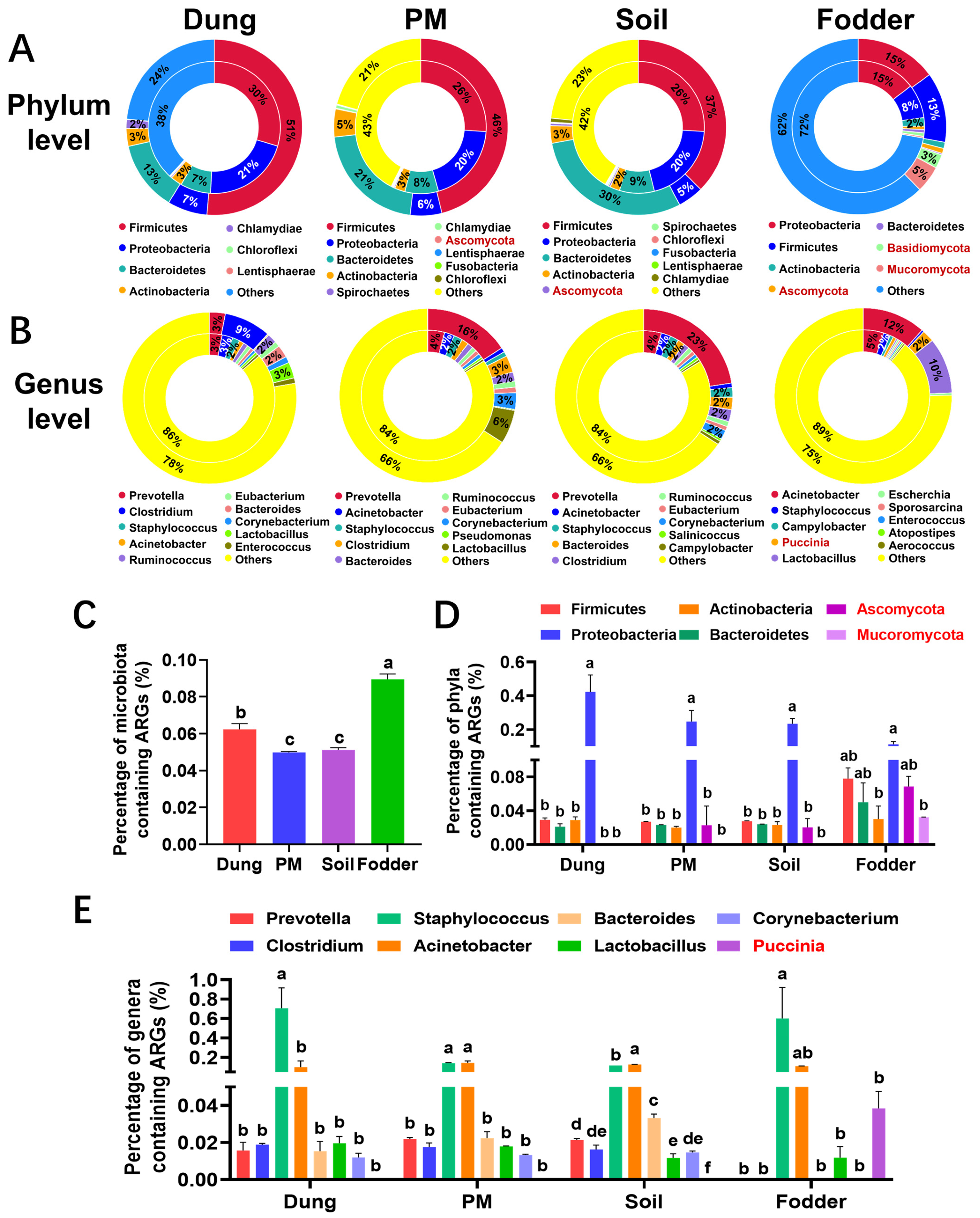
3.9. Absolute Quantification of Major ARGs

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Zhang, Y.; Li, J.; Lin, J.G.; Zhang, N.; Cao, W. Biogas energy generated from livestock manure in China: Current situation and future trends. J. Environ. Manag. 2021, 297, 113324. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Johnson, T.A.; Su, J.Q.; Qiao, M.; Guo, G.X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, G.; Lawlor, P.G.; Gutierrez, M.; Gardiner, G.E. Assessing the biosafety risks of pig manure for use as a feedstock for composting. Sci. Total Environ. 2013, 463–464, 712–719. [Google Scholar] [CrossRef]
- Tang, Q.; Huang, K.; Liu, J.; Shen, D.; Dai, P.; Li, Y.; Li, C. Seasonal variations of microbial assemblage in fine particulate matter from a nursery pig house. Sci. Total Environ. 2020, 708, 134921. [Google Scholar] [CrossRef]
- Reardon, S. Antibiotic use in farming set to soar despite drug-resistance fears. Nature 2023, 614, 397. [Google Scholar] [CrossRef]
- Luiken, R.E.; Heederik, D.J.; Scherpenisse, P.; Van Gompel, L.; van Heijnsbergen, E.; Greve, G.D.; Jongerius-Gortemaker, B.G.; Tersteeg-Zijderveld, M.H.; Fischer, J.; Juraschek, K.; et al. Determinants for antimicrobial resistance genes in farm dust on 333 poultry and pig farms in nine European countries. Environ. Res. 2022, 208, 112715. [Google Scholar] [CrossRef]
- Liu, T.; Li, G.; Liu, Z.; Xi, L.; Ma, W.; Gao, X. Characteristics of aerosols from swine farms: A review of the past two-decade progress. Environ. Int. 2023, 178, 108074. [Google Scholar] [CrossRef]
- Abdelrazik, E.; El-Hadidi, M. Tracking Antibiotic Resistance from the Environment to Human Health. Methods Mol. Biol. 2023, 2649, 289–301. [Google Scholar] [CrossRef]
- Xu, M.; Li, H.; Li, S.; Li, C.; Li, J.; Ma, Y. The presence of tetracyclines and sulfonamides in swine feeds and feces: Dependence on the antibiotic type and swine growth stages. Environ. Sci. Pollut. Res. 2020, 27, 43093–43102. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, Y.; Yuan, Y.; Xie, Y. A systematic review on antibiotics misuse in livestock and aquaculture and regulation implications in China. Sci. Total Environ. 2021, 798, 149205. [Google Scholar] [CrossRef]
- Ai-e, W.; Meng-Qi, Y.; De-Hai, W. Spatial-temporal characteristics and decoupling effect of carbon emissions in the major pig producing areas in China. J. Agric. Resour. Environ. 2018, 35, 269. [Google Scholar]
- Yan, Z.; Wang, C.; Liu, T. An analysis of the environmental efficiency of pig farms and its determinants—A field study from China. Environ. Sci. Pollut. Res. 2020, 27, 38084–38093. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zhang, H.; Song, D.; Chen, H.; Lin, X.; Wang, Y.; Ji, L. Distribution of antibiotic, heavy metals and antibiotic resistance genes in livestock and poultry feces from different scale of farms in Ningxia, China. J. Hazard. Mater. 2022, 440, 129719. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.Z.; He, L.Y.; Bai, H.; He, L.X.; Zhang, M.; Chen, Z.Y.; Liu, Y.S.; Ying, G.G. Airborne bacterial community and antibiotic resistome in the swine farming environment: Metagenomic insights into livestock relevance, pathogen hosts and public risks. Environ. Int. 2023, 172, 107751. [Google Scholar] [CrossRef]
- GB 13078-2017; Standardization Administration of the People’s Republic of China. Hygienical Standard for Feeds. Standards Press of China: Beijing, China, 2017.
- Wang, Y.; Li, Y.; Li, H.; Zhou, J.; Wang, T. Seasonal dissemination of antibiotic resistome from livestock farms to surrounding soil and air: Bacterial hosts and risks for human exposure. J. Environ. Manag. 2023, 325, 116638. [Google Scholar] [CrossRef]
- Shen, D.; Wu, S.; Li, Z.; Tang, Q.; Dai, P.; Li, Y.; Li, C. Distribution and physicochemical properties of particulate matter in swine confinement barns. Environ. Pollut. 2019, 250, 746–753. [Google Scholar] [CrossRef]
- Tett, A.; Pasolli, E.; Masetti, G.; Ercolini, D.; Segata, N. Prevotella diversity, niches and interactions with the human host. Nat. Rev. Microbiol. 2021, 19, 585–599. [Google Scholar] [CrossRef]
- Bonfante, P.; Venice, F. Mucoromycota: Going to the roots of plant-interacting fungi. Fungal Biol. Rev. 2020, 34, 100–113. [Google Scholar] [CrossRef]
- He, M.; Zhao, R.; Liu, D.; Denchev, T.T.; Begerow, D.; Yurkov, A.; Kemler, M.; Millanes, A.M.; Wedin, M.; McTaggart, A.R.; et al. Species diversity of Basidiomycota. Fungal Divers. 2022, 114, 281–325. [Google Scholar] [CrossRef]
- Challacombe, J.F.; Hesse, C.N.; Bramer, L.M.; McCue, L.A.; Lipton, M.; Purvine, S.; Nicora, C.; Gallegos-Graves, V.; Porras-Alfaro, A.; Kuske, C.R. Genomes and secretomes of Ascomycota fungi reveal diverse functions in plant biomass decomposition and pathogenesis. BMC Genom. 2019, 20, 976. [Google Scholar] [CrossRef]
- Oliveira, T.C.; Cabral, J.; Santana, L.R.; Tavares, G.G.; Santos, L.; Paim, T.P.; Muller, C.; Silva, F.G.; Costa, A.C.; Souchie, E.L.; et al. The arbuscular mycorrhizal fungus Rhizophagus clarus improves physiological tolerance to drought stress in soybean plants. Sci. Rep. 2022, 12, 9044. [Google Scholar] [CrossRef] [PubMed]
- Horz, H.P.; Conrads, G. The discussion goes on: What is the role of Euryarchaeota in humans? Archaea 2010, 2010, 967271. [Google Scholar] [CrossRef]
- Caruso, R.; Lo, B.C.; Nunez, G. Host-microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 2020, 20, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Hoegenauer, C.; Hammer, H.F.; Mahnert, A.; Moissl-Eichinger, C. Methanogenic archaea in the human gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 805–813. [Google Scholar] [CrossRef]
- Yue, Z.; Zhang, J.; Zhou, Z.; Ding, C.; Wan, L.; Liu, J.; Chen, L.; Wang, X. Pollution characteristics of livestock faeces and the key driver of the spread of antibiotic resistance genes. J. Hazard. Mater. 2021, 409, 124957. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, W.; Yang, L.; Stedtfeld, R.D.; Peng, A.; Gu, C.; Boyd, S.A.; Li, H. Antibiotic resistance genes and bacterial communities in cornfield and pasture soils receiving swine and dairy manures. Environ. Pollut. 2019, 248, 947–957. [Google Scholar] [CrossRef]
- Durso, L.M.; Cook, K.L. Impacts of antibiotic use in agriculture: What are the benefits and risks? Curr. Opin. Microbiol. 2014, 19, 37–44. [Google Scholar] [CrossRef]
- Zhou, L.J.; Ying, G.G.; Zhang, R.Q.; Liu, S.; Lai, H.J.; Chen, Z.F.; Yang, B.; Zhao, J.L. Use patterns, excretion masses and contamination profiles of antibiotics in a typical swine farm, south China. Environ. Sci. Process. Impacts 2013, 15, 802–813. [Google Scholar] [CrossRef]
- Kim, E.; Aoki, T. Sequence analysis of the florfenicol resistance gene encoded in the transferable R-plasmid of a fish pathogen, Pasteurella piscicida. Microbiol. Immunol. 1996, 40, 665–669. [Google Scholar] [CrossRef]
- Yang, F.; Han, B.; Gu, Y.; Zhang, K. Swine liquid manure: A hotspot of mobile genetic elements and antibiotic resistance genes. Sci. Rep. 2020, 10, 15037. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, Z.; Hu, B.; Zhu, L. Partitioning and migration of antibiotic resistance genes at soil-water-air interface mediated by plasmids. Environ. Pollut. 2023, 327, 121557. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, C.; Jiang, G.; Ma, J.; Li, Y.; Chen, H.; Guo, J. Bioaerosol is an important transmission route of antibiotic resistance genes in pig farms. Environ. Int. 2021, 154, 106559. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yan, Z.; Zhang, Z.; Zhou, Q.; Zhou, J.; Wakeland, E.K.; Fang, X.; Xuan, Z.; Shen, D.; Li, Q.Z. Complete genome analysis of three Acinetobacter baumannii clinical isolates in China for insight into the diversification of drug resistance elements. PLoS ONE 2013, 8, e66584. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic resistance genes in bacteria: Occurrence, spread, and control. J. Basic Microb. 2021, 61, 1049–1070. [Google Scholar] [CrossRef]
- Zalewska, M.; Blazejewska, A.; Czapko, A.; Popowska, M. Antibiotics and Antibiotic Resistance Genes in Animal Manure—Consequences of Its Application in Agriculture. Front. Microbiol. 2021, 12, 610656. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Shen, D.; Guo, Z.; Zhong, Q.; Huang, K. Contamination Characteristics of Antibiotic Resistance Genes in Multi-Vector Environment in Typical Regional Fattening House. Toxics 2024, 12, 916. https://doi.org/10.3390/toxics12120916
Wang K, Shen D, Guo Z, Zhong Q, Huang K. Contamination Characteristics of Antibiotic Resistance Genes in Multi-Vector Environment in Typical Regional Fattening House. Toxics. 2024; 12(12):916. https://doi.org/10.3390/toxics12120916
Chicago/Turabian StyleWang, Kai, Dan Shen, Zhendong Guo, Qiuming Zhong, and Kai Huang. 2024. "Contamination Characteristics of Antibiotic Resistance Genes in Multi-Vector Environment in Typical Regional Fattening House" Toxics 12, no. 12: 916. https://doi.org/10.3390/toxics12120916
APA StyleWang, K., Shen, D., Guo, Z., Zhong, Q., & Huang, K. (2024). Contamination Characteristics of Antibiotic Resistance Genes in Multi-Vector Environment in Typical Regional Fattening House. Toxics, 12(12), 916. https://doi.org/10.3390/toxics12120916