
A Novel Bacillus Velezensis for Efficient Degradation of Zearalenone

 ,
,
Abstract
1. Introduction
2. Materials and Methods
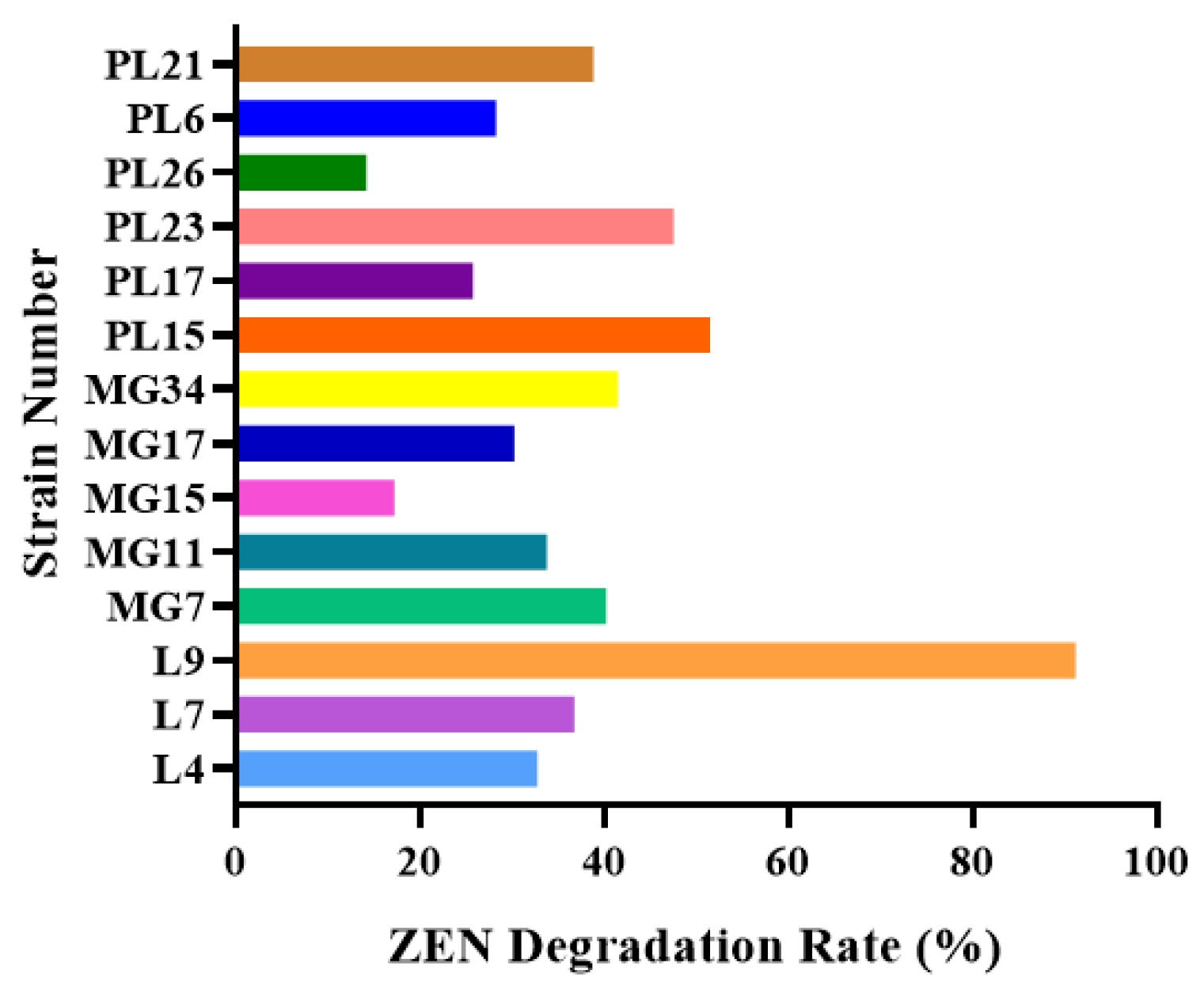
2.1. Isolation of Zearalenone-Degrading Strains
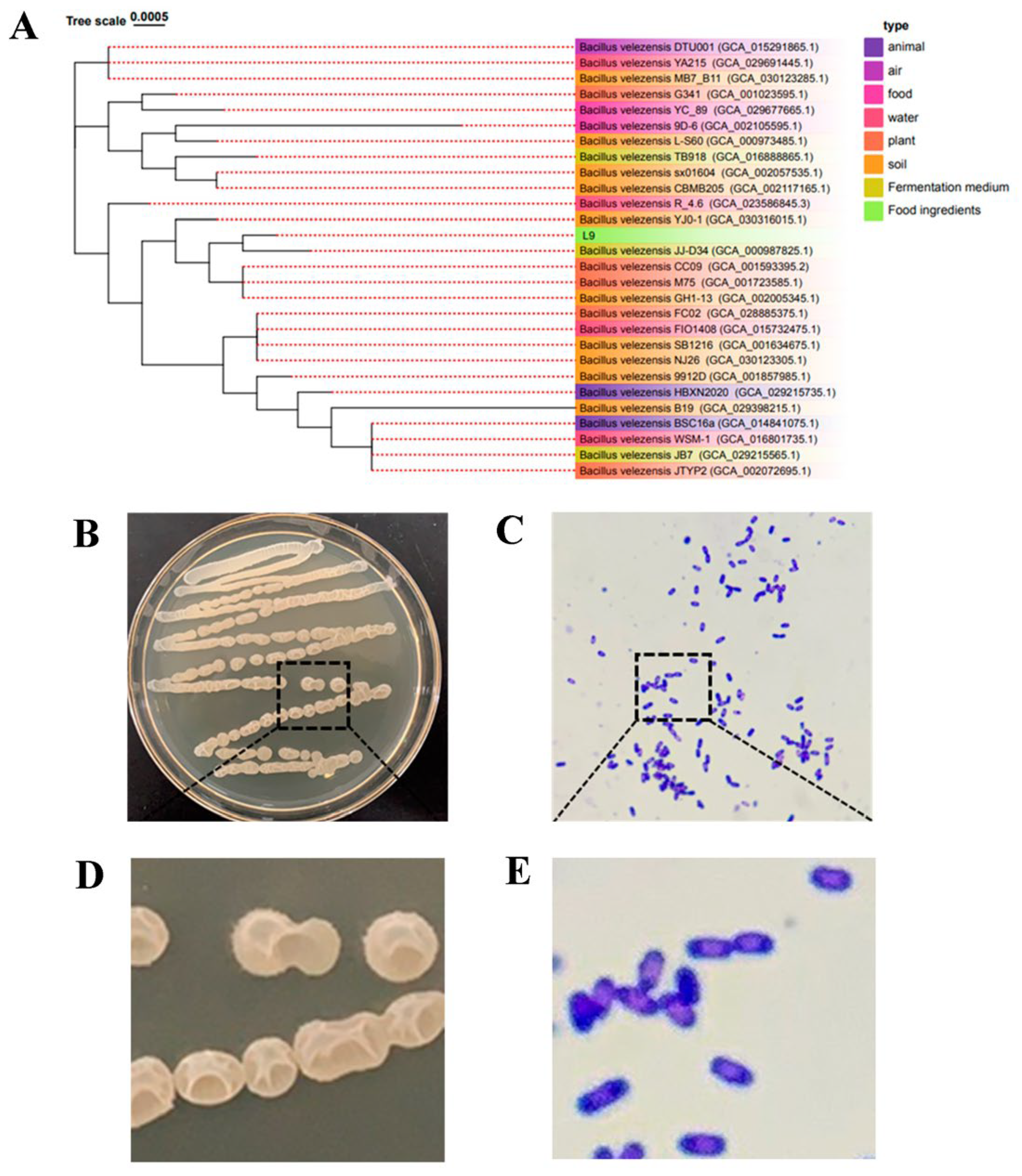
2.2. 16S rDNA Sequencing of L9 Strain
2.3. Morphological Identification of L9 Strain
2.4. The Adsorption Ability of L9 Strain to ZEN
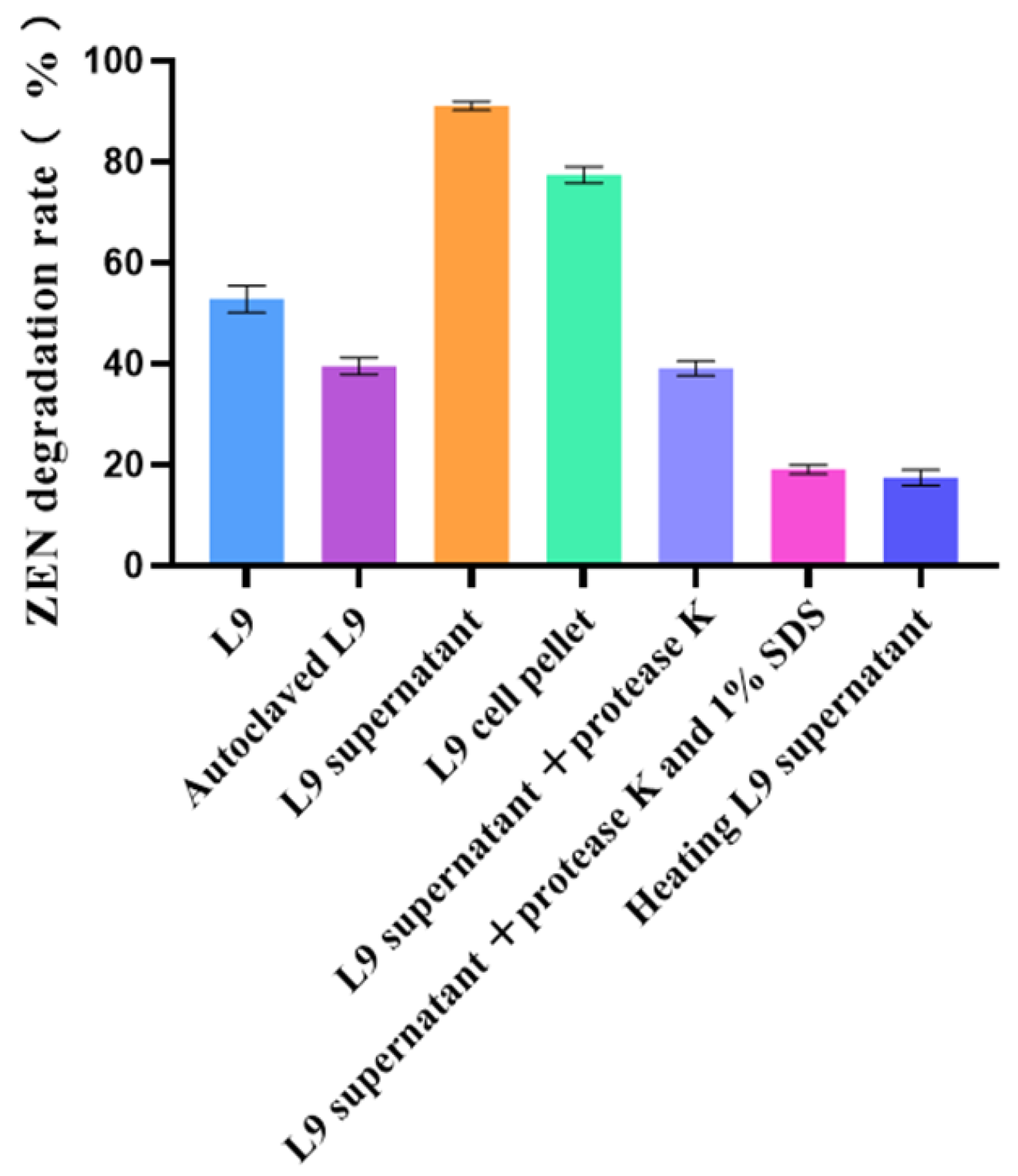
2.5. Validation of Degradation Mechanisms of L9 Strain
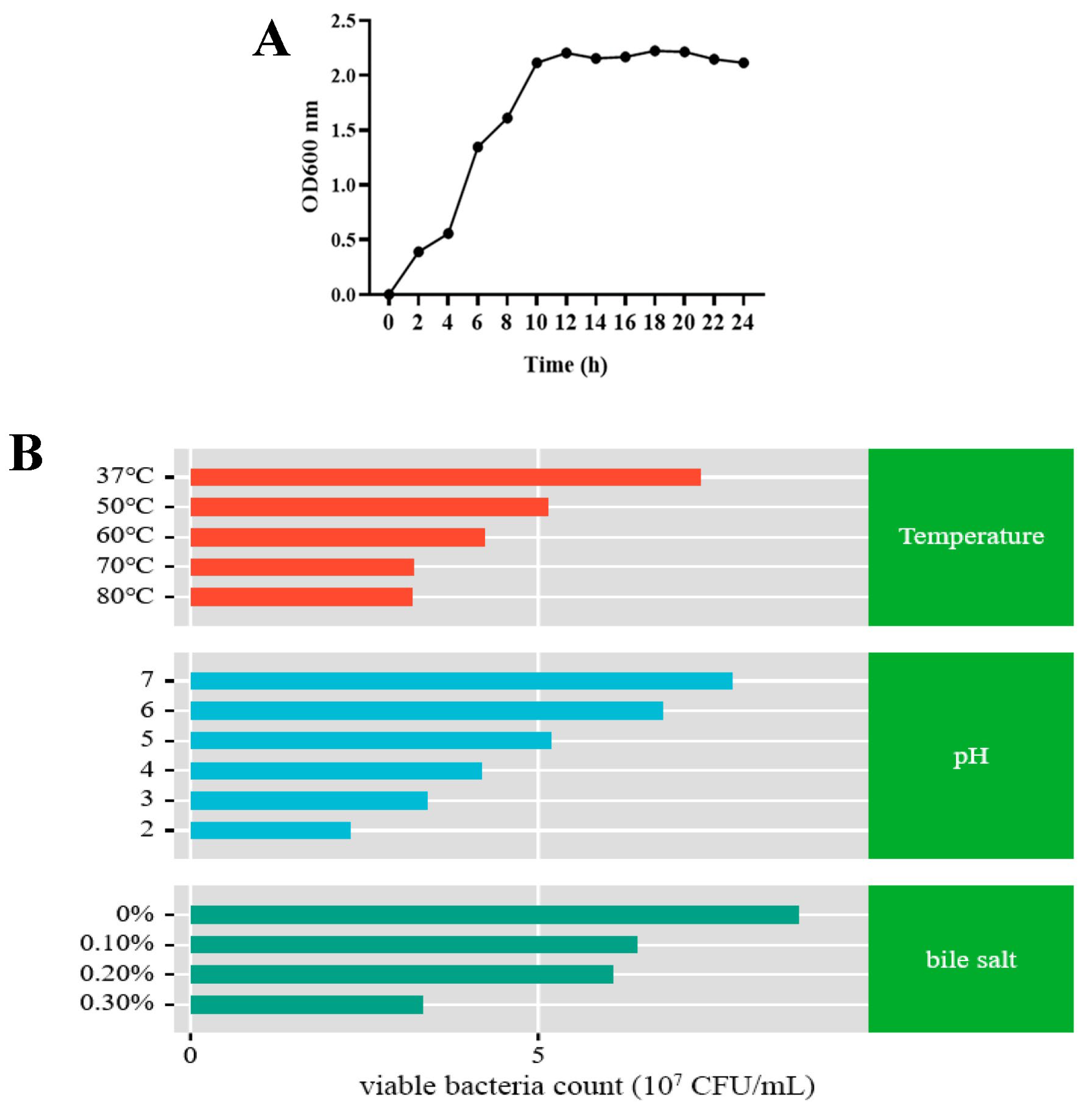
2.6. Temperature, Acid, and Bile Salt Tolerance Assay of L9 Strain
2.6.1. Determination of Strain Growth Curve
2.6.2. Effects of Temperature on L9 Strain
2.6.3. Effects of pH on L9 Strain
2.6.4. Effects of Bile Salts on L9 Strain
2.7. Genome Sequencing of L9 Strain
2.8. Quality Control and Assembly of Genome Sequencing Data of L9 Strain
2.9. Gene Component Prediction of L9 Strain
2.10. Basic Genome Annotation of L9 Strain
2.11. Prediction of Genes Encoding for CAZymes and Secondary Metabolites
2.12. Average Nucleotide Consistency (ANI) Analysis
2.13. Pan-Genome Analysis
3. Results
3.1. Isolation and Identification of L9 Strain
3.2. Analysis of Mechanism of Degradation of ZEN by L9 Strain
3.3. Temperature, Acid, and Bile Salt Tolerance of L9 Strain
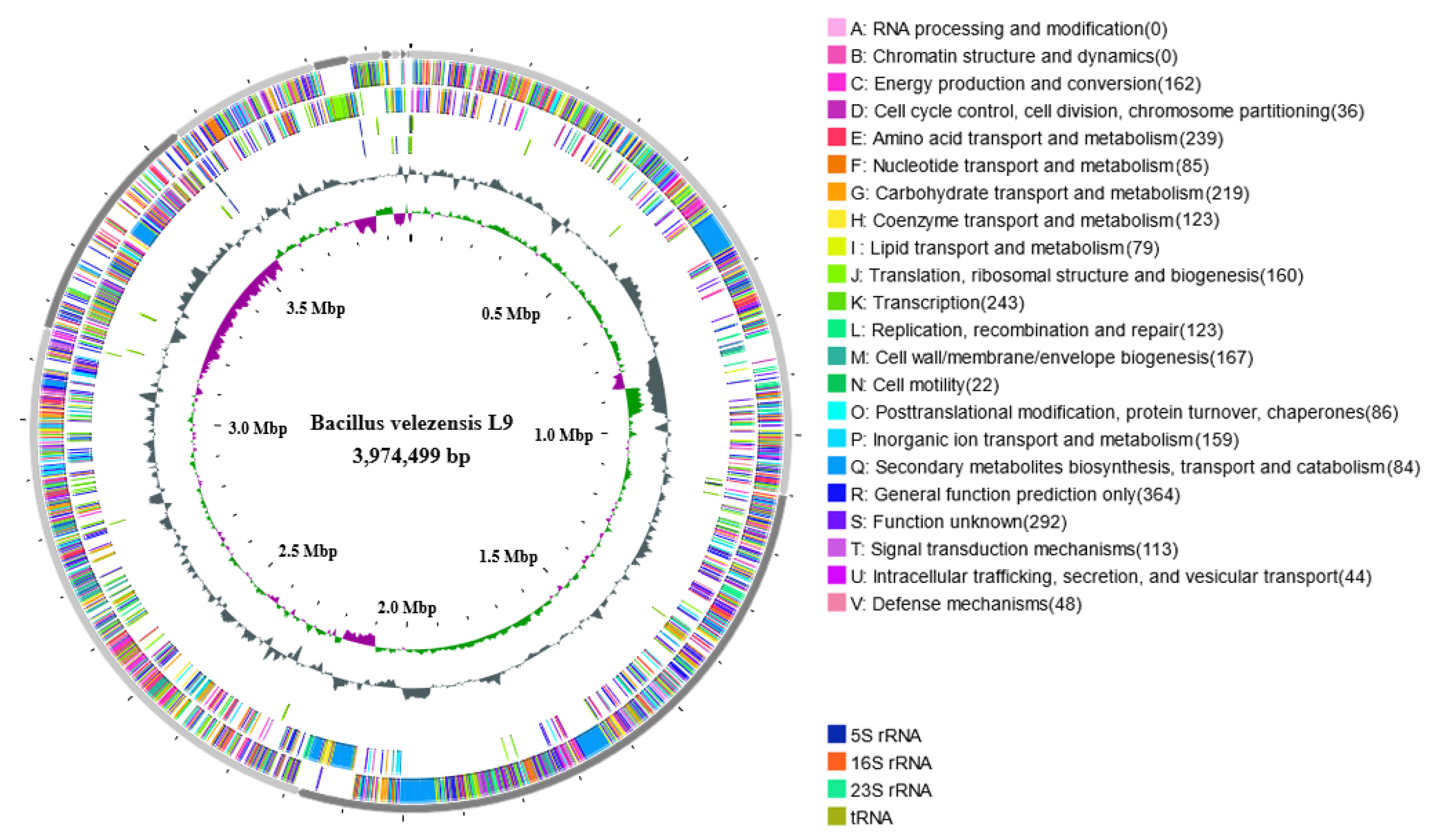
3.4. Genomic Overview of L9 Strain
3.5. Genomic Basis Functional Annotation Results
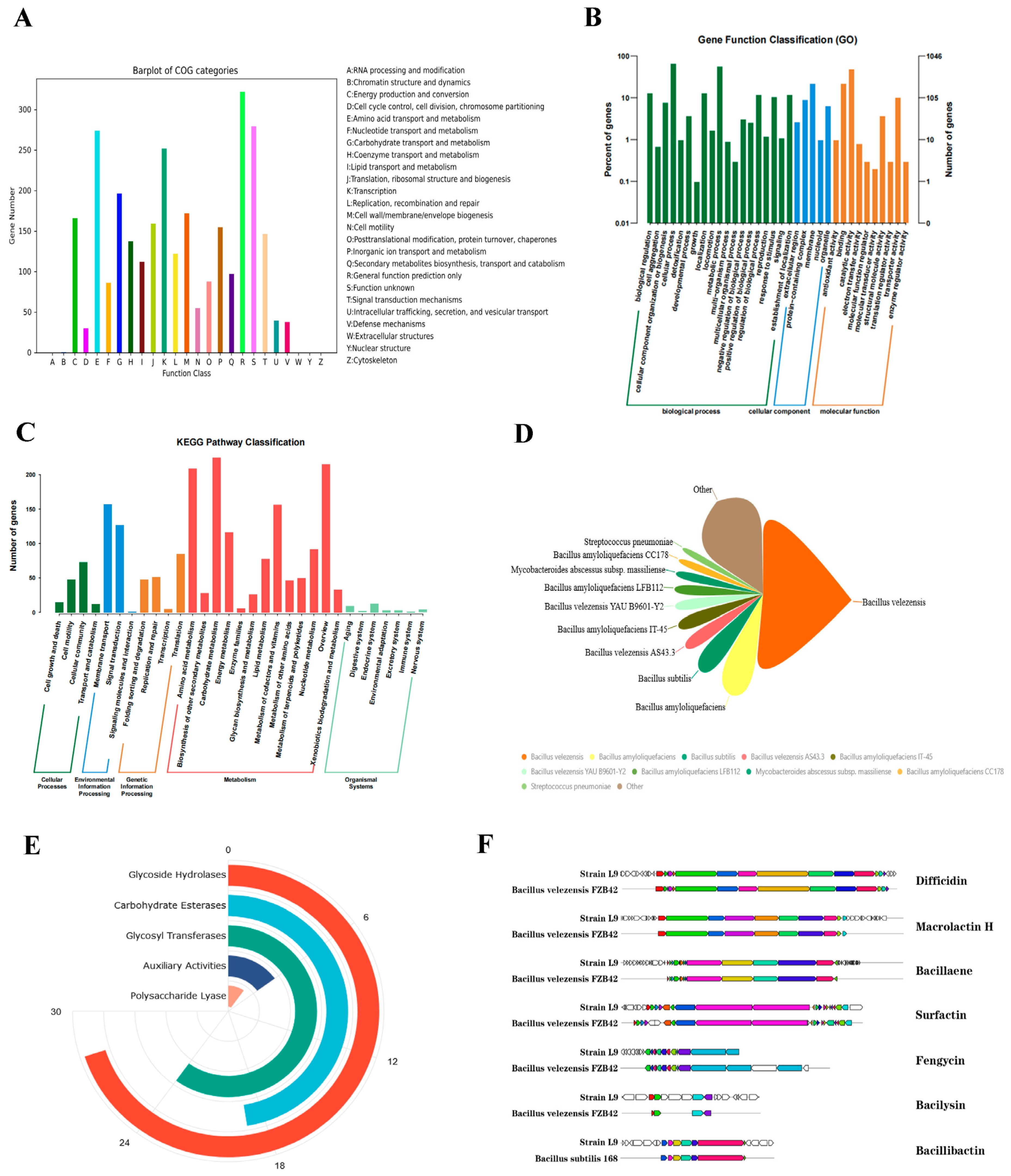
3.5.1. COG Database Annotation Results
3.5.2. GO Database Annotation Results
3.5.3. KEGG Database Annotation Results
3.5.4. NR Database Annotation Results
3.6. CAZy Classification Result
3.7. Secondary Metabolite Analysis
3.8. Average Nucleotide Identity (ANI) Analysis
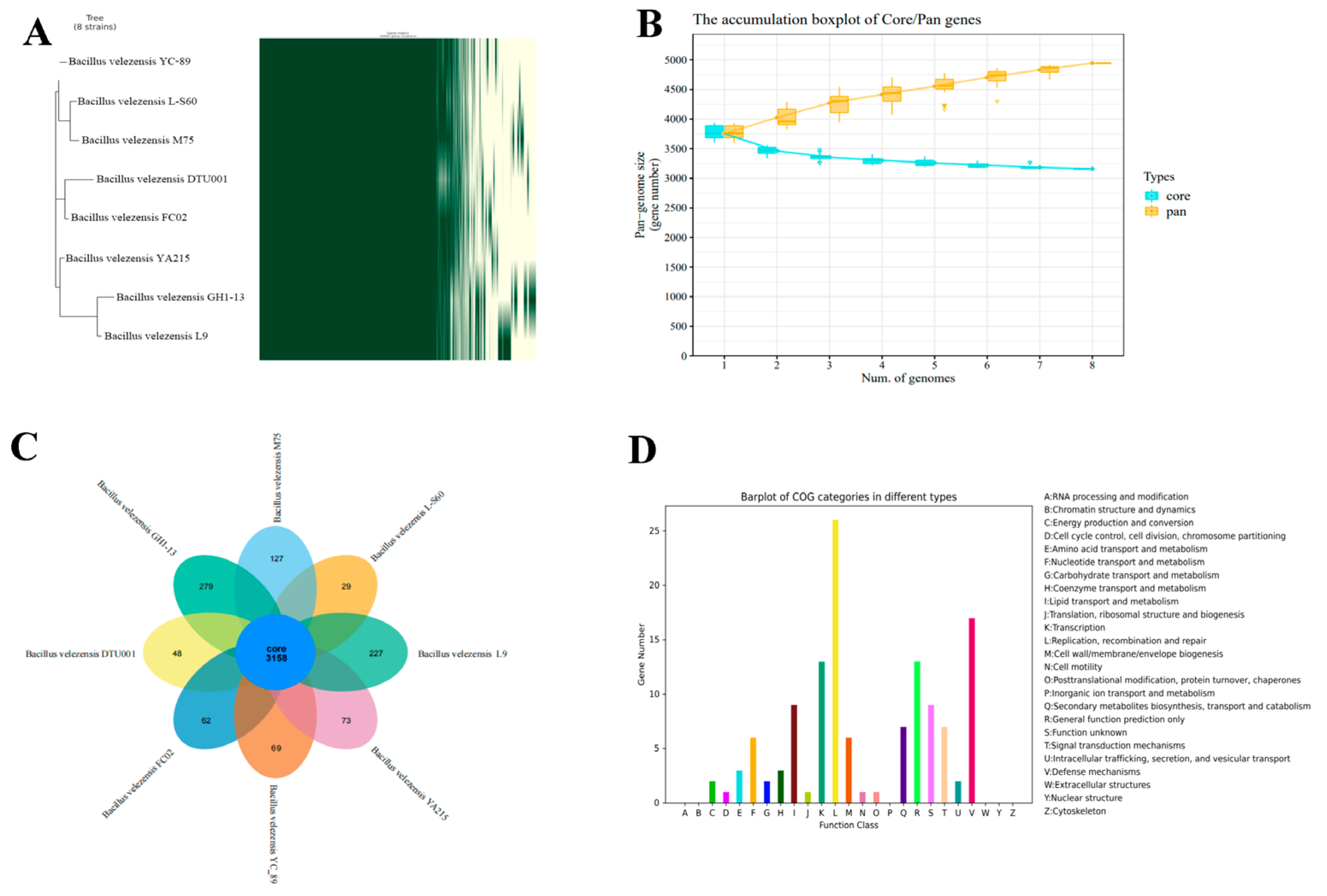
3.9. Pan-Genome Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mukhtar, K.; Nabi, B.; Ansar, S.; Bhat, Z.; Aadil, R.; Mousavi Khaneghah, A. Mycotoxins and consumers’ awareness: Recent progress and future challenges. Toxicon Off. J. Int. Soc. Toxinol. 2023, 232, 107227. [Google Scholar] [CrossRef] [PubMed]
- Lijalem, Y.; Gab-Allah, M.; Yu, H.; Choi, K.; Kim, B. Occurrence of zearalenone and its major metabolites in cereal flour from Korea. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2023, 40, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, F.; Schwake-Anduschus, C. Relevance of Zearalenone and its modified forms in bakery products. Mycotoxin Res. 2023, 39, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Balló, A.; Busznyákné Székvári, K.; Czétány, P.; Márk, L.; Török, A.; Szántó, Á.; Máté, G. Estrogenic and Non-Estrogenic Disruptor Effect of Zearalenone on Male Reproduction: A Review. Int. J. Mol. Sci. 2023, 24, 1578. [Google Scholar] [CrossRef] [PubMed]
- Güneş, B.; Yalçın, S.; Yalçın, S. Longitudinal follow-up of zearalenone and deoxynivalenol mycotoxins in breast milk in the first five months of life. BMC Pharmacol. Toxicol. 2023, 24, 37. [Google Scholar] [CrossRef] [PubMed]
- Gajęcki, M.; Gajęcka, M. The Multidirectional Influence of Feed-Borne Deoxynivalenol and Zearalenone on Animal Health. Toxins 2023, 15, 419. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Fu, W.; Zhao, X.; Chang, X.; Liu, H.; Zhou, L.; Li, J.; Cheng, R.; Wu, X.; Li, X.; et al. Zearalenone disturbs the reproductive-immune axis in pigs: The role of gut microbial metabolites. Microbiome 2022, 10, 234. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jiang, Y.; Lu, F.; Wang, S.; Liu, M.; Liu, F.; Huang, L.; Li, Y.; Jiao, N.; Jiang, S.; et al. Quantitative Proteomic Analysis of Zearalenone-Induced Intestinal Damage in Weaned Piglets. Toxins 2022, 14, 702. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, M.; Du, Y.; Li, P.; Huang, Z. Zearalenone induces the senescence of cardiovascular cells in vitro and in vivo. Environ. Sci. Pollut. Res. Int. 2023, 30, 56037–56053. [Google Scholar] [CrossRef]
- Eskola, M.; Kos, G.; Elliott, C.; Hajšlová, J.; Mayar, S.; Krska, R. Worldwide contamination of food-crops with mycotoxins: Validity of the widely cited ‘FAO estimate’ of 25. Crit. Rev. Food Sci. Nutr. 2020, 60, 2773–2789. [Google Scholar] [CrossRef]
- Mao, X.; Chen, W.; Wu, H.; Shao, Y.; Zhu, Y.; Guo, Q.; Li, Y.; Xia, L. Alternaria Mycotoxins Analysis and Exposure Investigation in Ruminant Feeds. Toxins 2023, 15, 495. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Guan, S.; Li, A.; Wang, J.; An, G.; Hofstetter, U.; Schatzmayr, G. Mycotoxin Occurrence in Feeds and Raw Materials in China: A Five-Year Investigation. Toxins 2023, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Zhang, W.; Xu, N.; Jiang, X.; Cheng, J.; Wang, R.; Wang, P. Efficient and simultaneous removal of aflatoxin B, B, G, G, and zearalenone from vegetable oil by use of a metal-organic framework absorbent. Food Chem. 2023, 418, 135881. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Li, K.; Pan, L.; Luo, X.; Xing, J.; Wang, J.; Wang, L.; Wang, R.; Zhai, Y.; Chen, Z. Effect of Ozone and Electron Beam Irradiation on Degradation of Zearalenone and Ochratoxin A. Toxins 2020, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Xu, H.; Ji, Q.; Xu, R.; Zhu, M.; Dang, Y.; Shi, X.; Zhang, L.; Xia, Y. Mutation, food-grade expression, and characterization of a lactonase for zearalenone degradation. Appl. Microbiol. Biotechnol. 2023, 107, 5107–5118. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, A.; Yu, Q.; Tang, Y.; Yu, Y. Acinetobacter sp. Induced Expression of the Oxa Gene in and Its Increased ZEN Degradation Stability by Immobilization. Toxins 2023, 15, 387. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zheng, H.; Xu, J.; Zhao, X.; Shu, W.; Li, X.; Song, H.; Ma, Y. New Biotransformation Mode of Zearalenone Identified in Bacillus subtilis Y816 Revealing a Novel ZEN Conjugate. J. Agric. Food Chem. 2021, 69, 7409–7419. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bai, Y.; Huang, H.; Tu, T.; Wang, Y.; Wang, Y.; Luo, H.; Yao, B.; Su, X. Degradation of Aflatoxin B and Zearalenone by Bacterial and Fungal Laccases in Presence of Structurally Defined Chemicals and Complex Natural Mediators. Toxins 2019, 11, 609. [Google Scholar] [CrossRef]
- Chen, S.; Pan, L.; Liu, S.; Pan, L.; Li, X.; Wang, B. Recombinant expression and surface display of a zearalenone lactonohydrolase from Trichoderma aggressivum in Escherichia coli. Protein Expr. Purif. 2021, 187, 105933. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization by One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Bianco, A.; Capozzi, L.; Monno, M.; Del Sambro, L.; Manzulli, V.; Pesole, G.; Loconsole, D.; Parisi, A. Characterization of Bacillus cereus Group Isolates From Human Bacteremia by Whole-Genome Sequencing. Front. Microbiol. 2020, 11, 599524. [Google Scholar] [CrossRef]
- Brown, J.; Pirrung, M.; McCue, L. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef]
- Bayat, A.; Deshpande, N.; Wilkins, M.; Parameswaran, S. Fast Short Read De-Novo Assembly Using Overlap-Layout-Consensus Approach. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ye, C.; Li, X.; Chen, Q.; Wu, Y.; Zhang, F.; Pan, R.; Zhang, S.; Chen, S.; Wang, X.; et al. quarTeT: A telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Hortic. Res. 2023, 10, uhad127. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Grant, J.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinform. 2019, 20, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.; Kristensen, D.; Makarova, K.; Wolf, Y.; Koonin, E. Microbial genome analysis: The COG approach. Brief. Bioinform. 2019, 20, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Garron, M.; Henrissat, B. The continuing expansion of CAZymes and their families. Curr. Opin. Chem. Biol. 2019, 53, 82–87. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.; Charlop-Powers, Z.; van Wezel, G.; Medema, M.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- de Albuquerque, N.; Haag, K. Using average nucleotide identity (ANI) to evaluate microsporidia species boundaries based on their genetic relatedness. J. Eukaryot. Microbiol. 2023, 70, e12944. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, C.; Zhao, D.; Zhang, Y.; You, Y.; Jia, X.; Yang, J.; Wang, L.; Wang, J.; Fu, H.; et al. PGAP-X: Extension on pan-genome analysis pipeline. BMC Genom. 2018, 19, 36. [Google Scholar] [CrossRef] [PubMed]
- Angiuoli, S.; Salzberg, S. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 2011, 27, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Nandi, T.; Ong, C.; Singh, A.; Boddey, J.; Atkins, T.; Sarkar-Tyson, M.; Essex-Lopresti, A.; Chua, H.; Pearson, T.; Kreisberg, J.; et al. A genomic survey of positive selection in Burkholderia pseudomallei provides insights into the evolution of accidental virulence. PLoS Pathog. 2010, 6, e1000845. [Google Scholar] [CrossRef] [PubMed]
- Cai, P.; Liu, S.; Tu, Y.; Shan, T. Toxicity, biodegradation, and nutritional intervention mechanism of zearalenone. Sci. Total Environ. 2024, 911, 168648. [Google Scholar] [CrossRef] [PubMed]
- Calado, T.; Abrunhosa, L.; Cabo Verde, S.; Alté, L.; Venâncio, A.; Fernández-Cruz, M. Effect of Gamma-Radiation on Zearalenone-Degradation, Cytotoxicity and Estrogenicity. Foods 2020, 9, 1687. [Google Scholar] [CrossRef]
- Adegoke, T.; Yang, B.; Tian, X.; Yang, S.; Gao, Y.; Ma, J.; Wang, G.; Si, P.; Li, R.; Xing, F. Simultaneous degradation of aflatoxin B and zearalenone by Porin and Peroxiredoxin enzymes cloned from Acinetobacter nosocomialis Y1. J. Hazard. Mater. 2023, 459, 132105. [Google Scholar] [CrossRef]
- Wang, N.; Li, P.; Pan, J.; Wang, M.; Long, M.; Zang, J.; Yang, S. Bacillus velezensis A2 fermentation exerts a protective effect on renal injury induced by Zearalenone in mice. Sci. Rep. 2018, 8, 13646. [Google Scholar] [CrossRef]
- Chen, S.; Wang, H.; Shih, W.; Ciou, Y.; Chang, Y.; Ananda, L.; Wang, S.; Hsu, J. Application of Zearalenone (ZEN)-Detoxifying Bacillus in Animal Feed Decontamination through Fermentation. Toxins 2019, 11, 330. [Google Scholar] [CrossRef]
- Qin, X.; Su, X.; Tu, T.; Zhang, J.; Wang, X.; Wang, Y.; Wang, Y.; Bai, Y.; Yao, B.; Luo, H.; et al. Enzymatic Degradation of Multiple Major Mycotoxins by Dye-Decolorizing Peroxidase from Bacillus subtilis. Toxins 2021, 13, 429. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ao, X.; Lei, Y.; Ji, C.; Ma, Q. Bacillus subtilis ANSB01G culture alleviates oxidative stress and cell apoptosis induced by dietary zearalenone in first-parity gestation sows. Anim. Nutr. 2020, 6, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Huo, X.; Zhao, L.; Ma, Q.; Zhang, J.; Ji, C.; Zhao, L. Protective Effects of Bacillus subtilis ANSB060, Bacillus subtilis ANSB01G, and Devosia sp. ANSB714-Based Mycotoxin Biodegradation Agent on Mice Fed with Naturally moldy Diets. Probiotics Antimicrob. Proteins 2020, 12, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.; Yu, D.; Liu, N.; Wu, A. Degrading Ochratoxin A and Zearalenone Mycotoxins Using a Multifunctional Recombinant Enzyme. Toxins 2019, 11, 301. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wu, T.; Zhang, H.; Sun, Z.; Mwabulili, F.; Xie, Y.; Sun, S.; Ma, W.; Li, Q.; Yang, Y.; et al. Mining Lactonase Gene from Aflatoxin B-Degrading Strain Bacillus megaterium and Degrading Properties of the Recombinant Enzyme. J. Agric. Food Chem. 2023, 71, 20762–20771. [Google Scholar] [CrossRef] [PubMed]
- Gari, J.; Abdella, R. Degradation of zearalenone by microorganisms and enzymes. PeerJ 2023, 11, e15808. [Google Scholar] [CrossRef] [PubMed]
- Złoch, M.; Rogowska, A.; Pomastowski, P.; Railean-Plugaru, V.; Walczak-Skierska, J.; Rudnicka, J.; Buszewski, B. Use of Lactobacillus paracasei strain for zearalenone binding and metabolization. Toxicon Off. J. Int. Soc. Toxinol. 2020, 181, 9–18. [Google Scholar] [CrossRef]
- Xu, L.; Sun, X.; Wan, X.; Li, H.; Yan, F.; Han, R.; Li, H.; Li, Z.; Tian, Y.; Liu, X.; et al. Identification of a Bacillus amyloliquefaciens H6 Thioesterase Involved in Zearalenone Detoxification by Transcriptomic Analysis. J. Agric. Food Chem. 2020, 68, 10071–10080. [Google Scholar] [CrossRef]
- Taş, N.; de Jong, A.; Li, Y.; Trubl, G.; Xue, Y.; Dove, N. Metagenomic tools in microbial ecology research. Curr. Opin. Biotechnol. 2021, 67, 184–191. [Google Scholar] [CrossRef]
- Cortés, J.; Curto, M.; Carvalho, V.; Pérez, P.; Ribas, J. The fungal cell wall as a target for the development of new antifungal therapies. Biotechnol. Adv. 2019, 37, 107352. [Google Scholar] [CrossRef]
- Nagy, V.; Seidl, V.; Szakacs, G.; Komoń-Zelazowska, M.; Kubicek, C.; Druzhinina, I. Application of DNA bar codes for screening of industrially important fungi: The haplotype of Trichoderma harzianum sensu stricto indicates superior chitinase formation. Appl. Environ. Microbiol. 2007, 73, 7048–7058. [Google Scholar] [CrossRef] [PubMed]
- Latgé, J. Tasting the fungal cell wall. Cell. Microbiol. 2010, 12, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Lim, D.; Baek, K.; Jang, S.; Park, B.; Mayakrishnan, V. Production of chitinase from Escherichia fergusonii, chitosanase from Chryseobacterium indologenes, Comamonas koreensis and its application in N-acetylglucosamine production. Int. J. Biol. Macromol. 2018, 112, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Hasim, S.; Coleman, J. Targeting the fungal cell wall: Current therapies and implications for development of alternative antifungal agents. Future Med. Chem. 2019, 11, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Amos, B.; Pook, V.; Prates, E.; Stork, J.; Shah, M.; Jacobson, D.; DeBolt, S. Discovery and Characterization of Fluopipamine, a Putative Cellulose Synthase 1 Antagonist within Arabidopsis. J. Agric. Food Chem. 2024. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wajjiha, B.; Zhang, P.; Dang, Y.; Prasad, R.; Wei, Y.; Zhang, S. Serendipita indica chitinase protects rice from the blast and bakanae diseases. J. Basic Microbiol. 2023, 63, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Hong, M.; Cha, C. Fungal β-Glycosidase Belonging to Subfamily 4 of Glycoside Hydrolase Family 30 with Transglycosylation Activity. J. Agric. Food Chem. 2021, 69, 15261–15267. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Shen, D.; Xiong, Q.; Bao, B.; Zhang, W.; Dai, T.; Zhao, Y.; Borriss, R.; Fan, B. The Plant-Beneficial Rhizobacterium Bacillus velezensis FZB42 Controls the Soybean Pathogen Phytophthora sojae Due to Bacilysin Production. Appl. Environ. Microbiol. 2021, 87, e0160121. [Google Scholar] [CrossRef]
- Guimarães, C.; Pasqualino, I.; de Sousa, J.; Nogueira, F.; Seldin, L.; de Castilho, L.; Freire, D. Bacillus velezensis H2O-1 surfactin efficiently maintains its interfacial properties in extreme conditions found in post-salt and pre-salt oil reservoirs. Colloids Surfaces. B Biointerfaces 2021, 208, 112072. [Google Scholar] [CrossRef]
- Romero-Tabarez, M.; Jansen, R.; Sylla, M.; Lünsdorf, H.; Häussler, S.; Santosa, D.; Timmis, K.; Molinari, G. 7-O-malonyl macrolactin A, a new macrolactin antibiotic from Bacillus subtilis active against methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, and a small-colony variant of Burkholderia cepacia. Antimicrob. Agents Chemother. 2006, 50, 1701–1709. [Google Scholar] [CrossRef]
- Nannan, C.; Vu, H.; Gillis, A.; Caulier, S.; Nguyen, T.; Mahillon, J. Bacilysin within the Bacillus subtilis group: Gene prevalence versus antagonistic activity against Gram-negative foodborne pathogens. J. Biotechnol. 2021, 327, 28–35. [Google Scholar] [CrossRef]
- Li, H.; Han, X.; Dong, Y.; Xu, S.; Chen, C.; Feng, Y.; Cui, Q.; Li, W. Bacillaenes: Decomposition Trigger Point and Biofilm Enhancement in Bacillus. ACS Omega 2021, 6, 1093–1098. [Google Scholar] [CrossRef]
- Dimopoulou, A.; Theologidis, I.; Benaki, D.; Koukounia, M.; Zervakou, A.; Tzima, A.; Diallinas, G.; Hatzinikolaou, D.; Skandalis, N. Direct Antibiotic Activity of Bacillibactin Broadens the Biocontrol Range of Bacillus amyloliquefaciens MBI600. mSphere 2021, 6, e0037621. [Google Scholar] [CrossRef]
- Abdallah, D.; Krier, F.; Jacques, P.; Tounsi, S.; Frikha-Gargouri, O. Agrobacterium tumefaciens C58 presence affects Bacillus velezensis 32a ecological fitness in the tomato rhizosphere. Environ. Sci. Pollut. Res. Int. 2020, 27, 28429–28437. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Chromosome |
|---|---|
| Genome size (bp) | 3,974,499 |
| G+C content (%) | 46.25 |
| Protein-coding genes (CDS) | 4082 |
| Total gene length (bp) | 3,561,637 |
| Gene/genome (%) | 89.61 |
| 5S rRNA | 8 |
| 16S rRNA | 1 |
| 23S rRNA | 1 |
| tRNA | 81 |
| Cluster ID | Genomic Site | Product Type | Most Similar Known Gene Cluster | |||
|---|---|---|---|---|---|---|
| Initial Site | Terminal Site | Most Similar Product | Similarity, % | Source | ||
| Cluster 1 | 382,141 | 427,027 | NRPS | — | — | — |
| Cluster 2 | 573,274 | 667,047 | transAT-PKS | difficidin | 100 | Bacillus velezensis FZB42 |
| Cluster 3 | 928,854 | 969,954 | T3PKS | — | — | — |
| Cluster 4 | 1,033,593 | 1,055,476 | terpene | — | — | — |
| Cluster 5 | 1,078,046 | 1,101,559 | NRPS | plipastatin | 38 | — |
| Cluster 6 | 64,361 | 105,605 | PKS-like | butirosin A/ butirosin B | 7 | — |
| Cluster 7 | 187,671 | 208,411 | terpene | — | — | — |
| Cluster 8 | 504,977 | 591,316 | transAT-PKS | macrolactin H | 100 | Bacillus velezensis FZB42 |
| Cluster 9 | 814,801 | 915,529 | transAT-PKS | bacillaene | 100 | Bacillus velezensis FZB42 |
| Cluster 10 | 980,011 | 1,078,369 | NRPS | fengycin | 86 | Bacillus velezensis FZB42 |
| Cluster 11 | 298,436 | 339,854 | other | bacilysin | 100 | Bacillus subtilis 168 |
| Cluster 12 | 859,024 | 910,818 | NRPS | bacillibactin | 100 | Bacillus velezensis FZB42 |
| Cluster 13 | 194,589 | 259,996 | NRPS | surfactin | 82 | Bacillus velezensis FZB42 |
| Cluster 14 | 70,747 | 111,868 | ladderane | — | — | Bacillus velezensis FZB42 |
| Cluster 15 | 1 | 14,773 | NRPS | fengycin | 20 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Chen, S.; Yu, Z.; Yao, J.; Jia, Y.; Liao, C.; Chen, J.; Wei, Y.; Guo, R.; He, L.; et al. A Novel Bacillus Velezensis for Efficient Degradation of Zearalenone. Foods 2024, 13, 530. https://doi.org/10.3390/foods13040530
Li Y, Chen S, Yu Z, Yao J, Jia Y, Liao C, Chen J, Wei Y, Guo R, He L, et al. A Novel Bacillus Velezensis for Efficient Degradation of Zearalenone. Foods. 2024; 13(4):530. https://doi.org/10.3390/foods13040530
Chicago/Turabian StyleLi, Yijia, Songbiao Chen, Zuhua Yu, Jie Yao, Yanyan Jia, Chengshui Liao, Jian Chen, Ying Wei, Rongxian Guo, Lei He, and et al. 2024. "A Novel Bacillus Velezensis for Efficient Degradation of Zearalenone" Foods 13, no. 4: 530. https://doi.org/10.3390/foods13040530
APA StyleLi, Y., Chen, S., Yu, Z., Yao, J., Jia, Y., Liao, C., Chen, J., Wei, Y., Guo, R., He, L., & Ding, K. (2024). A Novel Bacillus Velezensis for Efficient Degradation of Zearalenone. Foods, 13(4), 530. https://doi.org/10.3390/foods13040530