Steroid Glycosides Hyrcanoside and Deglucohyrcanoside: On Isolation, Structural Identification, and Anticancer Activity

 ,
,  ,
,  , , ,
, , ,  , and
, and 
Abstract
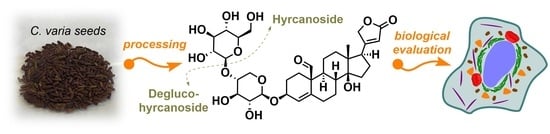
1. Introduction
2. Materials and Methods
2.1. Materials
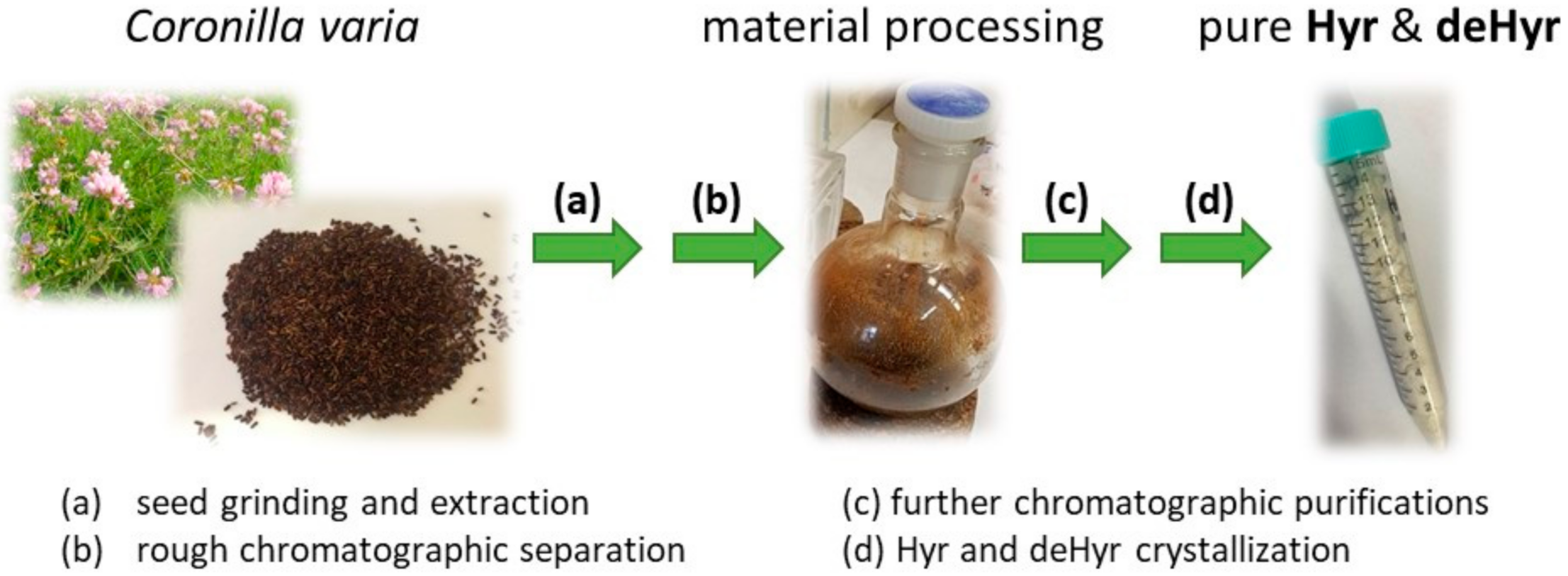
2.2. Extraction of C. varia Seeds and Isolation of Compounds
2.3. In Silico Modeling
2.4. Cell Lines
2.5. MTS Cytotoxic Assay
2.6. Mice and Peritoneal Primary Cells
2.7. Viability of Mouse Primary Cells
2.8. Analysis of Cell Cycle Arrest and Cell Death
2.9. BrDU Incorporation Analysis
2.10. BrU Incorporation Analysis
3. Results and Discussion
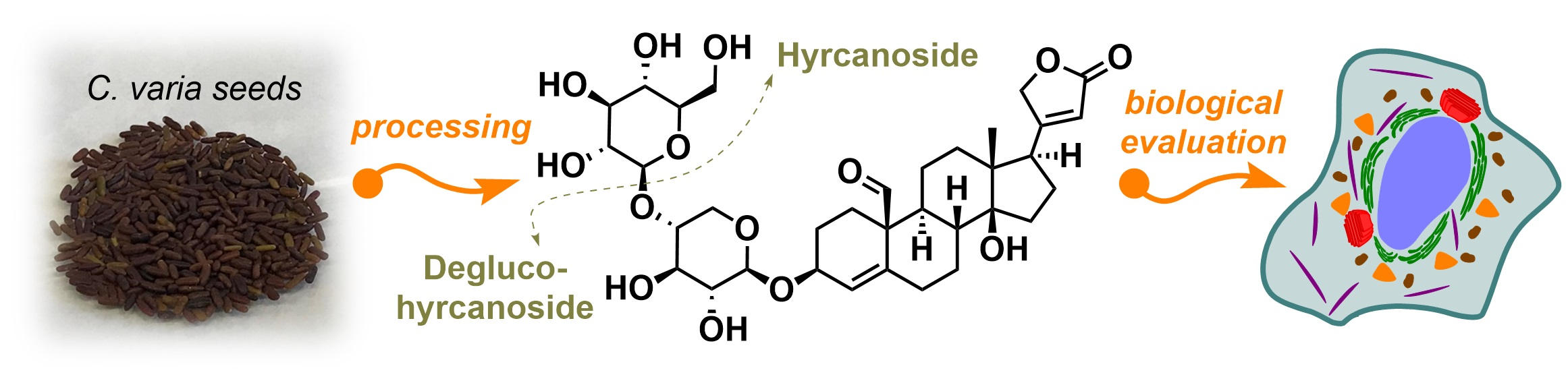
3.1. Isolation and Identification of Hyr and deHyr
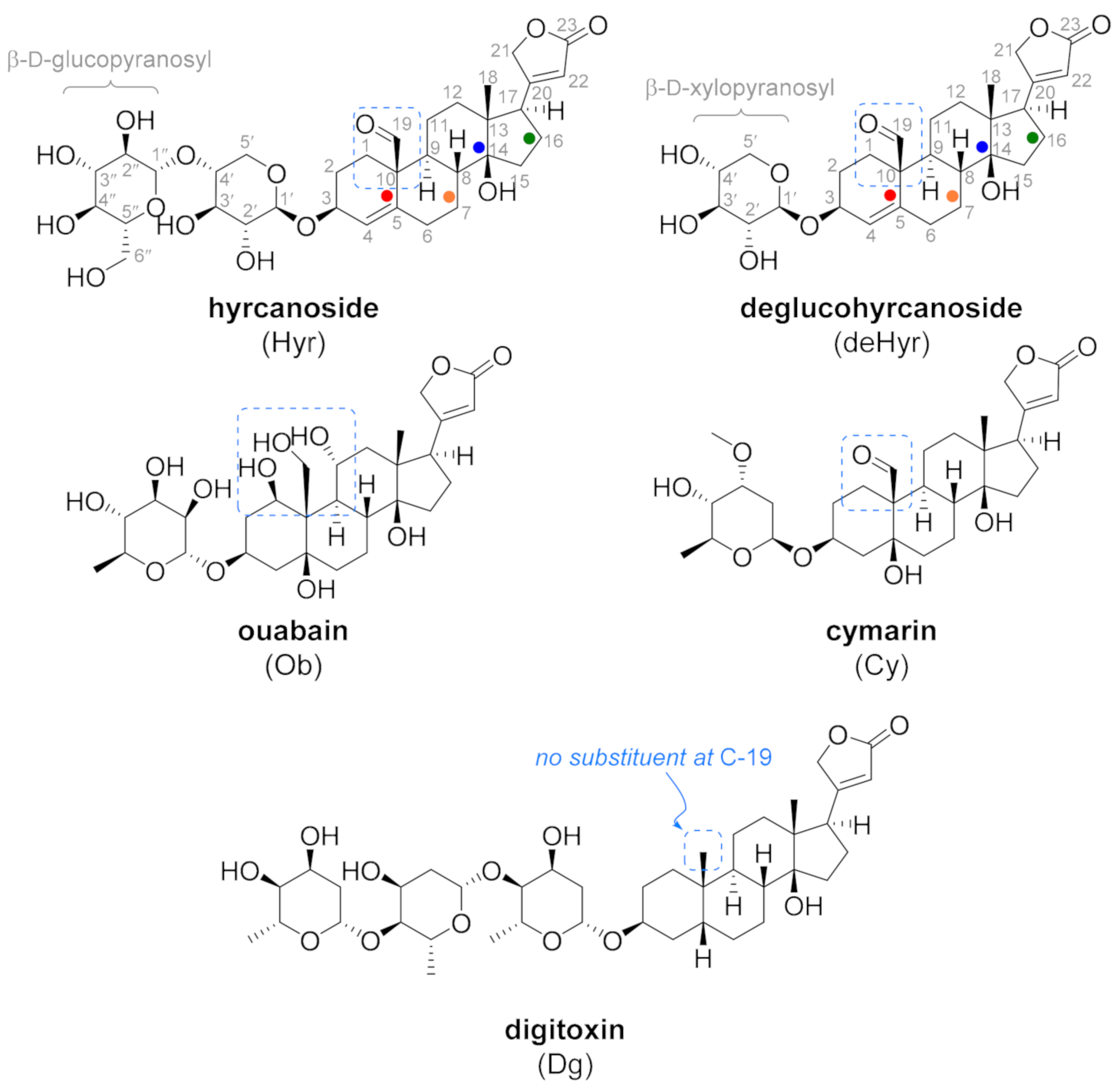
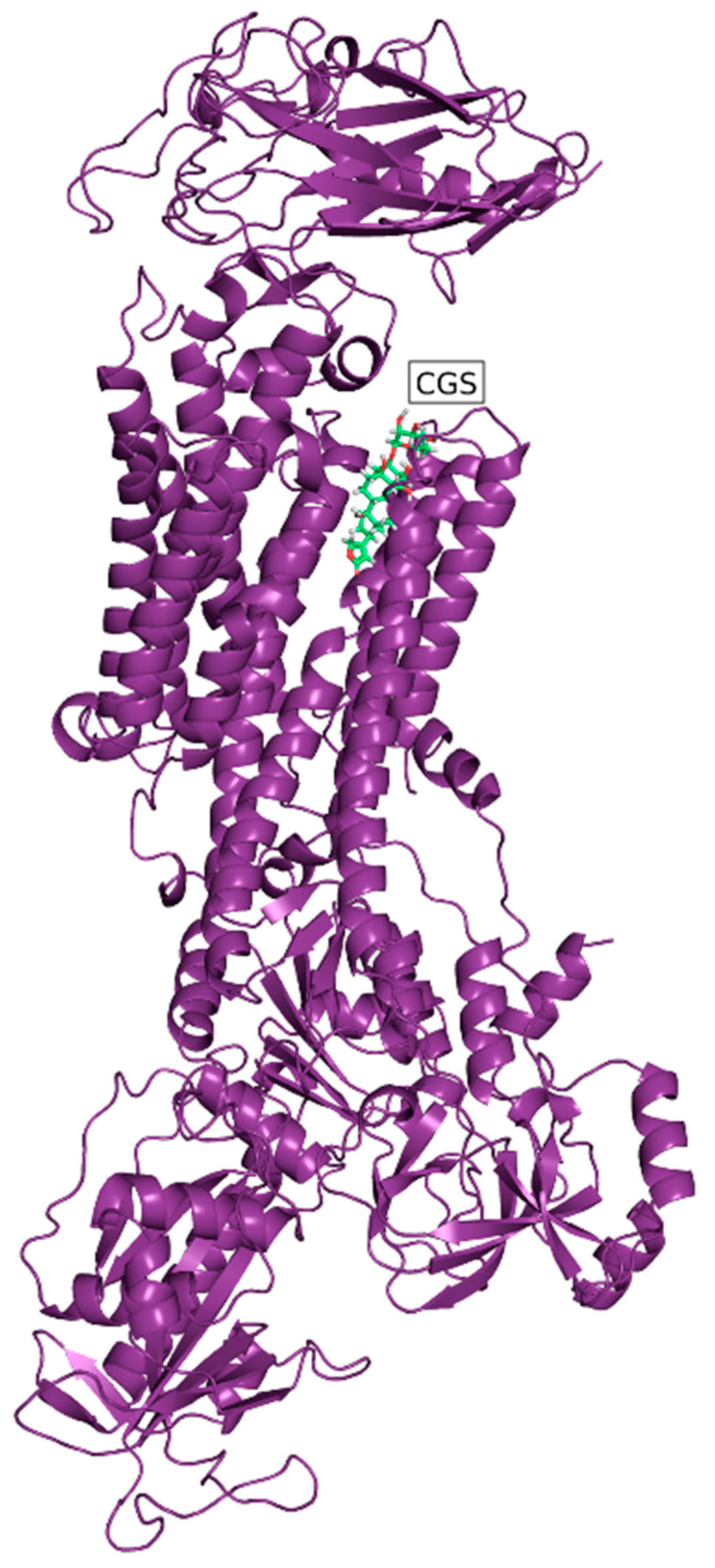
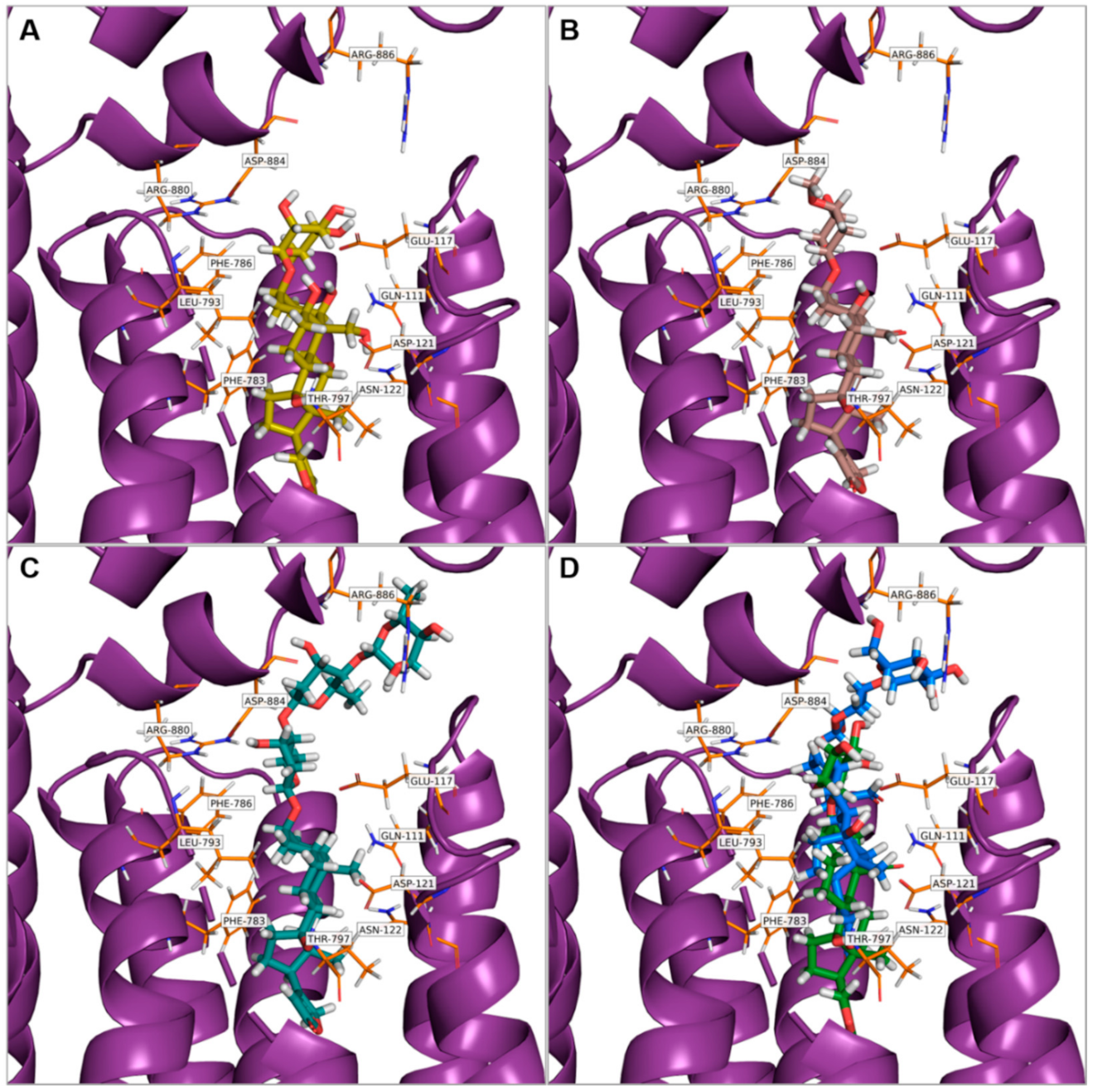
3.2. In Silico Modeling
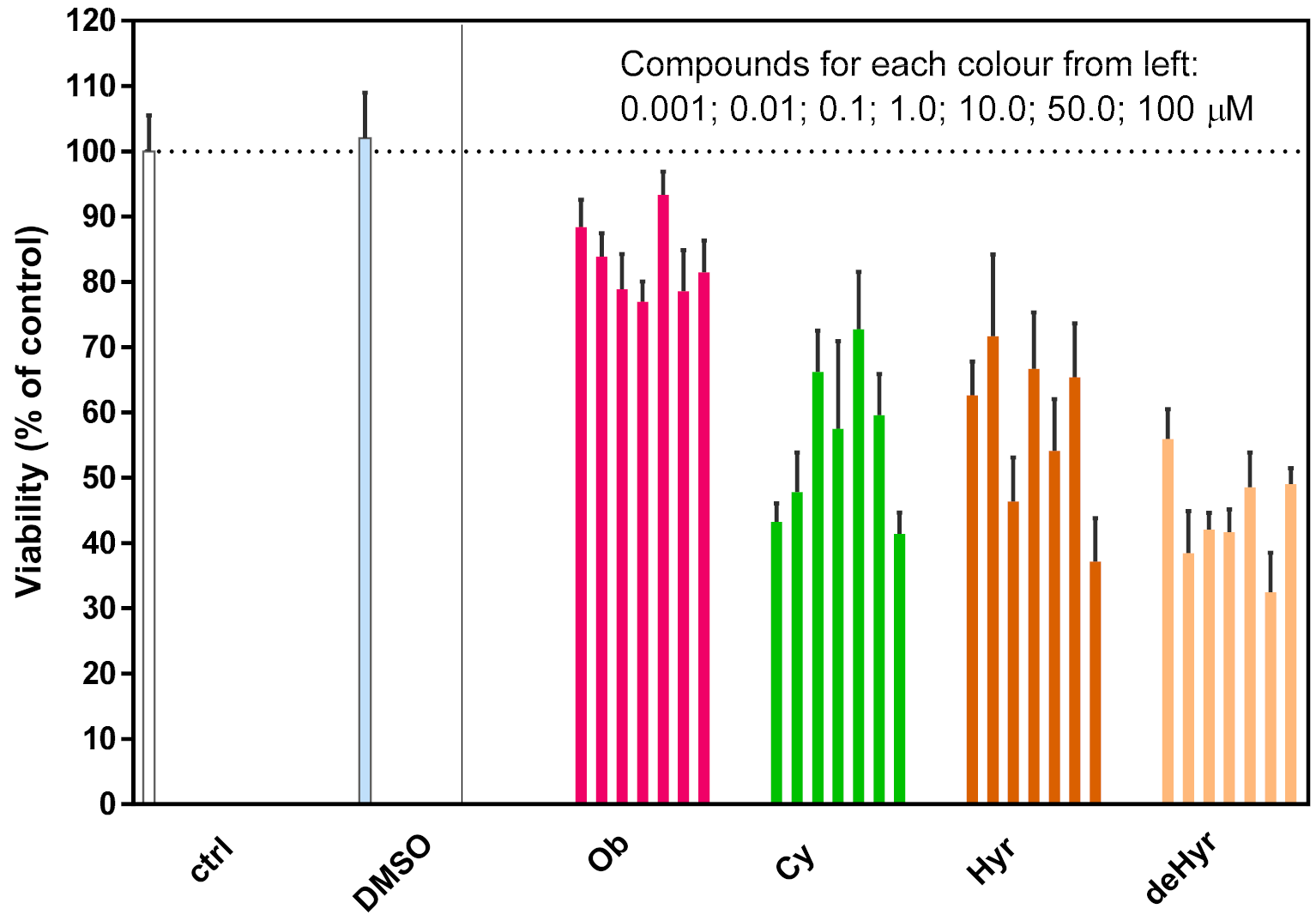
3.3. Anticancer Potential of the Evaluated Steroid Glycosides
3.4. Steroid Glycoside Toxicity to Mouse Macrophages
3.5. Cell Cycle and Cell Death Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 7 December 2020).
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials. Available online: ClinicalTrials.gov (accessed on 7 December 2020).
- Platz, E.A.; Yegnasubramanian, S.; Liu, J.O.; Chong, C.R.; Shim, J.S.; Kenfield, S.A.; Stampfer, M.J.; Willett, W.C.; Giovannucci, E.; Nelson, W.G. A novel two-stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discov. 2011, 1, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.H.; Farrag, E.; Selim, M.; Osman, M.S.; Hasanine, A.; Selim, A. Cardiac glycosides use and the risk and mortality of cancer; systematic review and meta-analysis of observational studies. PLoS ONE 2017, 12, e0178611. [Google Scholar] [CrossRef] [PubMed]
- Couraud, S.; Azoulay, L.; Dell’Aniello, S.; Suissa, S. Cardiac glycosides use and the risk of lung cancer: A nested case–control study. BMC Cancer 2014, 14, 573. [Google Scholar] [CrossRef]
- Karasneh, R.A.; Murray, L.J.; Cardwell, C.R. Cardiac glycosides and breast cancer risk: A systematic review and meta-analysis of observational studies. Int. J. Cancer 2017, 140, 1035–1041. [Google Scholar] [CrossRef]
- Kepp, O.; Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Sukkurwala, A.Q.; Michaud, M.; Galluzzi, L.; Zitvogel, L.; et al. Anticancer activity of cardiac glycosides. At the frontier between cell-autonomous and immunological effects. Oncoimmunology 2012, 1, 1640–1642. [Google Scholar] [CrossRef]
- Schönfeld, W.; Weiland, J.; Lindig, C.; Masnyk, M.; Kabat, M.M.; Kurek, A.; Wicha, J.; Repke, K.R.H. The lead structure in cardiac glycosides is 5β,14β-androstane-3β,14-diol. Naunyn Schmiedebergs Arch. Pharmacol. 1985, 329, 414–426. [Google Scholar] [CrossRef]
- Morsy, N. References. In Aromatic and Medicinal Plants—Back to Nature; El-Shemy, H., Ed.; IntechOpen: London, UK, 2017; pp. 29–45. ISBN 978-953-51-7348-9. [Google Scholar]
- Manunta, P.; Hamilton, B.P.; Hamlyn, J.M. Structure-activity relationships for the hypertensinogenic activity of ouabain. Hypertension 2001, 37, 472–477. [Google Scholar] [CrossRef]
- Magpusao, A.N.; Omolloh, G.; Johnson, J.; Gascón, J.; Peczuh, M.W.; Fenteany, G. Cardiac glycoside activities link Na+/K+ ATPase ion-transport to breast cancer cell migration via correlative SAR. ACS Chem. Biol. 2015, 10, 561–569. [Google Scholar] [CrossRef]
- Wang, H.Y.; Xin, W.; Zhou, M.; Stueckle, T.A.; Rojanasakul, Y.; O’Doherty, G.A. Stereochemical survey of digitoxin monosaccharides: New anticancer analogues with enhanced apoptotic activity and growth inhibitory effect on human non-small cell lung cancer cell. ACS Med. Chem. Lett. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Iyer, A.K.V.; Zhou, M.; Azad, N.; Elbaz, H.; Wang, L.; Rogalsky, D.K.; Rojanasakul, Y.; O’Doherty, G.A.; Langenhan, J.M. A direct comparison of the anticancer activities of digitoxin MeON-neoglycosides and O-glycosides. ACS Med. Chem. Lett. 2010, 1, 326–330. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M.; Pastor, N.; Azrak, S.S.; Ayuso, M.J.; Austin, C.A.; Cortés, F. Digitoxin inhibits the growth of cancer cell lines at concentrations commonly found in cardiac patients. J. Nat. Prod. 2005, 68, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Ayogu, J.I.; Odoh, A.S. Prospects and therapeutic applications of cardiac glycosides in cancer remediation. ACS Comb. Sci. 2020, 22, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Reuter, H.; Henderson, S.A.; Han, T.; Ross, R.S.; Goldhaber, J.I.; Philipson, K.D. The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ. Res. 2002, 90, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/endoplasmic reticulum calcium ATPase inhibitors: Beyond anticancer perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef]
- Danen, E.H.J.; Sonneveld, P.; Sonnenberg, A.; Yamada, K.M. Dual stimulation of Ras/mitogen-activated protein kinase and Rhoa by cell adhesion to fibronectin supports growth factor–stimulated cell cycle progression. J. Cell Biol. 2000, 151, 1413–1422. [Google Scholar] [CrossRef]
- Prassas, I.; Karagiannis, G.S.; Batruch, I.; Dimitromanolakis, A.; Datti, A.; Diamandis, E.P. Digitoxin-induced cytotoxicity in cancer cells is mediated through distinct kinase and interferon signaling networks. Mol. Cancer Ther. 2011, 10, 2083–2093. [Google Scholar] [CrossRef]
- McConkey, D.J.; Lin, Y.; Nutt, L.K.; Ozel, H.Z.; Newman, R.A. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 2000, 60, 3807–3812. [Google Scholar]
- Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Shen, S.; Yamazaki, T.; Sukkurwala, A.Q.; Michaud, M.; Mignot, G.; et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci. Transl. Med. 2012, 4, 143–199. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.; Lifshitz, Y.; Bab-Dinitz, E.; Kapri-Pardes, E.; Goldshleger, R.; Tal, D.M.; Karlish, S.J.D. Selectivity of digitalis glycosides for isoforms of human Na,K-ATPase. J. Biol. Chem. 2010, 285, 19582–19592. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, H.A.; Stueckle, T.A.; Wang, H.Y.L.; O’Doherty, G.A.; Lowry, D.T.; Sargent, L.M.; Wang, L.; Dinu, C.Z.; Rojanasakul, Y. Digitoxin and a synthetic monosaccharide analog inhibit cell viability in lung cancer cells. Toxicol. Appl. Pharmacol. 2012, 258, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, J.; Chen, B.; Zhou, M.; Zeng, Y.; Zhang, Q.; Guo, Y.; Chen, J.; Ouyang, J. Cardiac glycosides inhibit proliferation and induce apoptosis of human hematological malignant cells. Int. J. Clin. Exp. Pathol. 2016, 9, 9268–9275. [Google Scholar]
- Zhang, Y.Z.; Chen, X.; Fan, X.X.; He, J.X.; Huang, J.; Xiao, D.K.; Zhou, Y.L.; Zheng, S.Y.; Xu, J.H.; Yao, X.J.; et al. Compound library screening identified cardiac glycoside digitoxin as an effective growth inhibitor of gefitinib-resistant non-small cell lung cancer via downregulation of alpha-tubulin and inhibition of microtubule formation. Molecules 2016, 21, 374. [Google Scholar] [CrossRef]
- Williams, L.M.; Cassady, J.M. Potential antitumor agents: A cytotoxic cardenolide from Coronilla varia L. J. Pharm. Sci. 1976, 65, 912–914. [Google Scholar] [CrossRef]
- Hembree, J.A.; Chang, C.J.; McLaughlin, L.J.; Peck, G.; Cassady, J.M. Potential antitumor agents: A cytotoxic cardenolide from Coronilla varia. J. Nat. Prod. 1979, 42, 293–298. [Google Scholar] [CrossRef]
- Slavík, J.; Zácková, P.; Michlová, J.; Opletal, L.; Sovová, M. Phytotherapeutic aspects of diseases of the circulatory system. III. Cardiotonic and cardiotoxic effects of hyrcanoside and deglucohyrcanoside isolated from Coronilla varia L. Ceska Slov. Farm. 1994, 43, 298–302. [Google Scholar]
- Zácková, P.; Sovová, M.; Horáková, M.; Opletalová, V. Study of Coronilla varia L. III. Pharmacological evaluation of its effects on heart function. Ceskoslovenska Farm. 1982, 31, 242–246. [Google Scholar]
- Gersl, V. Effects of Coronilla varia Linné extract and lanatoside C in rabbits with experimental acute heart overloading in vivo. Sb. Ved. Pr. Lek. Fak. Karlov. Univerzity Hradci Kral. Suppl. 1980, 23, 445–457. [Google Scholar]
- Jurášek, M.; Džubák, P.; Rimpelová, S.; Sedlák, D.; Konečný, P.; Frydrych, I.; Gurská, S.; Hajdúch, M.; Bogdanová, K.; Kolář, M.; et al. Trilobolide-steroid hybrids: Synthesis, cytotoxic and antimycobacterial activity. Steroids 2017, 117, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Řehulka, J.; Vychodilová, K.; Krejčí, P.; Gurská, S.; Hradil, P.; Hajdúch, M.; Džubák, P.; Hlaváč, J. Fluorinated derivatives of 2-phenyl-3-hydroxy-4(1H)-quinolinone as tubulin polymerization inhibitors. Eur. J. Med. Chem. 2020, 192, 112176. [Google Scholar] [CrossRef] [PubMed]
- Bagirov, R.B.; Komissarenko, N.F. New cardenolides from seeds of Coronilla hyrcana. Khimiya Prir. Soedin. 1966, 2, 251–257. [Google Scholar]
- Nurmukhamedova, M.R.; Nikonov, G.K. Glycosides from Dorema hyrcanum. Khimiya Prir. Soedin. 1976, 3, 101–102. [Google Scholar]
- Khushbaktova, Z.A.; Mukhtasimova, R.; Syrov, V.N.; Sultanov, M.B. O farmakologicheskikh svoistvach novogo fenolglykozida—girkanozida [Pharmacological properties of a new phenolglycoside—hyrcanoside]. Dokl. Akad. Nauk. 1983, 39, 54–55. [Google Scholar]
- Abubakirov, N.K. The chemistry of cardiac glycosides in the Soviet union. Khimiya Prir. Soedin. 1971, 7, 553–571. [Google Scholar] [CrossRef]
- Zatula, V.V.; Maksyutina, N.P.; Kolesnikov, D.G. Cardenolides of Securigera securidaca. Khimiya Prir. Soedin. 1965, 1, 153–156. [Google Scholar]
- Zatula, V.V.; Chernobrovaya, N.V.; Kolesnikov, D.G. A chemical study of the structure of securigenin and its bioside securidaside. Khimiya Prir. Soedin. 1966, 2, 438–439. [Google Scholar] [CrossRef]
- Zatula, V.V. Kil’kisne vyznachennia sekurydazydu v nasinni sekuryhery mechovydnoi [Quantitative determination of securidazide in seeds of Securigera securidaca]. Farmatsevtychnyi Zhurnal (Kiev) 1968, 23, 85–88. [Google Scholar]
- Zatula, V.V.; Kovalev, I.P.; Kolesnikov, D.G. The structure of securigenin and securigenol. Khimiya Prir. Soedin. 1969, 5, 127–128. [Google Scholar] [CrossRef]
- Tofighi, Z.; Moradi-Afrapoli, F.; Ebrahimi, S.N.; Goodarzi, S.; Hadjiakhoondi, A.; Neuburger, M.; Hamburger, M.; Abdollahi, M.; Yassa, N. Securigenin glycosides as hypoglycemic principles of Securigera securidaca seeds. J. Nat. Med. 2017, 71, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.; Yatimea, L.; Nissena, P.; Fedosova, N.U. Crystal structure of the high-affinity Na+,K+-ATPase–ouabain complex with Mg2+ bound in the cation binding site 1. Proc. Natl. Acad. Sci. USA 2013, 110, 10958–10963. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.; Gregersena, J.L.; Yatimea, L.; Nissena, P.; Fedosova, N.U. Structures and characterization of digoxin- and bufalin-bound Na+,K+-ATPase compared with the ouabain-bound complex. Proc. Natl. Acad. Sci. USA 2015, 112, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Ren, Y.; Ren, J.; Erxleben, C.; Johnson, M.E.; Gentile, S.; Kinghorn, A.D.; Swanson, S.M.; Burdette, J.E. (+)-Strebloside-induced cytotoxicity in ovarian cancer cells is mediated through cardiac glycoside signaling networks. J. Nat. Prod. 2017, 80, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Paula, S.; Tabet, M.R.; Ball, W.J. Interactions between cardiac glycosides and sodium/potassium-ATPase: Three-dimensional structure-activity relationship models for ligand binding to the E2-Pi form of the enzyme versus activity inhibition. Biochemistry 2005, 44, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Levrier, C.; Kiremire, B.; Guéritte, F.; Litaudon, M. Toxicarioside M, a new cytotoxic 10β-hydroxy-19-nor-cardenolide from Antiaris toxicaria. Fitoterapia 2012, 83, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Perne, A.; Muellner, M.K.; Steinrueck, M.; Craig-Mueller, N.; Mayerhofer, J.; Schwarzinger, I.; Sloane, M.; Uras, I.Z.; Hoermann, G.; Nijman, S.M.B.; et al. Cardiac glycosides induce cell death in human cells by inhibiting general protein synthesis. PLoS ONE 2009, 4, e8292. [Google Scholar] [CrossRef]
- Price, E.M.; Lingrel, J.B. Structure-function relationships in the Na,K-ATPase alpha subunit: Site-directed mutagenesis of glutamine-111 to arginine and asparagine-122 to aspartic acid generates a ouabain-resistant enzyme. Biochemistry 1988, 27, 8400–8408. [Google Scholar] [CrossRef]
- Calderon-Montano, J.M.; Burgos-Moron, E.; Lopez-Lazaro, M. The in vivo antitumor activity of cardiac glycosides in mice xenografted with human cancer cells is probably an experimental artifact. Oncogene 2014, 33, 2947–2948. [Google Scholar] [CrossRef]
- Zhang, X.J.; Mei, W.L.; Tan, G.H.; Wang, C.C.; Zhou, S.L.; Huang, F.R.; Chen, B.; Dai, H.F.; Huang, F.Y. Strophalloside induces apoptosis of SGC-7901 cells through the mitochondrion-dependent caspase-3 pathway. Molecules 2015, 20, 5714–5728. [Google Scholar] [CrossRef]
- Akimova, O.A.; Tverskoi, A.M.; Smolyaninova, L.V.; Mongin, A.A.; Lopina, O.D.; La, J.; Dulin, N.O.; Orlov, S.N. Critical role of the α1-Na(+), K(+)-ATPase subunit in insensitivity of rodent cells to cytotoxic action of ouabain. Apoptosis 2015, 20, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.Y.; Chen, Y.Y.; Deng, C.M.; Zhang, C.Q.; Jiang, M.M. Nerigoside suppresses colorectal cancer cell growth and metastatic potential through inhibition of ERK/GSK3β/β-catenin signaling pathway. Phytomedicine 2019, 57, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Gan, H.; Huang, Y.; Chen, Y.; Chen, L.; Shan, A.; Zhao, H.; Wu, M.; Li, X.; Ma, Q.; et al. Digitoxin inhibits proliferation of multidrug-resistant HepG2 cells through G2/M cell cycle arrest and apoptosis. Oncol. Lett. 2020, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Hiyoshi, H.; Abdelhady, S.; Segerström, L.; Sveinbjörnsson, B.; Nuriya, M.; Lundgren, T.K.; Desfrere, L.; Miyakawa, A.; Yasui, M.; Kogner, P.; et al. Quiescence and γH2AX in neuroblastoma are regulated by ouabain/Na,K-ATPase. Br. J. Cancer 2012, 106, 1807–1815. [Google Scholar] [CrossRef]
- Newman, R.A.; Kondo, Y.; Yokoyama, T.; Dixon, S.; Cartwright, C.; Chan, D.; Johansen, M.; Yang, P. Autophagic cell death of human pancreatic tumor cells mediated by oleandrin, a lipid-soluble cardiac glycoside. Integr. Cancer Ther. 2007, 6, 354–364. [Google Scholar] [CrossRef]
- Wang, T.; Xu, P.; Wang, F.; Zhou, D.; Wang, R.; Meng, L.; Wang, X.; Zhou, M.; Chen, B.; Ouyang, J. Effects of digoxin on cell cycle, apoptosis and NF-κB pathway in Burkitt’s lymphoma cells and animal model. Leuk. Lymphoma 2017, 58, 1673–1685. [Google Scholar] [CrossRef]
- Škubník, J.; Jurášek, M.; Ruml, T.; Rimpelová, S. Mitotic poisons in research and medicine. Molecules 2020, 25, 4632. [Google Scholar] [CrossRef]
 dot, B with
dot, B with  , C with
, C with  , and D with
, and D with  dot).
dot, B with , C with , and D with dot).
dot).
dot, B with , C with , and D with dot).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand * | Binding Energies (kcal·mol−1) |
|---|---|
| Ob | −9.85 |
| deHyr | −8.48 |
| Dg | −7.47 |
| Cy | −6.42 |
| Hyr | −5.85 |
| Compound | Ob | Cy | Dg | Hyr | deHyr |
|---|---|---|---|---|---|
| Cell Line | IC50 [nM] a | ||||
| CCRF-CEM | 32 ± 5.8 | 14 ± 2.1 | 20 ± 3.5 | 660 ± 38 | 110 ± 21 |
| K562 | 57 ± 6.2 | 25 ± 4.5 | 19 ± 2.2 | 800 ± 52 | 130 ± 21 |
| A549 | 22 ± 0.94 | 15 ± 1.8 | 19 ± 2.0 | 550 ± 38 | 90 ± 14 |
| HCT116 | 30 ± 3.6 | 21 ± 2.5 | 20 ± 1.5 | 670 ± 37 | 140 ± 16 |
| HCT116p53-/- | 28 ± 7.6 | 19 ± 3.8 | 27 ± 9.6 | 730 ± 98 | 130 ± 23 |
| MiaPaCa-2 | 49 ± 2.01 | 44 ± 8.6 | 79 ± 2.8 | 491 ± 23 | 120 ± 5.2 |
| MCF-7 | 52 ± 5.6 | 29 ± 8.1 | 78 ± 1.1 | 566 ± 29 | 119 ± 6.4 |
| U-2 OS | 59 ± 1.1 | 43 ± 1.8 | 45 ± 2.1 | 1104 ± 59 | 165 ± 11 |
| 5637 | 110 ± 11 | 89 ± 5.5 | 58 ± 1.6 | 1511 ± 51 | 398 ± 46 |
| PC-3 | 49 ± 1.4 | 26 ± 2.9 | 69 ± 2.2 | 756 ± 29 | 122 ± 4.7 |
| HEK 293T | 35 ± 1.2 | 3.02 ± 1.4 | 69 ± 2.1 | 383 ± 8.9 | 38 ± 0.97 |
| MRC-5 | 39 ± 1.6 | 26 ± 2.4 | 78 ± 1.9 | 302 ± 43 | 77 ± 12 |
| BJ | 54 ± 9.9 | 54 ± 10 | 47 ± 12 | 1450 ± 250 | 230 ± 35 |
| L929 | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| Compound * | < G1 | G0/G1 | S | G2/M | M a | BrDU b | BrU c |
|---|---|---|---|---|---|---|---|
| Control | 2.74 | 43.36 | 33.86 | 22.34 | 1.69 | 33.10 | 34.51 |
| Ob 1 × IC50 | 20.68 | 40.42 | 25.78 | 33.80 | 1.73 | 40.53 | 39.77 |
| Ob 5 × IC50 | 33.31 | 33.94 | 40.80 | 25.27 | 0.63 | 19.29 | 6.56 |
| Cy 1 × IC50 | 2.89 | 35.78 | 39.87 | 24.35 | 1.43 | 32.34 | 35.63 |
| Cy 5 × IC50 | 7.01 | 40.34 | 31.17 | 28.49 | 1.08 | 21.37 | 27.52 |
| Dg 1 × IC50 | 7.56 | 33.21 | 36.28 | 30.51 | 1.97 | 35.91 | 42.99 |
| Dg 5 × IC50 | 10.35 | 33.71 | 22.26 | 44.03 | 1.28 | 23.52 | 3.88 |
| Hyr 1 × IC50 | 2.82 | 38.48 | 35.22 | 26.30 | 1.41 | 30.58 | 28.80 |
| Hyr 5 × IC50 | 5.82 | 42.13 | 23.01 | 34.86 | 0.95 | 23.58 | 3.82 |
| deHyr 1 × IC50 | 22.02 | 44.90 | 17.60 | 37.50 | 1.44 | 37.40 | 38.91 |
| deHyr 5 × IC50 | 27.43 | 39.12 | 34.53 | 26.34 | 1.56 | 6.08 | 12.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rimpelová, S.; Zimmermann, T.; Drašar, P.B.; Dolenský, B.; Bejček, J.; Kmoníčková, E.; Cihlářová, P.; Gurská, S.; Kuklíková, L.; Hajdůch, M.; et al. Steroid Glycosides Hyrcanoside and Deglucohyrcanoside: On Isolation, Structural Identification, and Anticancer Activity. Foods 2021, 10, 136. https://doi.org/10.3390/foods10010136
Rimpelová S, Zimmermann T, Drašar PB, Dolenský B, Bejček J, Kmoníčková E, Cihlářová P, Gurská S, Kuklíková L, Hajdůch M, et al. Steroid Glycosides Hyrcanoside and Deglucohyrcanoside: On Isolation, Structural Identification, and Anticancer Activity. Foods. 2021; 10(1):136. https://doi.org/10.3390/foods10010136
Chicago/Turabian StyleRimpelová, Silvie, Tomáš Zimmermann, Pavel B. Drašar, Bohumil Dolenský, Jiří Bejček, Eva Kmoníčková, Petra Cihlářová, Soňa Gurská, Lucie Kuklíková, Marián Hajdůch, and et al. 2021. "Steroid Glycosides Hyrcanoside and Deglucohyrcanoside: On Isolation, Structural Identification, and Anticancer Activity" Foods 10, no. 1: 136. https://doi.org/10.3390/foods10010136
APA StyleRimpelová, S., Zimmermann, T., Drašar, P. B., Dolenský, B., Bejček, J., Kmoníčková, E., Cihlářová, P., Gurská, S., Kuklíková, L., Hajdůch, M., Ruml, T., Opletal, L., Džubák, P., & Jurášek, M. (2021). Steroid Glycosides Hyrcanoside and Deglucohyrcanoside: On Isolation, Structural Identification, and Anticancer Activity. Foods, 10(1), 136. https://doi.org/10.3390/foods10010136