Probing the Effect of Six-Membered N-Heterocyclic Carbene—6-Mes—on the Synthesis, Structure and Reactivity of Me2MOR(NHC) (M = Ga, In) Complexes

 , and
, and
Abstract
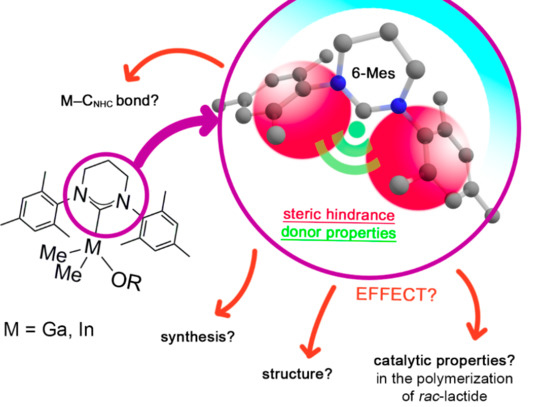
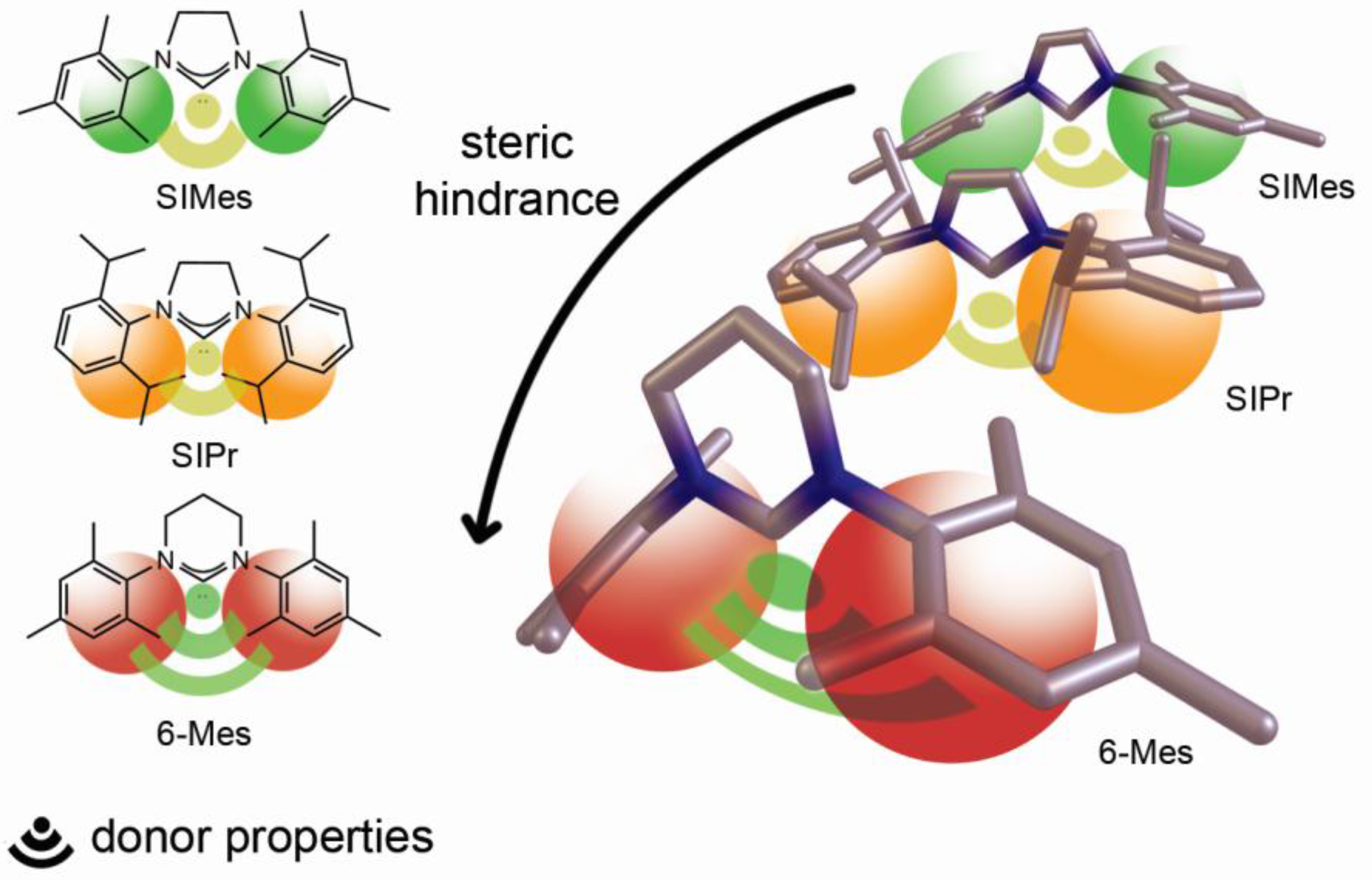
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
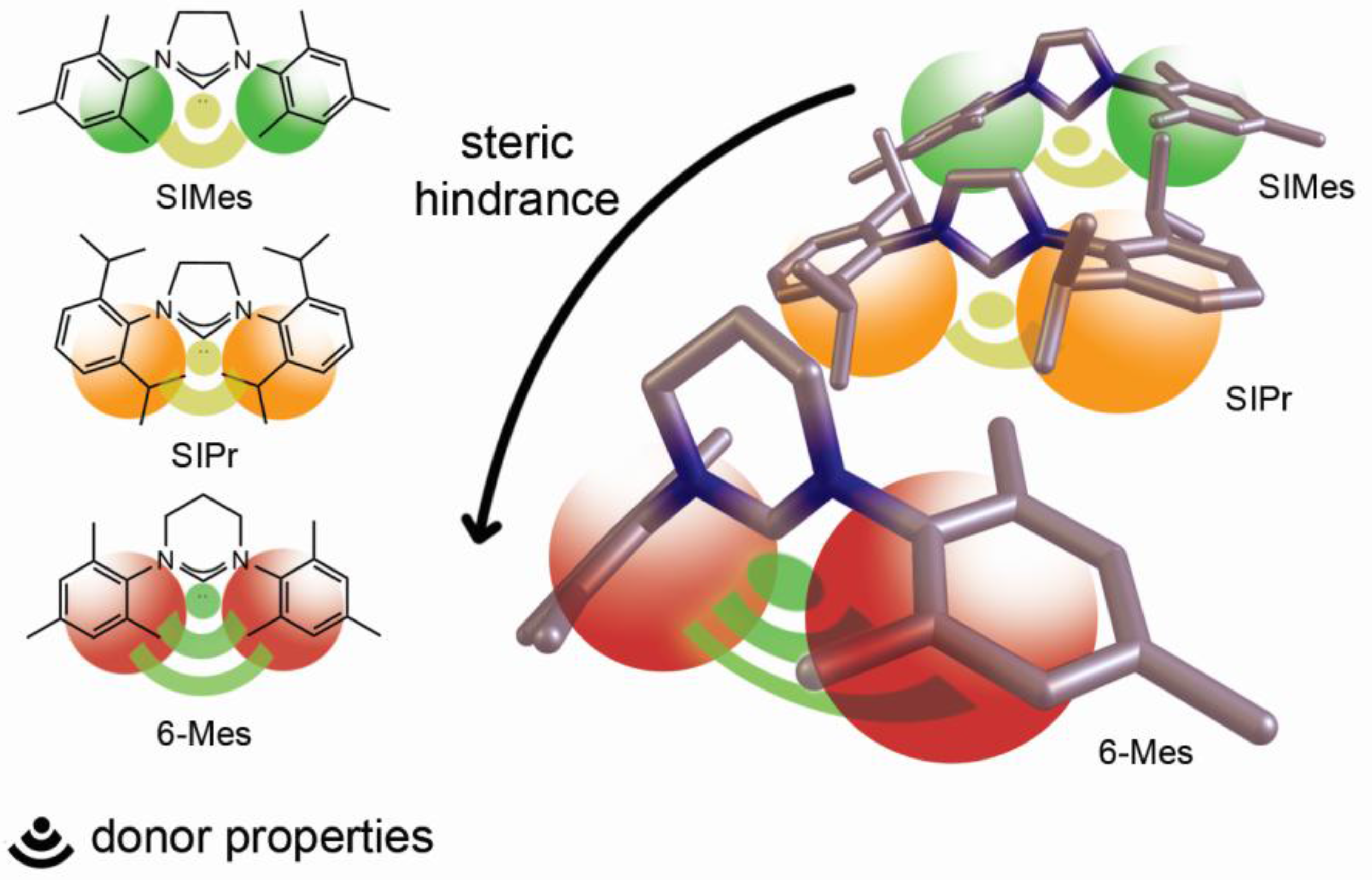
2. Results and Discussion
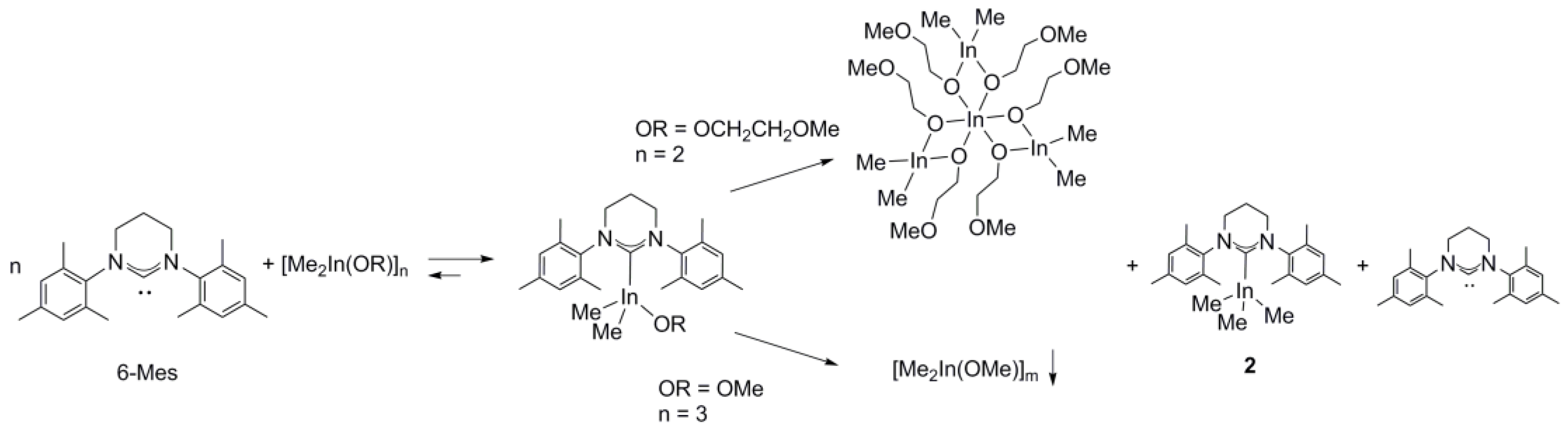
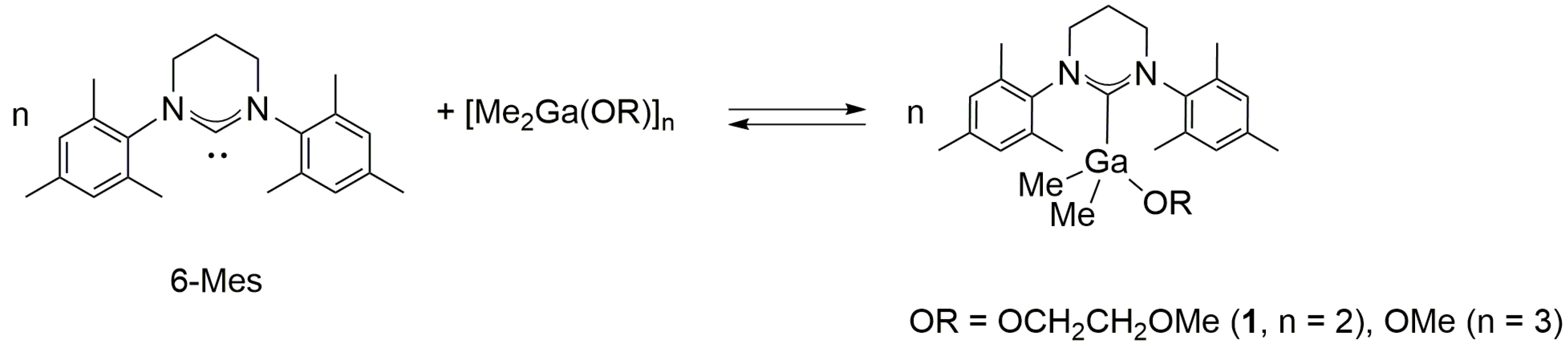
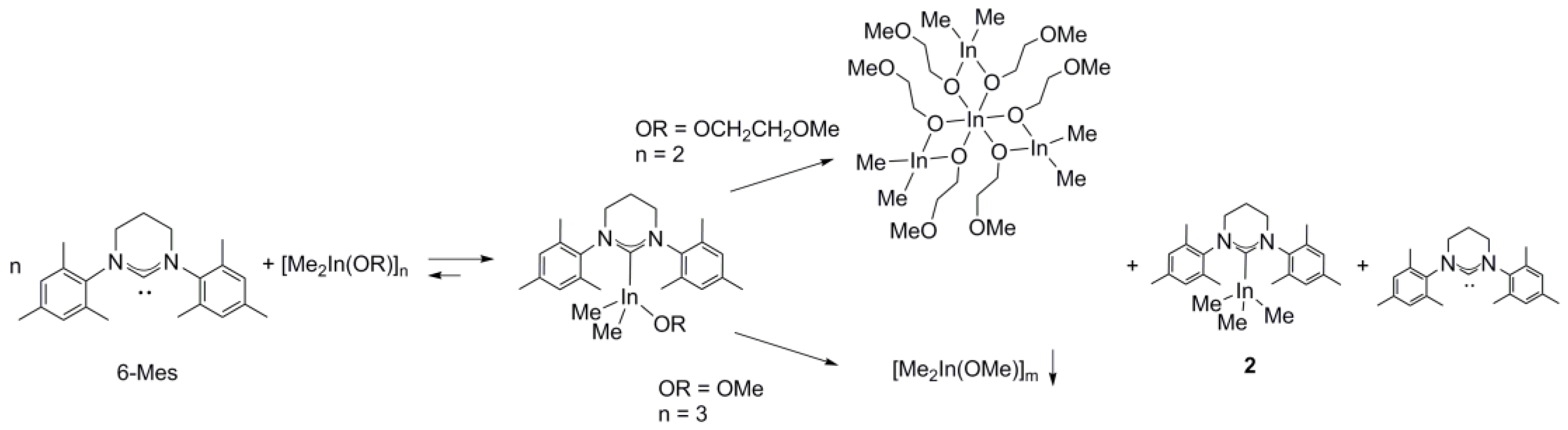
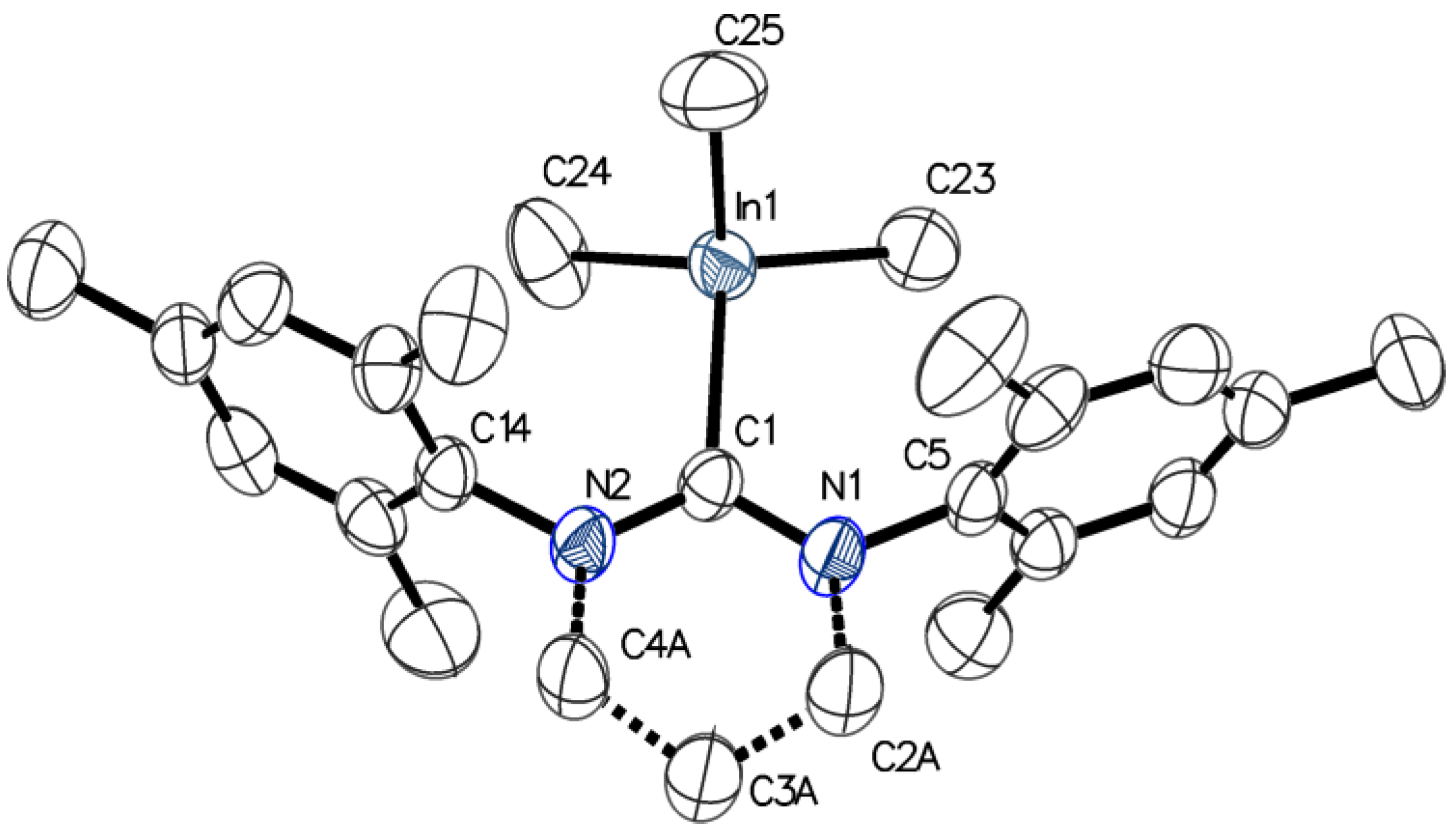
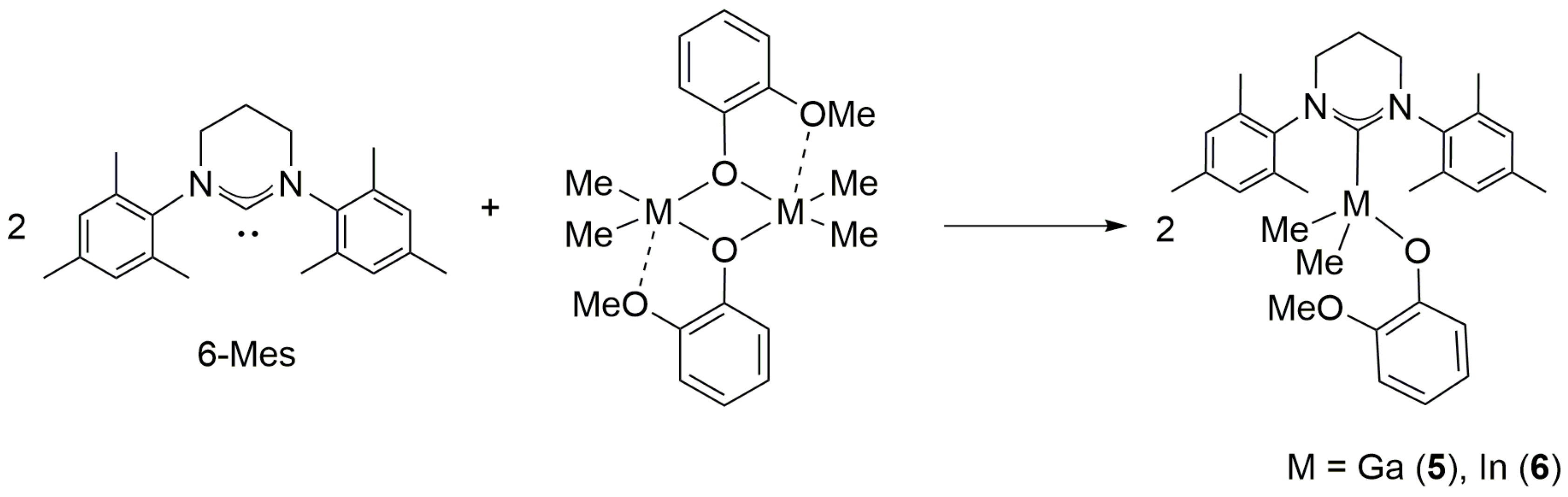
2.1. Reactivity of [Me2M(μ-OR)]n (M= Ga, In ; OR = OCH2CH2OMe, OMe ; n = 2, 3) towards 6-Mes
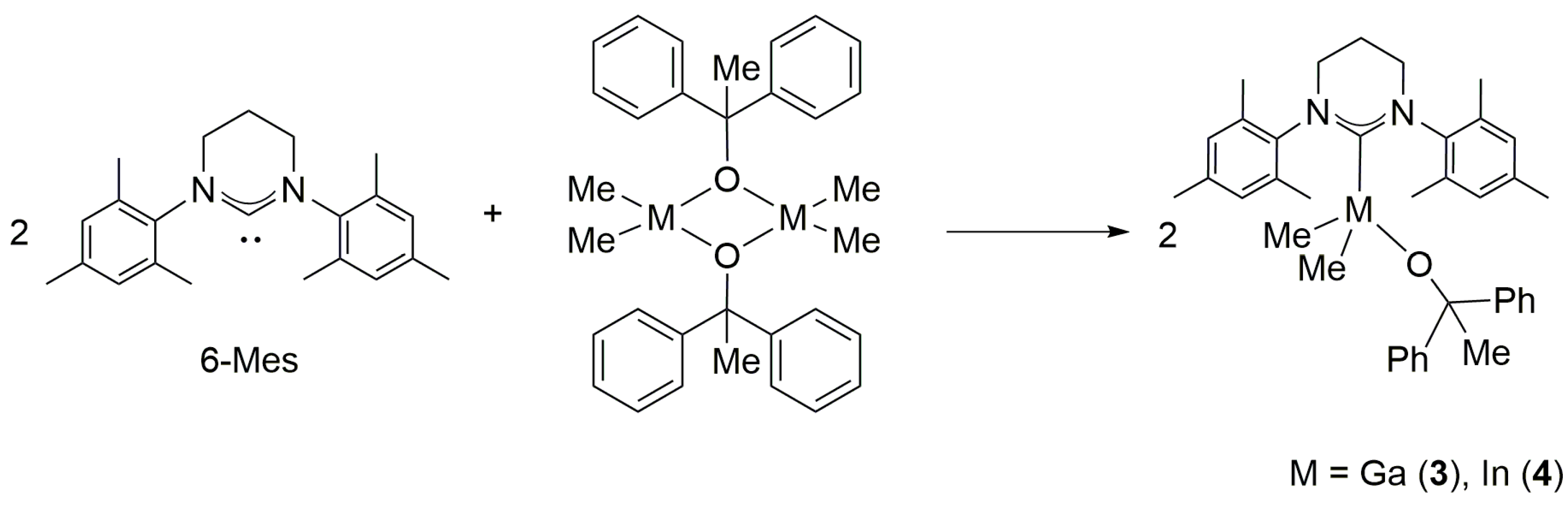
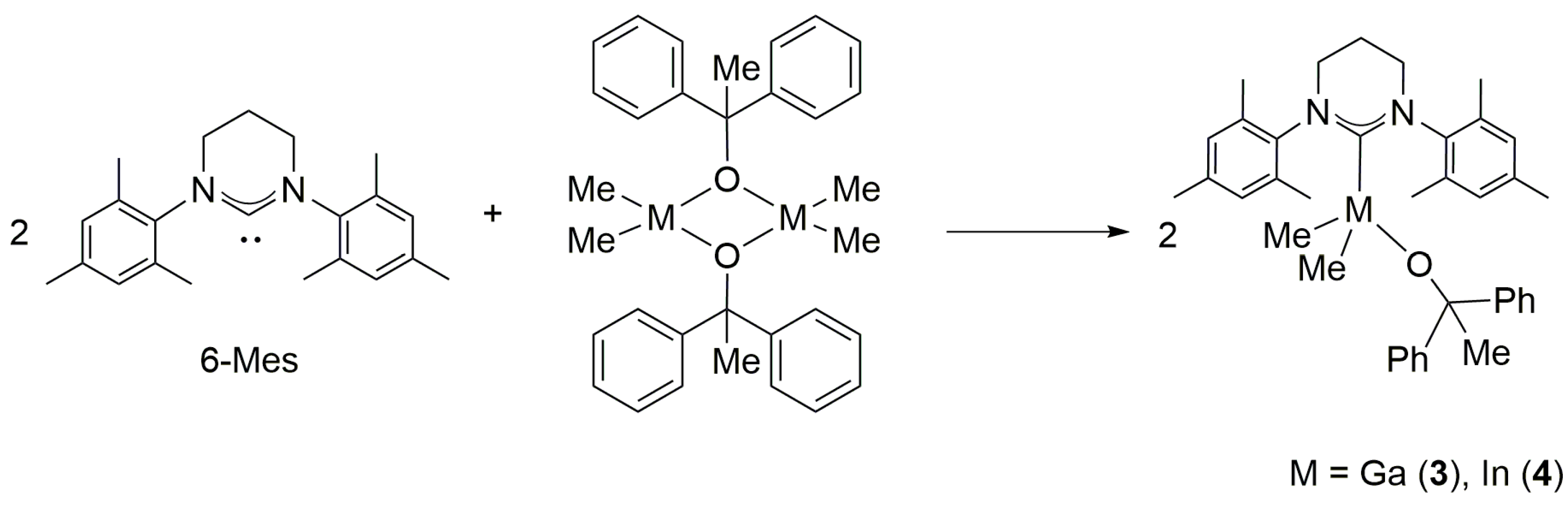
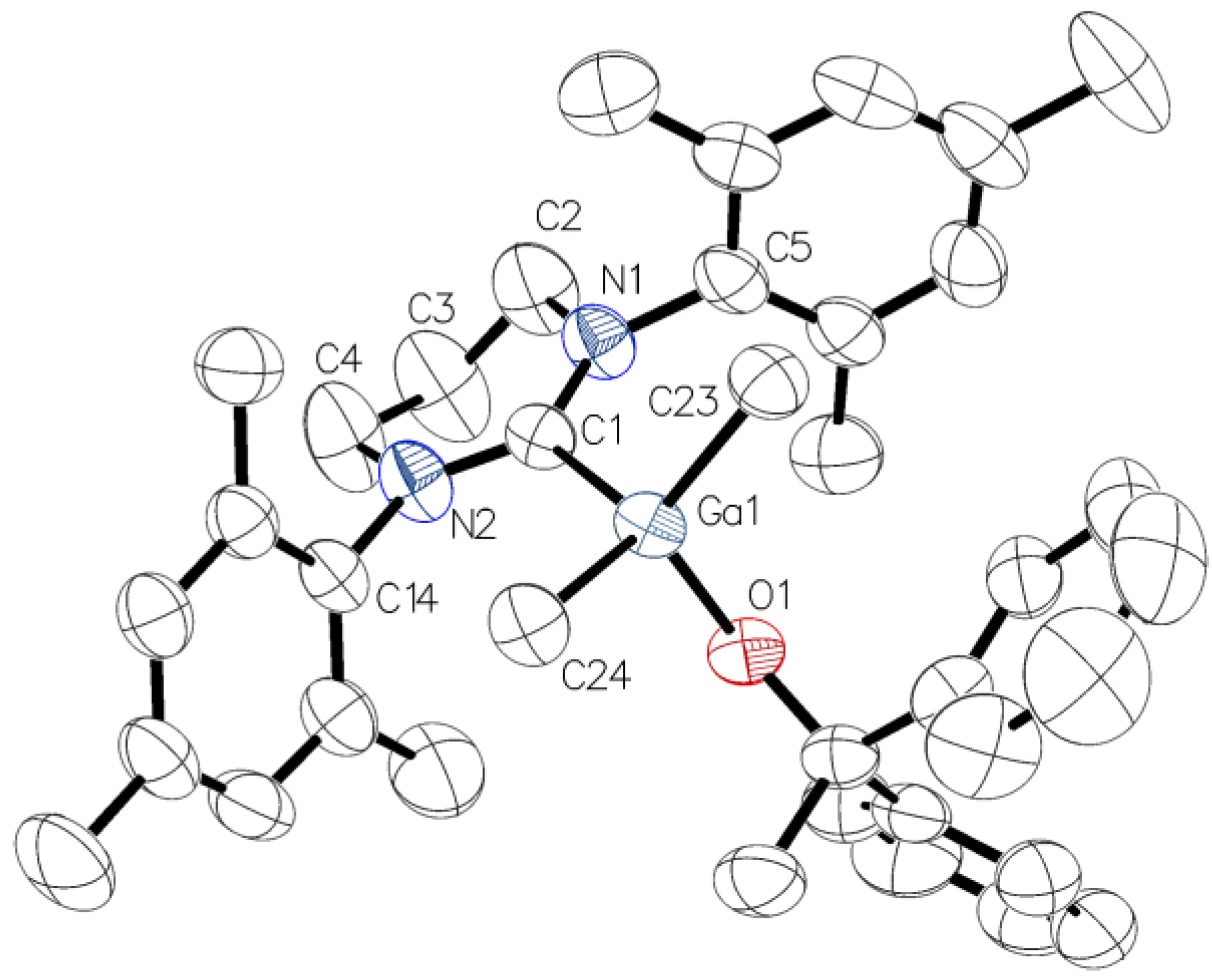
2.2. Reactivity of [Me2M(μ-OCPh2Me)]2 (M = Ga, In) towards 6-Mes
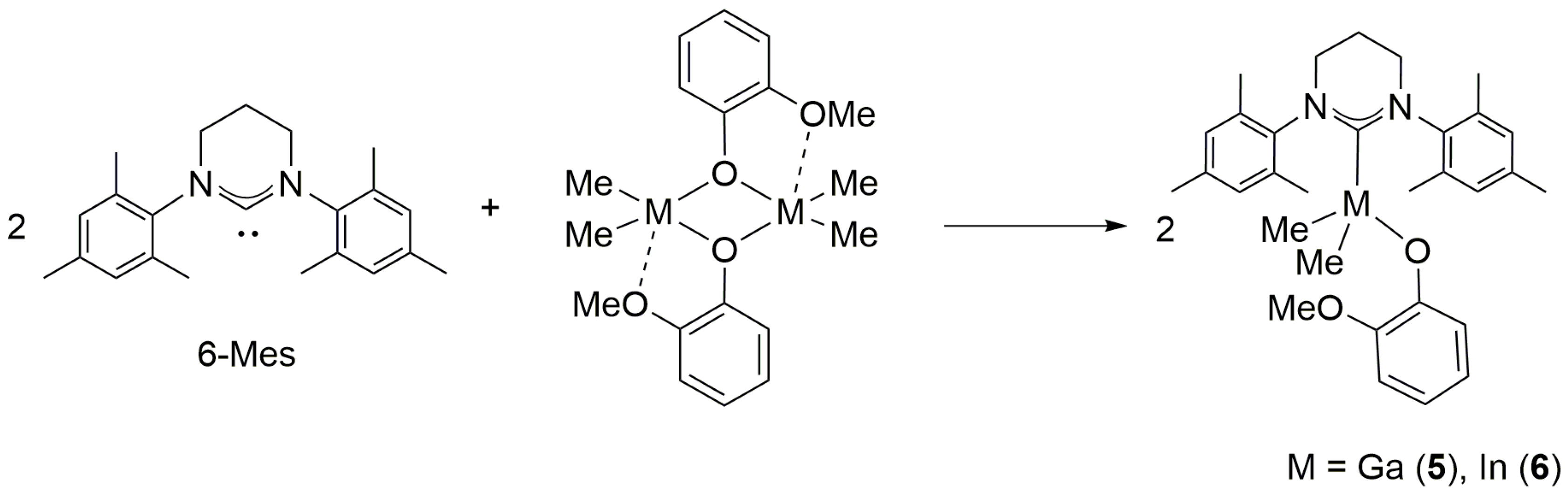
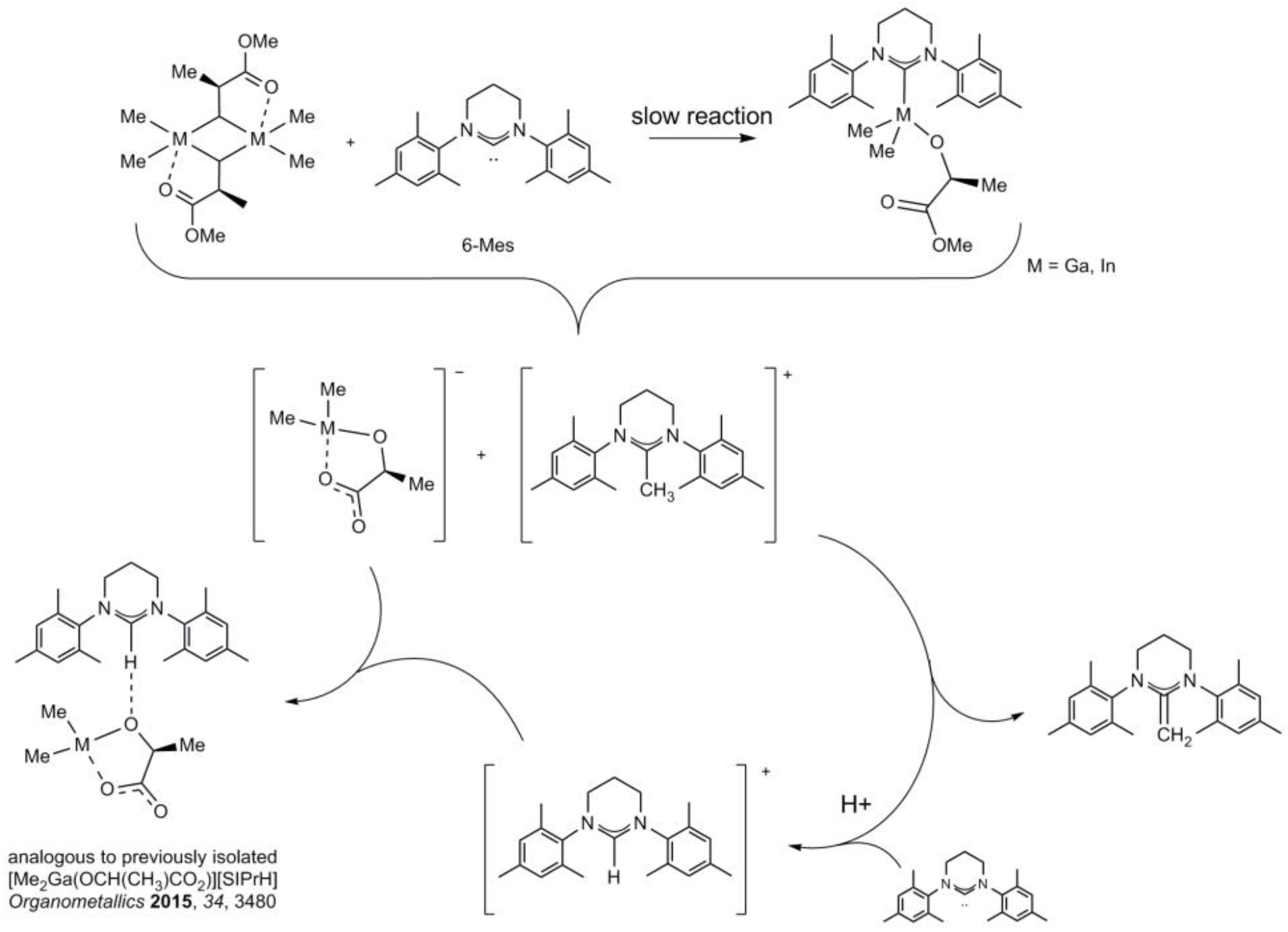
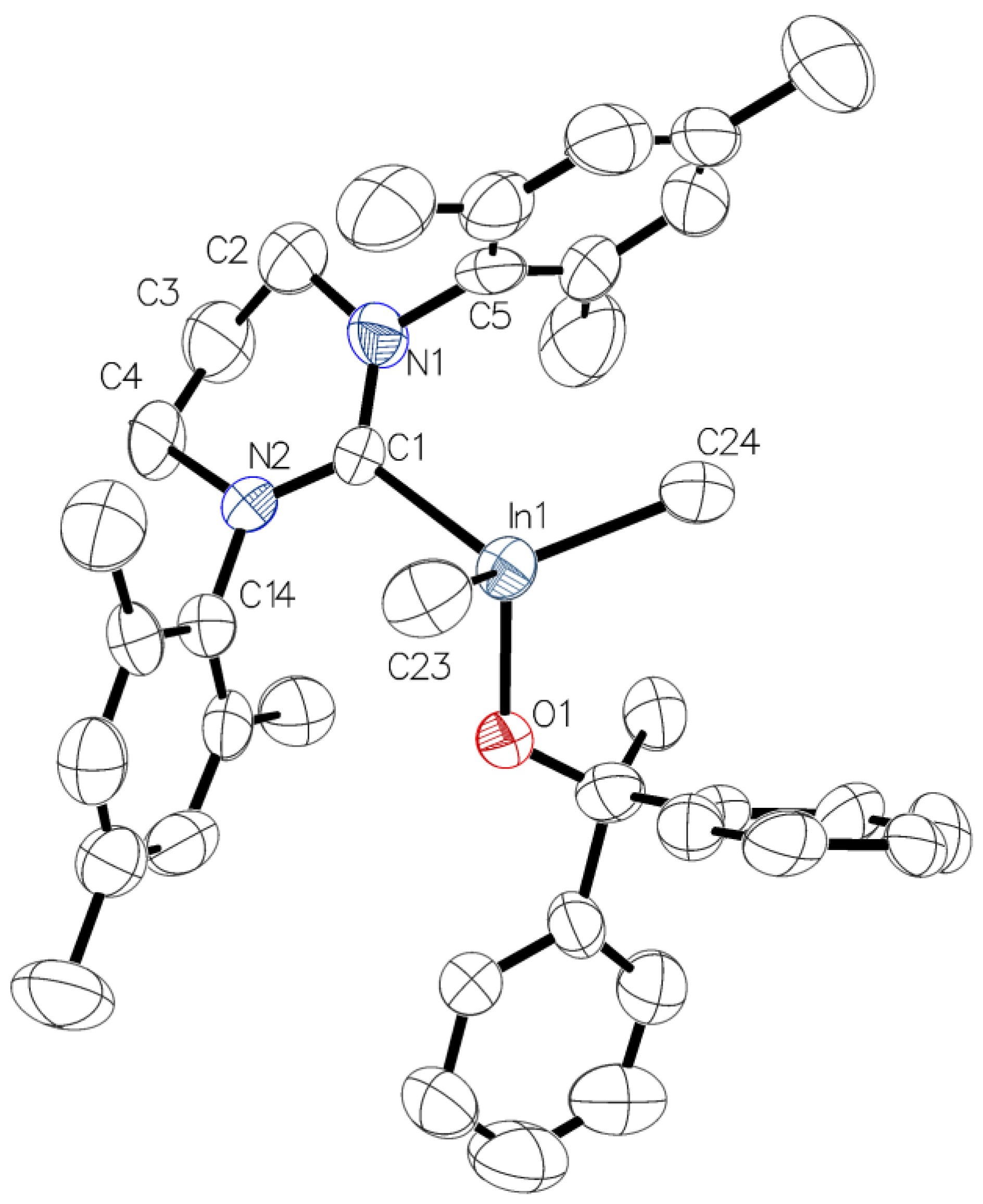
2.3. Reactivity of [Me2M(μ-OC6H4OMe)]2 (M = Ga, In) towards 6-Mes
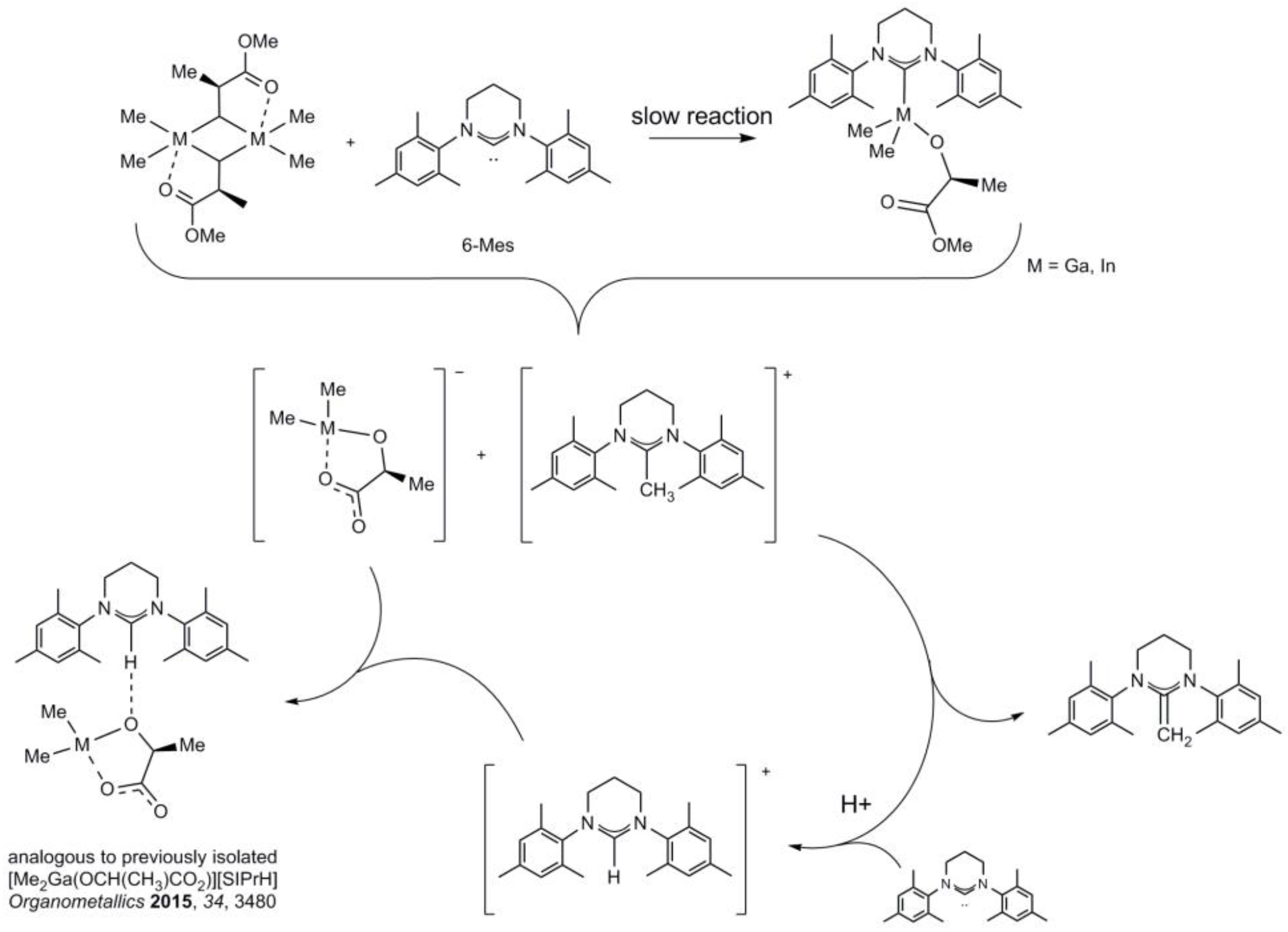
2.4. Reactivity of [Me2M(μ-(S)-OCH(Me)CO2Me)]2 (M = Ga, In) towards 6-Mes
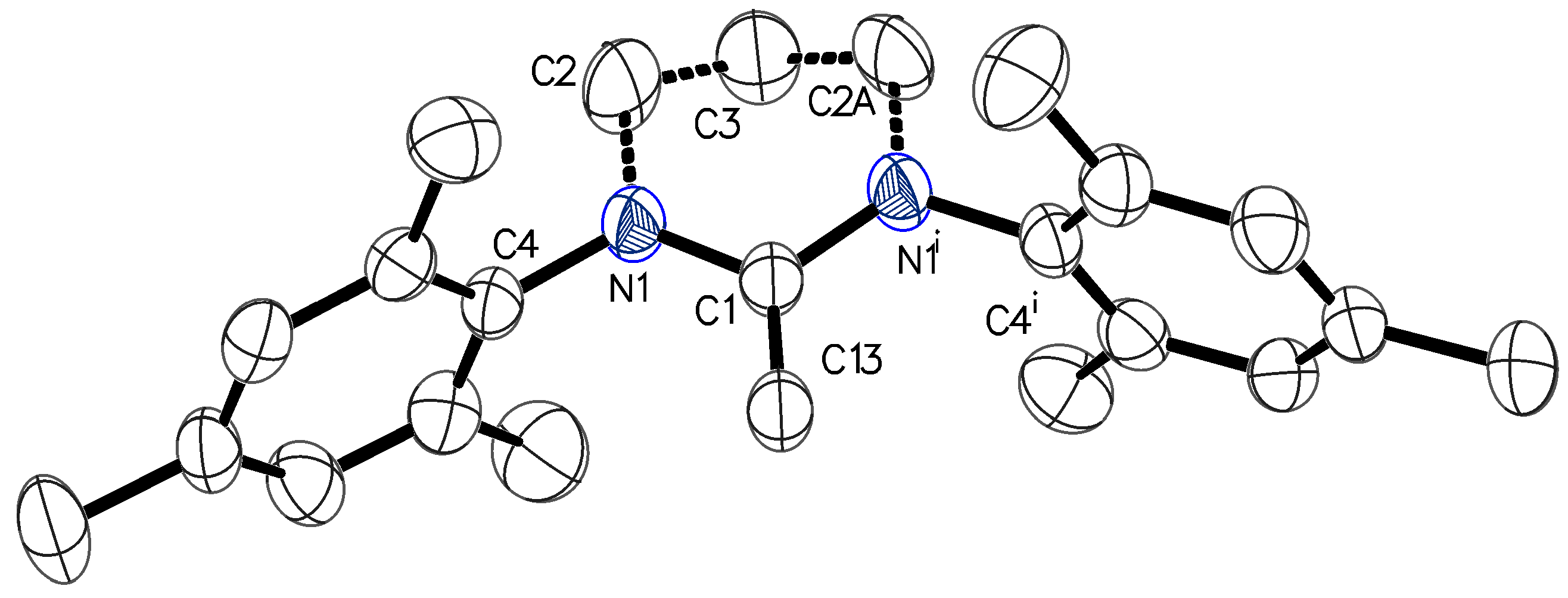
2.5. Activity of Me2Ga(OCPh2Me)(6-Mes) (3 and 4), Me2M(OC6H4OMe)(6-Mes) (5 and 6) (M = Ga, In) and Me2Ga(OCH2CH2OMe)(6-Mes) (1) in the ROP of rac-Lactide
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis of Indium and Gallium Complexes
3.3. General Procedure for the ROP of rac-LA with 1 and 3–6
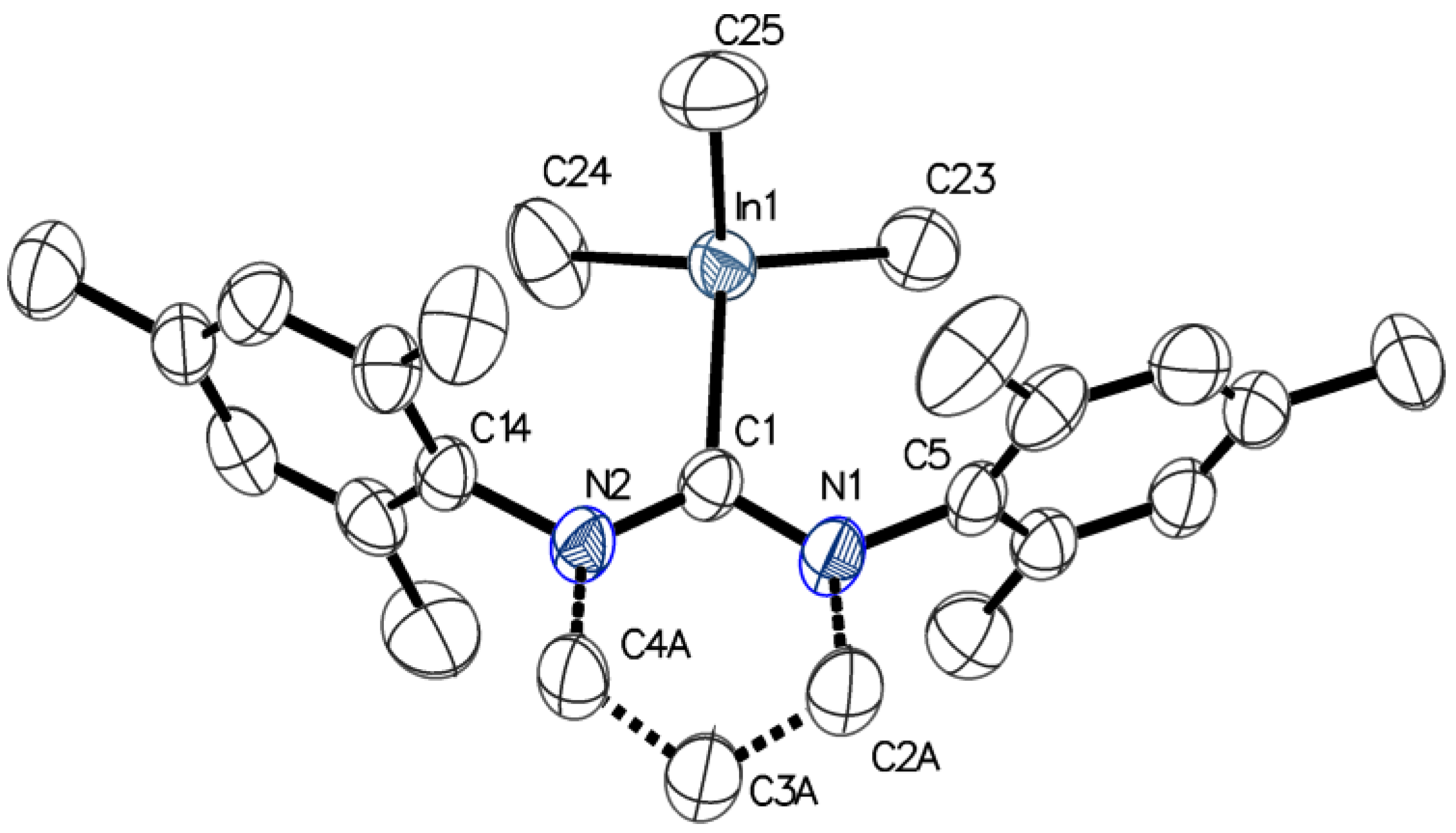
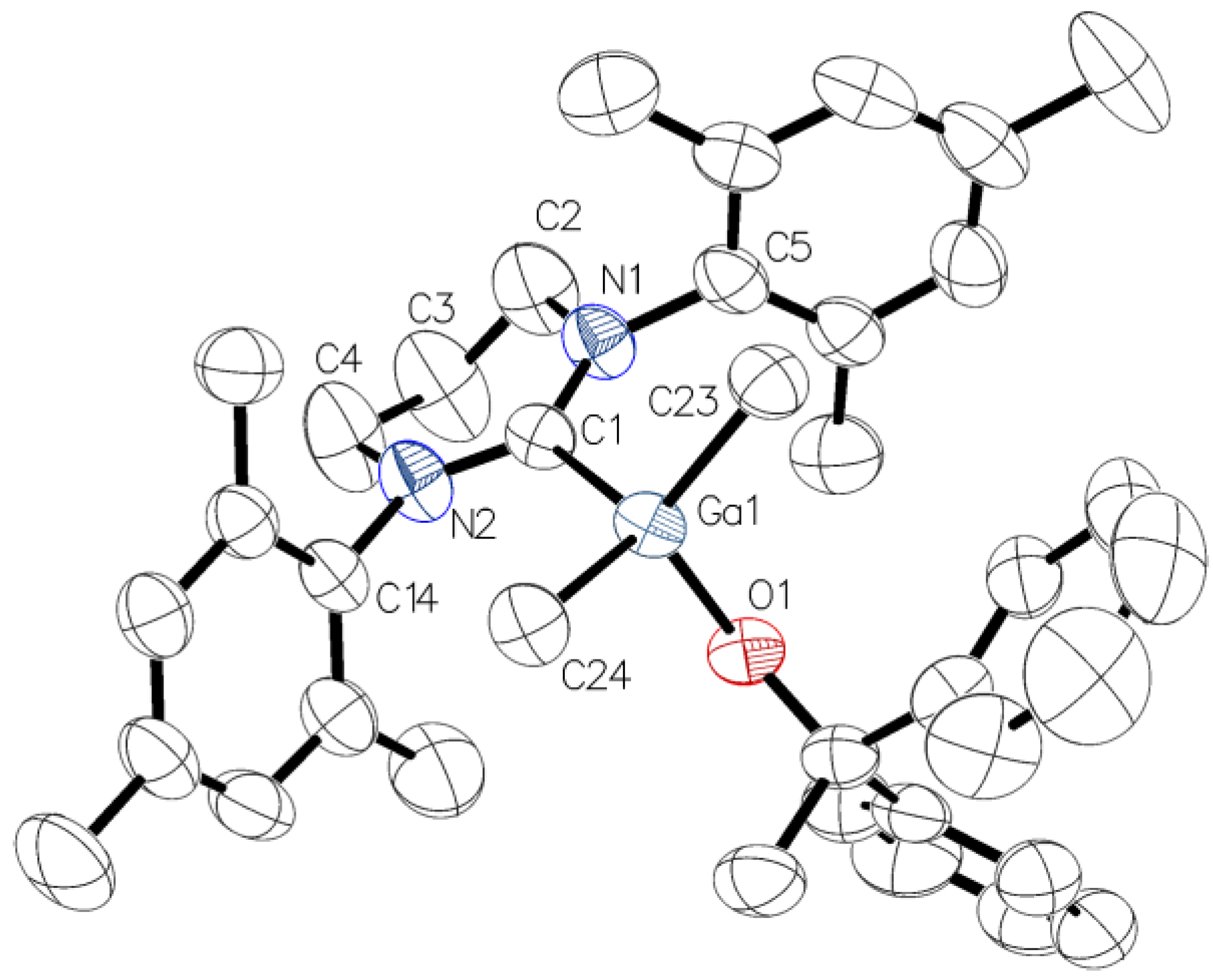
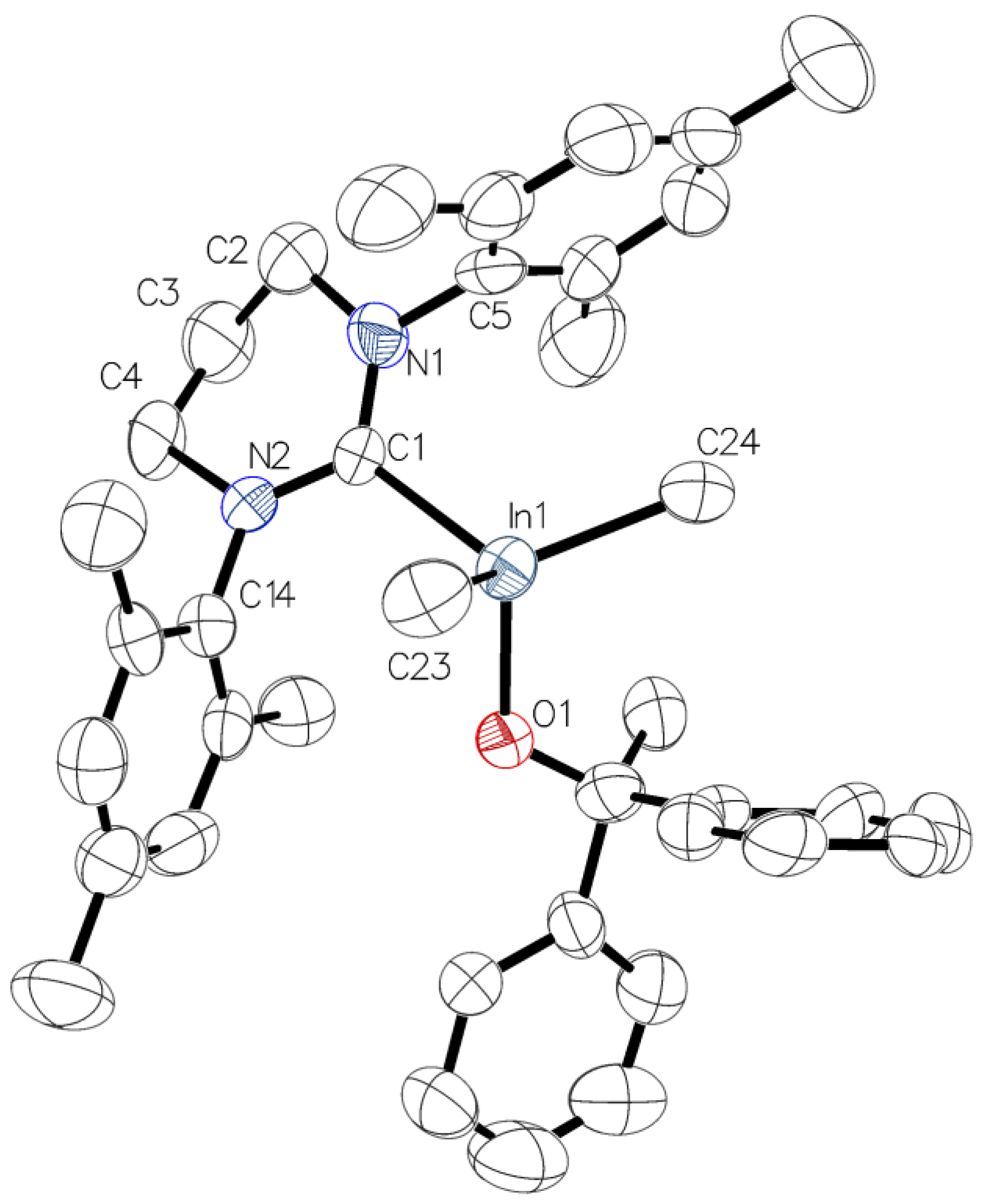
3.4. X-Ray Structure Determination
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Horeglad, P.; Szczepaniak, G.; Dranka, M.; Zachara, J. The first facile stereoselectivity switch in the polymerization of rac-lactide—From heteroselective to isoselective dialkylgallium alkoxides with the help of N-heterocyclic carbenes. Chem. Commun. 2012, 48, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Horeglad, P.; Cybularczyk, M.; Trzaskowski, B.; Żukowska, G.Z.; Dranka, M.; Zachara, J. Dialkylgallium Alkoxides Stabilized with N-Heterocyclic Carbenes: Opportunities and Limitations for the Controlled and Stereoselective Polymerization of rac-Lactide. Organometallics 2015, 34, 3480–3496. [Google Scholar] [CrossRef]
- Cybularczyk, M.; Dranka, M.; Zachara, J.; Horeglad, P. Effect of In–CNHC Bonds on the Synthesis, Structure, and Reactivity of Dialkylindium Alkoxides: How Indium Compares to Gallium. Organometallics 2016, 35, 3311–3322. [Google Scholar] [CrossRef]
- Fliedel, C.; Schnee, G.; Avilés, T.; Dagorne, S. Group 13 metal (Al, Ga, In, Tl) complexes supported by heteroatom-bonded carbene ligands. Coord. Chem. Rev. 2014, 275, 63–86. [Google Scholar] [CrossRef]
- Osten, K.M.; Mehrkhodavandi, P. Indium Catalysts for Ring Opening Polymerization: Exploring the Importance of Catalyst Aggregation. Acc. Chem. Res. 2017, 50, 2861–2869. [Google Scholar] [CrossRef] [PubMed]
- Myers, D.; White, A.J.P.; Forsyth, C.M.; Williams, C.K. Phosphasalen Indium Complexes Showing High Rates and Isoselectivities in rac-Lactide Polymerizations. Angew. Chem. Int. Ed. 2017, 56, 5277–5282. [Google Scholar] [CrossRef] [PubMed]
- Aluthge, D.C.; Ahn, J.M.; Mehrkhodavandi, P. Overcoming aggregation in indium salen catalysts for isoselective lactide polymerization. Chem. Sci. 2015, 6, 5284–5292. [Google Scholar] [CrossRef]
- Normand, M.; Dorcet, V.; Kirillov, E.; Carpentier, J.-F. {Phenoxy-imine}aluminum versus -indium Complexes for the Immortal ROP of Lactide: Different Stereocontrol, Different Mechanisms. Organometallics 2013, 32, 1694–1709. [Google Scholar] [CrossRef]
- Horeglad, P.; Litwińska, A.; Żukowska, G.Z.; Kubicki, D.; Szczepaniak, G.; Dranka, M.; Zachara, J. The influence of organosuperbases on the structure and activity of dialkylgallium alkoxides in the polymerization of rac-lactide: The road to stereo diblock PLA copolymers. Appl. Organometal. Chem. 2013, 27, 328–336. [Google Scholar] [CrossRef]
- Dai, Z.; Sun, Y.; Xiong, J.; Pan, X.; Tang, N.; Wu, J. Simple sodium and potassium phenolates as catalysts for highly isoselective polymerization of rac-lactide. Catal. Sci. Technol. 2016, 6, 515–520. [Google Scholar] [CrossRef]
- Bakewell, C.; White, A.J.P.; Long, N.J.; Williams, C.K. Scandium and Yttrium Phosphasalen Complexes as Initiators for Ring-Opening Polymerization of Cyclic Esters. Inorg. Chem. 2015, 54, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Abbina, S.; Du, G. Zinc-Catalyzed Highly Isoselective Ring Opening Polymerization of rac-Lactide. ACS Macro Lett. 2014, 3, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Buffet, J.-C.; Blaudeck, R.P.; Sujecki, S.; Blake, A.J.; Wilson, C. C3-Symmetric Lanthanide Tris(alkoxide) Complexes Formed by Preferential Complexation and Their Stereoselective Polymerization of rac-Lactide. Angew. Chem., Int. Ed. 2008, 47, 6033–6036. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.R.; Breyfogle, L.E.; Hillmyer, M.A.; Tolman, W.B. Stereoelective polymerization of D,L-lactide using N-heterocyclic carbene based compounds. Chem. Commun. 2004, 2504–2505. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.R.; Schaller, C.P.; Hillmyer, M.A.; Tolman, W.B. Zinc N-heterocyclic carbene complexes and their polymerization of D,L-lactide. J. Organomet. Chem. 2005, 690, 5881–5891. [Google Scholar] [CrossRef]
- Arnold, P.L.; Casely, I.J.; Turner, Z.R.; Bellabarba, R.; Tooze, R.B. Magnesium and zinc complexes of functionalised, saturated N-heterocyclic carbene ligands: Carbene lability and functionalisation, and lactide polymerisation catalysis. Dalton Trans. 2009, 7236–7247. [Google Scholar] [CrossRef] [PubMed]
- Romain, C.; Fliedel, C.; Bellemin-Laponnaz, S.; Dagorne, S. NHC Bis-Phenolate Aluminum Chelates: Synthesis, Structure, and Use in Lactide and Trimethylene Carbonate Polymerization. Organometallics 2014, 33, 5730–5739. [Google Scholar] [CrossRef]
- Zhao, N.; Hou, G.; Deng, X.; Zi, G.; Walter, M.D. Group 4 metal complexes with new chiral pincer NHC-ligands: Synthesis, structureand catalytic activity. Dalton Trans. 2014, 43, 8261–8272. [Google Scholar] [CrossRef] [PubMed]
- Romain, C.; Heinrich, B.; Bellemin-Laponnaz, S.; Dagorne, S. A robust zirconium N-heterocyclic carbene complex for the living and highly stereoselective ring-opening polymerization of rac-lactide. Chem. Commun. 2012, 48, 2213–2215. [Google Scholar] [CrossRef] [PubMed]
- Romain, C.; Brelot, L.; Bellemin-Laponnaz, S.; Dagorne, S. Synthesis and Structural Characterization of a Novel Family of Titanium Complexes Bearing a Tridentate Bis-phenolate-N-heterocyclic Carbene Dianionic Ligand and Their Use in the Controlled ROP of rac-Lactide. Organometallics 2010, 29, 1191–1198. [Google Scholar] [CrossRef]
- Schnee, G.; Bolley, A.; Hild, F.; Specklin, D.; Dagorne, S. Group 13 metal (Al, Ga, In) alkyls supported by N-heterocyclic carbenes for use in lactide ring-opening polymerization catalysis. Catal. Today 2017, 289, 204–210. [Google Scholar] [CrossRef]
- Patel, D.; Liddle, S.T.; Mungur, S.A.; Rodden, M.; Blake, A.J.; Arnold, P. Bifunctional yttrium(III) and titanium(IV) NHC catalysts for lactide polymerisation. Chem. Commun. 2006, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Gill, A.M.; Yunpeng, L.; Yongxin, L.; Ganguly, R.; Falivene, L.; Garcia, F. Aryl-NHC-group 13 trimethyl complexes: Structural, stability and bonding insights. Dalton Trans. 2017, 46, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, M.; Beetstra, D.J.; Knight, J.C.; Ooi, L.-L.; Stasch, A.; Coles, S.; Male, L.; Hursthouse, M.B.; Cavell, K.J.; Dervisi, A.; Fallis, I.A. Novel Expanded Ring N-Heterocyclic Carbenes: Free Carbenes, Silver Complexes, And Structures. Organometallics 2008, 27, 3279–3289. [Google Scholar] [CrossRef]
- Dröge, T.; Glorius, F. The Measure of All Rings—N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. 2010, 49, 6940–6953. [Google Scholar] [CrossRef] [PubMed]
- Bourissou, D.; Guerret, O.; Gabbaï, F.P.; Bertrand, G. Stable Carbenes. Chem. Rev. 2000, 100, 39–91. [Google Scholar] [CrossRef] [PubMed]
- Chitsaz, S.; Neumüller, B.Z. Compounds with Organometallic Alkoxo–Indium Cages. Anorg. Allg. Chem. 2001, 627, 2451–2459. [Google Scholar] [CrossRef]
- Chitsaz, S.; Iravani, E.; Neumüller, B.Z. Sesquialkoxides of Gallium and Indium. Anorg. Allg. Chem. 2002, 628, 2279–2285. [Google Scholar] [CrossRef]
- Brown, I.D.; Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. Sect. B Struct. Sci. 1985, 41, 244–247. [Google Scholar] [CrossRef]
- Kaupp, M.; Metz, B.; Stoll, H. Breakdown of Bond Length-Bond Strength Correlation: A Case Study. Angew. Chem. Int. Ed. 2000, 39, 4607–4609. [Google Scholar] [CrossRef]
- Higelin, A.; Keller, S.; Göhringer, C.; Jones, C.; Krossing, I. Unusual Tilted Carbene Coordination in Carbene Complexes of Gallium(I) and Indium(I). Angew. Chem. Int. Ed. 2013, 52, 4941–4944. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Liddle, S.T. f-block N-heterocyclic carbene complexes. Chem. Commun. 2006, 3959–3971. [Google Scholar] [CrossRef] [PubMed]
- Horeglad, P.; Ablialimov, O.; Szczepaniak, G.; Dąbrowska, A.M.; Dranka, M.; Zachara, J. Dialkylgallium Complexes with Alkoxide and Aryloxide Ligands Possessing N-Heterocyclic Carbene Functionalities: Synthesis and Structure. Organometallics 2014, 33, 100–111. [Google Scholar] [CrossRef]
- Dąbrowska, A.M.; Hurko, A.; Dranka, M.; Varga, V.; Urbańczyk, M.; Horeglad, P. Towards NHC stabilized alkylgallium alkoxide/aryloxide cations—The advances, the limitations and the challenges. J. Organomet. Chem. 2017, 840, 63–69. [Google Scholar] [CrossRef]
- Horeglad, P.; Kruk, P.; Pécaut, J. Heteroselective Polymerization of rac-Lactide in the Presenceof Dialkylgallium Alkoxides: The Effect of Lewis Base on Polymerization Stereoselectivity. Organometallics 2010, 29, 3729–3734. [Google Scholar] [CrossRef]
- Lewiński, J.; Horeglad, P.; Wójcik, K.; Justyniak, I. Chelation Effect in Polymerization of Cyclic Esters by Metal Alkoxides: Structure Characterization of the Intermediate Formed by Primary Insertion of Lactide into the Al–OR Bond of an Organometallic Initiator. Organometallics 2005, 24, 4588–4593. [Google Scholar] [CrossRef]
- Dagorne, S.; Le Bideau, F.; Welter, R.; Bellemin-Laponnaz, S.; Maisse-François, A. Well-Defined Cationic Alkyl- and Alkoxide-Aluminum Complexes and Their Reactivity with ε-Caprolactone and Lactides. Chem. Eur. J. 2007, 13, 3202–3217. [Google Scholar] [CrossRef] [PubMed]
- Kolychev, E.L.; Asachenko, A.F.; Dzhevakov, P.B.; Bush, A.A.; Shuntikov, V.V.; Khrustalev, V.N.; Nechaev, M.S. Expanded ring diaminocarbene palladium complexes: Synthesis, structure, and Suzuki–Miyaura cross-coupling of heteroaryl chlorides in water. Dalton Trans. 2013, 42, 6859–6866. [Google Scholar] [CrossRef] [PubMed]
- CRYSALISPRO Software System, version 1.171.38.43 (1–3 and (6-Mes)=CH2) and 1.171.38.46 (4); Agilent Technologies: Oxford, UK, 2015.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cybularczyk-Cecotka, M.; Dąbrowska, A.M.; Guńka, P.A.; Horeglad, P. Probing the Effect of Six-Membered N-Heterocyclic Carbene—6-Mes—on the Synthesis, Structure and Reactivity of Me2MOR(NHC) (M = Ga, In) Complexes. Inorganics 2018, 6, 28. https://doi.org/10.3390/inorganics6010028
Cybularczyk-Cecotka M, Dąbrowska AM, Guńka PA, Horeglad P. Probing the Effect of Six-Membered N-Heterocyclic Carbene—6-Mes—on the Synthesis, Structure and Reactivity of Me2MOR(NHC) (M = Ga, In) Complexes. Inorganics. 2018; 6(1):28. https://doi.org/10.3390/inorganics6010028
Chicago/Turabian StyleCybularczyk-Cecotka, Martyna, Anna Maria Dąbrowska, Piotr A. Guńka, and Paweł Horeglad. 2018. "Probing the Effect of Six-Membered N-Heterocyclic Carbene—6-Mes—on the Synthesis, Structure and Reactivity of Me2MOR(NHC) (M = Ga, In) Complexes" Inorganics 6, no. 1: 28. https://doi.org/10.3390/inorganics6010028
APA StyleCybularczyk-Cecotka, M., Dąbrowska, A. M., Guńka, P. A., & Horeglad, P. (2018). Probing the Effect of Six-Membered N-Heterocyclic Carbene—6-Mes—on the Synthesis, Structure and Reactivity of Me2MOR(NHC) (M = Ga, In) Complexes. Inorganics, 6(1), 28. https://doi.org/10.3390/inorganics6010028