Effect of Low Spin Excited States for Magnetic Anisotropy of Transition Metal Mononuclear Single Molecule Magnets

Abstract
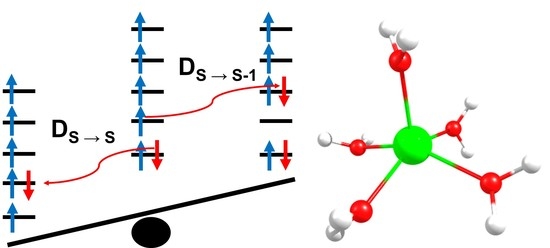
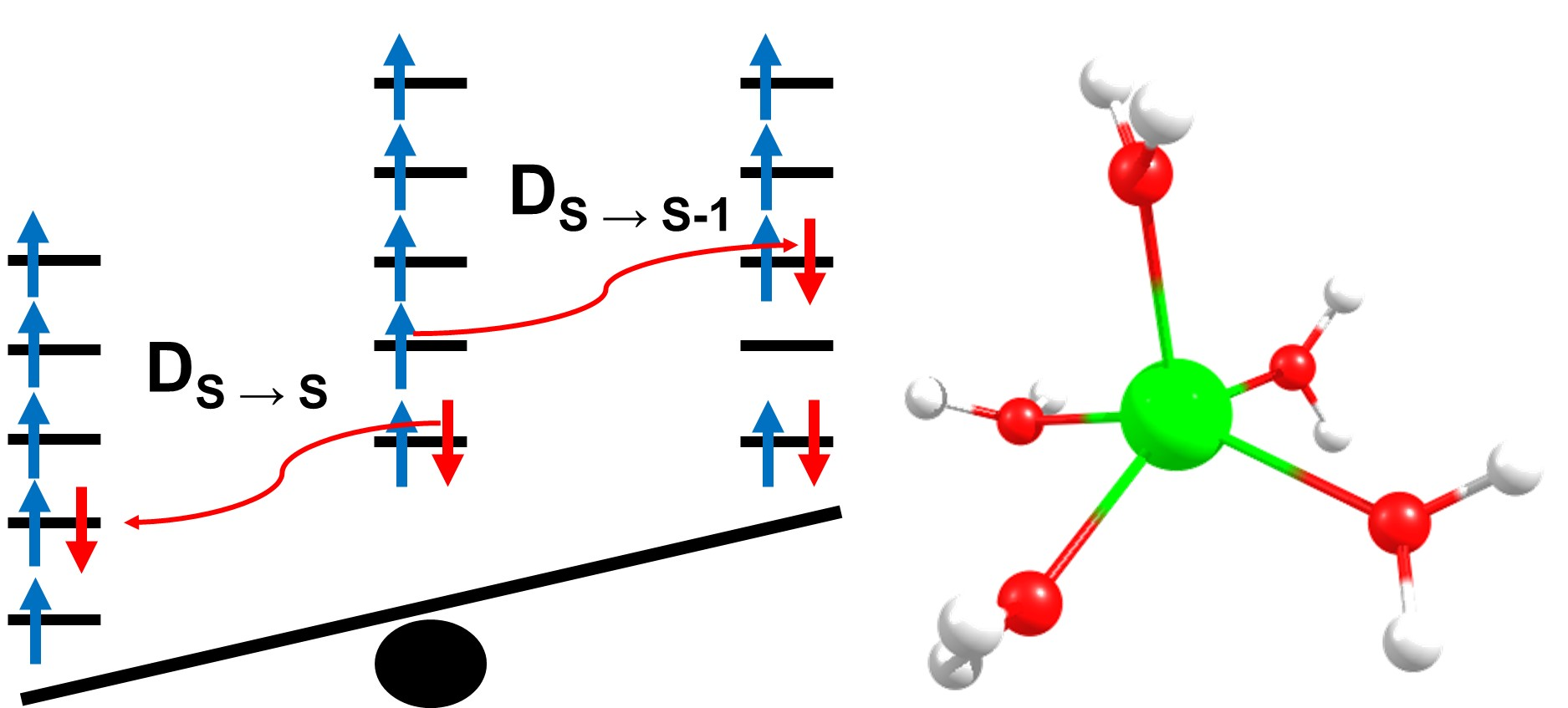
:
1. Introduction
2. Results and Discussion
2.1. d-Orbital Splitting
2.2. Effect of Jahn–Teller Distortion
2.3. Adequacy of Zero-Field Splitting Parameters
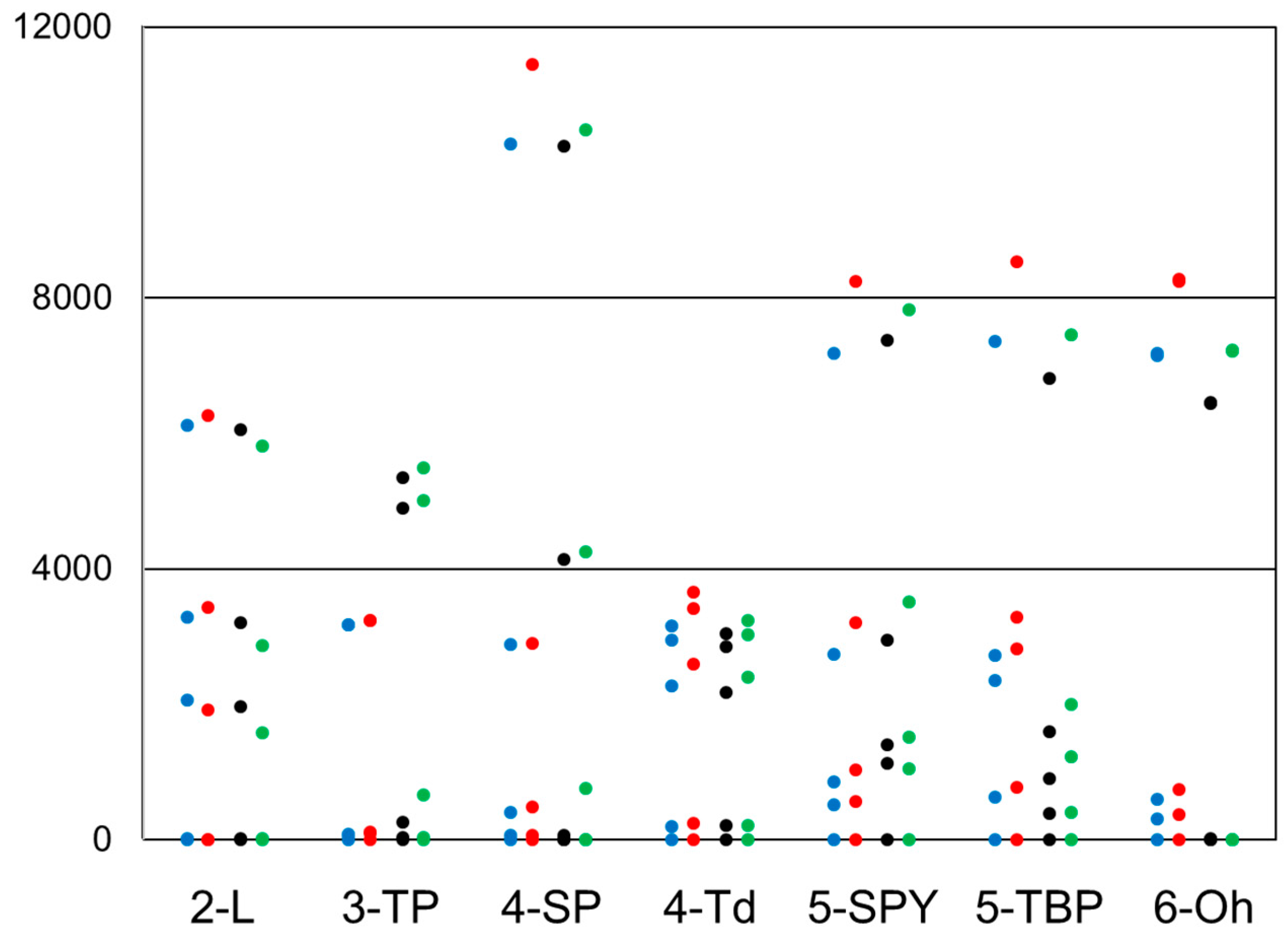
2.4. Inclusion of Lower Multiplicity Roots
2.5. Degenerate Ground States
3. Materials and Methods
Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.Y.; Kaizu, Y. Lanthanide double-decker complexes functioning as magnets at the single-molecular level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef] [PubMed]
- Habib, F.; Luca, O.R.; Vieru, V.; Shiddiq, M.; Korobkov, I.; Gorelsky, S.I.; Takase, M.K.; Chibotaru, L.F.; Hill, S.; Crabtree, R.H.; et al. Influence of the ligand field on slow magnetization relaxation versus spin crossover in mononuclear cobalt complexes. Angew. Chem. Int. Ed. 2013, 52, 11290–11293. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, J.-L.; Ungur, L.; Liu, J.; Li, Q.; Wang, L.; Ni, Z.; Chibotaru, L.F.; Chen, X.-M.; Tong, M. Symmetry-Supported Magnetic Blocking at 20 K in Pentagonal Bipyramidal Dy(III) Single-Ion Magnets. J. Am. Chem. Soc. 2016, 138, 2829–2837. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-S.; Chilton, N.F.; Winpenny, R.E.P.; Zheng, Y.-Z. On Approaching the Limit of Molecular Magnetic Anisotropy: A Near-Perfect Pentagonal Bipyramidal Dysprosium(III) Single-Molecule Magnet. Angew. Chem. Int. Ed. 2016, 55, 16071–16074. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.A.P.; Ortu, F.; Reta, D.; Chilton, N.F.; Mills, D.P. Molecular magnetic hysteresis at 60 kelvin in dysprosocenium. Nature 2017, 548, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Torres, R.; Menelaou, M.; Roubeau, O.; Sorrenti, A.; Brandariz-de-Pedro, G.; Sañudo, E.C.; Teat, S.J.; Fraxedas, J.; Ruiz, E.; Aliaga-Alcalde, N. Multiscale study of mononuclear Co(II) SMMs based on curcuminoid ligands. Chem. Sci. 2016, 7, 2793–2803. [Google Scholar] [CrossRef]
- Aravena, D.; Ruiz, E. Shedding Light on the Single-Molecule Magnet Behavior of Mononuclear Dy(III) Complexes. Inorg. Chem. 2013, 52, 13770–13778. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, M.; Zadrozny, J.M.; Long, J.R.; Neese, F. A theoretical analysis of chemical bonding, vibronic coupling, and magnetic anisotropy in linear iron(II) complexes with single-molecule magnet behavior. Chem. Sci. 2013, 4, 139–156. [Google Scholar] [CrossRef]
- Rechkemmer, Y.; Breitgoff, F.D.; Van Der Meer, M.; Atanasov, M.; Hakl, M.; Orlita, M.; Neugebauer, P.; Neese, F.; Sarkar, B.; Slageren, J. Van A four-coordinate cobalt(II) single-ion magnet with coercivity and a very high energy barrier. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cahier, B.; Maurice, R.; Bolvin, H.; Mallah, T.; Guihéry, N. Tools for Predicting the Nature and Magnitude of Magnetic Anisotropy in Transition Metal Complexes: Application to Co(II) Complexes. Magnetochemistry 2016, 2, 31. [Google Scholar] [CrossRef]
- Campbell, V.E.; Bolvin, H.; Rivière, E.; Guillot, R.; Wernsdorfer, W.; Mallah, T. Structural and electronic dependence of the single-molecule-magnet behavior of dysprosium(III) complexes. Inorg. Chem. 2014, 53, 2598–2605. [Google Scholar] [CrossRef] [PubMed]
- El-Khatib, F.; Cahier, B.; López-Jordà, M.; Guillot, R.; Rivière, E.; Hafez, H.; Saad, Z.; Girerd, J.-J.; Guihéry, N.; Mallah, T. Design of a Binuclear Ni(II) Complex with Large Ising-type Anisotropy and Weak Anti-Ferromagnetic Coupling. Inorg. Chem. 2017, 56, 10655–10663. [Google Scholar] [CrossRef] [PubMed]
- Cahier, B.; Perfetti, M.; Zakhia, G.; Naoufal, D.; El-Khatib, F.; Guillot, R.; Rivière, E.; Sessoli, R.; Barra, A.L.; Guihéry, N.; et al. Magnetic anisotropy in pentacoordinate Ni(II) and Co(II) complexes: Unraveling electronic and geometrical contributions. Chem. A Eur. J. 2017, 23, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, S.; Upadhyay, A.; Singh, S.K.; Gupta, T.; Tewary, S.; Langley, S.K.; Walsh, J.P.S.; Murray, K.S.; Rajaraman, G.; Shanmugam, M. A synthetic strategy for switching the single ion anisotropy in tetrahedral Co(II) complexes. Chem. Commun. 2015, 51, 3739–3742. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Rajeshkumar, T.; Rajaraman, G.; Murugavel, R. An air-stable Dy(III) single-ion magnet with high anisotropy barrier and blocking temperature. Chem. Sci. 2016, 7, 5181–5191. [Google Scholar] [CrossRef]
- Belio Castro, A.; Jung, J.; Golhen, S.; Le Guennic, B.; Ouahab, L.; Cador, O.; Pointillart, F. Slow Magnetic Relaxation in Unprecedented Mono-Dimensional Coordination Polymer of Ytterbium Involving Tetrathiafulvalene-Dicarboxylate Linker. Magnetochemistry 2016, 2, 26. [Google Scholar] [CrossRef]
- Zhang, P.; Jung, J.; Zhang, L.; Tang, J.; Le Guennic, B. Elucidating the Magnetic Anisotropy and Relaxation Dynamics of Low-Coordinate Lanthanide Compounds. Inorg. Chem. 2016, 55, 1905–1911. [Google Scholar] [CrossRef] [PubMed]
- Ribbing, C.; Pierloot, K.; Ceulemans, A. Ab Initio Calculations of the Trigonal and Zero-Field Splittings in Trischelated Diketonato Complexes of Trivalent Chromium. Inorg. Chem. 1998, 37, 5227–5232. [Google Scholar] [CrossRef]
- Atanasov, M.; Ganyushin, D.; Sivalingam, K.; Neese, F.; Atanasov, M.; Ganyushin, D.; Sivalingam, K.; Neese, F. A Modern First-Principles View on Ligand Field Theory Through the Eyes of Correlated Multireference Wavefunctions. In Molecular Electronic Structures of Transition Metal Complexes II; Mingos, D.M.P., Day, P., Dahl, J.P., Eds.; Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2012; ISBN 978-3-642-27377-3. [Google Scholar]
- Chibotaru, L.F. Ab Initio Methodology for Pseudospin Hamiltonians of Anisotropic Magnetic Complexes. In Advances in Chemical Physics; Rice, S.A., Dinner, A.R., Eds.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2013; Volume 153, ISBN 978-1-118-57176-7. [Google Scholar]
- Chibotaru, L.F.; Ungur, L. Ab initio calculation of anisotropic magnetic properties of complexes. I. Unique definition of pseudospin Hamiltonians and their derivation. J. Chem. Phys. 2012, 137, 64112. [Google Scholar] [CrossRef] [PubMed]
- Aravena, D.; Atanasov, M.; Neese, F. Periodic Trends in Lanthanide Compounds through the Eyes of Multireference ab Initio Theory. Inorg. Chem. 2016, 55, 4457–4469. [Google Scholar] [CrossRef] [PubMed]
- Schweinfurth, D.; Sommer, M.G.; Atanasov, M.; Demeshko, S.; Hohloch, S.; Meyer, F.; Neese, F.; Sarkar, B. The Ligand Field of the Azido Ligand: Insights into Bonding Parameters and Magnetic Anisotropy in a Co(II)–Azido Complex. J. Am. Chem. Soc. 2015, 137, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- García-Fuente, A.; Cimpoesu, F.; Ramanantoanina, H.; Herden, B.; Daul, C.; Suta, M.; Wickleder, C.; Urland, W. A ligand field theory-based methodology for the characterization of the Eu2+[Xe]4f65d1 excited states in solid state compounds. Chem. Phys. Lett. 2015, 622, 120–123. [Google Scholar] [CrossRef]
- Gatteschi, D.; Sessoli, R. Quantum Tunneling of Magnetization and Related Phenomena in Molecular Materials. Angew. Chem. Int. Ed. 2003, 42, 268–297. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Coca, S.; Urtizberea, A.; Cremades, E.; Alonso, P.J.; Camón, A.; Ruiz, E.; Luis, F. Origin of slow magnetic relaxation in Kramers ions with non-uniaxial anisotropy. Nat. Commun. 2014, 5, 4300. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.S.; Dreiser, J.; Weihe, H.; Sibille, R.; Johannesen, H.V.; Sørensen, M.A.; Nielsen, B.E.; Sigrist, M.; Mutka, H.; Rols, S.; et al. Design of Single-Molecule Magnets: Insufficiency of the Anisotropy Barrier as the Sole Criterion. Inorg. Chem. 2015, 54, 7600–7606. [Google Scholar] [CrossRef] [PubMed]
- Lunghi, A.; Totti, F.; Sanvito, S.; Sessoli, R. Intra-molecular origin of the spin-phonon coupling in slow-relaxing molecular magnets. Chem. Sci. 2017, 8, 6051–6059. [Google Scholar] [CrossRef] [PubMed]
- Woods, T.J.; Ballesteros-Rivas, M.F.; Gómez-Coca, S.; Ruiz, E.; Dunbar, K.R. Relaxation Dynamics of Identical Trigonal Bipyramidal Cobalt Molecules with Different Local Symmetries and Packing Arrangements: Magnetostructural Correlations and ab inito Calculations. J. Am. Chem. Soc. 2016, 138, 16407–16416. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, M.; Comba, P.; Helmle, S.; Müller, D.; Neese, F. Zero-Field Splitting in a Series of Structurally Related Mononuclear Ni(II)–Bispidine Complexes. Inorg. Chem. 2012, 51, 12324–12335. [Google Scholar] [CrossRef] [PubMed]
- Duboc, C.; Collomb, M.-N.; Neese, F. Understanding the Zero-Field Splitting of Mononuclear Manganese(II) Complexes from Combined EPR Spectroscopy and Quantum Chemistry. Appl. Magn. Reson. 2010, 37, 229–245. [Google Scholar] [CrossRef]
- Maurice, R.; de Graaf, C.; Guihéry, N. Magnetostructural relations from a combined ab initio and ligand field analysis for the nonintuitive zero-field splitting in Mn(III) complexes. J. Chem. Phys. 2010, 133, 84307. [Google Scholar] [CrossRef] [PubMed]
- Perić, M.; García-Fuente, A.; Zlatar, M.; Daul, C.; Stepanović, S.; García-Fernández, P.; Gruden-Pavlović, M. Magnetic anisotropy in “scorpionate” first-row transition-metal complexes: A theoretical investigation. Chem. A Eur. J. 2015, 21, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Coca, S.; Cremades, E.; Aliaga-Alcalde, N.; Ruiz, E. Mononuclear Single-Molecule Magnets: Tailoring the Magnetic Anisotropy of First-Row Transition-Metal Complexes. J. Am. Chem. Soc. 2013, 135, 7010–7018. [Google Scholar] [CrossRef] [PubMed]
- Stavretis, S.E.; Atanasov, M.; Podlesnyak, A.A.; Hunter, S.C.; Neese, F.; Xue, Z.-L. Magnetic Transitions in Iron Porphyrin Halides by Inelastic Neutron Scattering and Ab Initio Studies of Zero-Field Splittings. Inorg. Chem. 2015, 54, 9790–9801. [Google Scholar] [CrossRef] [PubMed]
- Aravena, D.; Venegas-Yazigi, D.; Ruiz, E. Single-Molecule Magnet Properties of Transition-Metal Ions Encapsulated in Lacunary Polyoxometalates: A Theoretical Study. Inorg. Chem. 2016, 55, 6405–6413. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Solomon, E.I. Calculation of Zero-Field Splittings, g-Values, and the Relativistic Nephelauxetic Effect in Transition Metal Complexes. Application to High-Spin Ferric Complexes. Inorg. Chem. 1998, 37, 6568–6582. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Suzuki, K.; Minato, T.; Shinoe, M.; Yamaguchi, K.; Mizuno, N. Field-induced slow magnetic relaxation of octahedrally coordinated mononuclear Fe(III)-, Co(II)-, and Mn(III)-containing polyoxometalates. Chem. Commun. 2015, 51, 4081–4084. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Coca, S.; Aravena, D.; Morales, R.; Ruiz, E. Large magnetic anisotropy in mononuclear metal complexes. Coord. Chem. Rev. 2015, 289–290, 379–392. [Google Scholar] [CrossRef]
- Freedman, D.E.; Harman, W.H.; Harris, T.D.; Long, G.J.; Chang, C.J.; Long, J.R. Slow magnetic relaxation in a high-spin iron(II) complex. J. Am. Chem. Soc. 2010, 132, 1224–1225. [Google Scholar] [CrossRef] [PubMed]
- Weismann, D.; Sun, Y.; Lan, Y.; Wolmershäuser, G.; Powell, A.K.; Sitzmann, H. High-Spin Cyclopentadienyl Complexes: A Single-Molecule Magnet Based on the Aryl–Iron(II) Cyclopentadienyl Type. Chem. A Eur. J. 2011, 17, 4700–4704. [Google Scholar] [CrossRef] [PubMed]
- Zadrozny, J.M.; Liu, J.; Piro, N.A.; Chang, C.J.; Hill, S.; Long, J.R. Slow magnetic relaxation in a pseudotetrahedral cobalt(II) complex with easy-plane anisotropy. Chem. Commun. 2012, 48, 3927–3929. [Google Scholar] [CrossRef] [PubMed]
- Mathonière, C.; Lin, H.-J.; Siretanu, D.; Clérac, R.; Smith, J.M. Photoinduced Single-Molecule Magnet Properties in a Four-Coordinate Iron(II) Spin Crossover Complex. J. Am. Chem. Soc. 2013, 135, 19083–19086. [Google Scholar] [CrossRef] [PubMed]
- Eichhöfer, A.; Lan, Y.; Mereacre, V.; Bodenstein, T.; Weigend, F. Slow magnetic relaxation in trigonal-planar mononuclear Fe(II) and Co(II) bis(trimethylsilyl)amido complexes—A comparative study. Inorg. Chem. 2014, 53, 1962–1974. [Google Scholar] [CrossRef] [PubMed]
- Novikov, V.V.; Pavlov, A.A.; Nelyubina, Y.V.; Boulon, M.-E.; Varzatskii, O.A.; Voloshin, Y.Z.; Winpenny, R.E.P. A Trigonal Prismatic Mononuclear Cobalt(II) Complex Showing Single-Molecule Magnet Behavior. J. Am. Chem. Soc. 2015, 137, 9792–9795. [Google Scholar] [CrossRef] [PubMed]
- Craig, G.A.; Murrie, M. 3D Single-Ion Magnets. Chem. Soc. Rev. 2015, 44, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Habib, F.; Korobkov, I.; Murugesu, M. Exposing the intermolecular nature of the second relaxation pathway in a mononuclear cobalt(II) single-molecule magnet with positive anisotropy. Dalt. Trans. 2015, 44, 6368–6373. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.K.; Khatua, S.; Tomar, K.; Konar, S. Field-Induced Single-Ion-Magnetic Behavior of Octahedral CoII in a Two-Dimensional Coordination Polymer. Eur. J. Inorg. Chem. 2016, 2016, 3525–3552. [Google Scholar] [CrossRef]
- Frost, J.M.; Harriman, K.L.M.; Murugesu, M. The rise of 3-d single-ion magnets in molecular magnetism: Towards materials from molecules? Chem. Sci. 2016, 7, 2470–2491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z.; Gómez-Coca, S.; Brown, A.J.; Saber, M.R.; Zhang, X.; Dunbar, K.R. Trigonal antiprismatic Co(II) single molecule magnets with large uniaxial anisotropies: Importance of Raman and tunneling mechanisms. Chem. Sci. 2016, 7, 6519–6527. [Google Scholar] [CrossRef] [PubMed]
- Zadrozny, J.M.; Greer, S.M.; Hill, S.; Freedman, D.E. A flexible iron(II) complex in which zero-field splitting is resistant to structural variation. Chem. Sci. 2016, 7, 416–423. [Google Scholar] [CrossRef]
- Atanasov, M.; Aravena, D.; Suturina, E.; Bill, E.; Maganas, D.; Neese, F. First principles approach to the electronic structure, magnetic anisotropy and spin relaxation in mononuclear 3d-transition metal single molecule magnets. Coord. Chem. Rev. 2015, 289, 177–214. [Google Scholar] [CrossRef]
- Ganyushin, D.; Neese, F. First-principles calculations of zero-field splitting parameters. J. Chem. Phys. 2006, 125, 24103. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Malmqvist, P.-Å.; Roos, B.O. The CASSCF state interaction method. Chem. Phys. Lett. 1989, 155, 189–194. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of n -electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.-P. N-electron valence state perturbation theory: A fast implementation of the strongly contracted variant. Chem. Phys. Lett. 2001, 350, 297–305. [Google Scholar] [CrossRef]
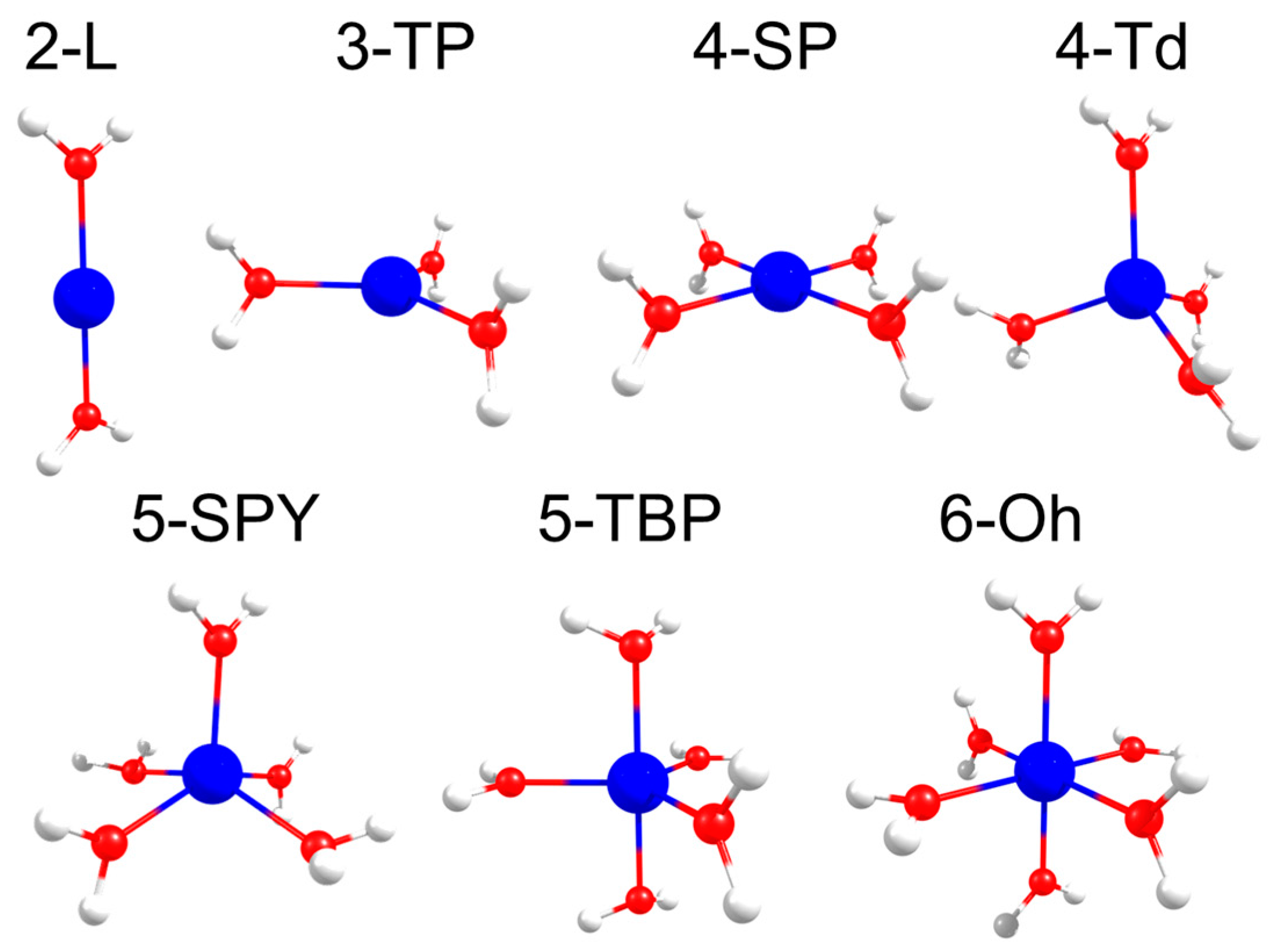

{kind=link}
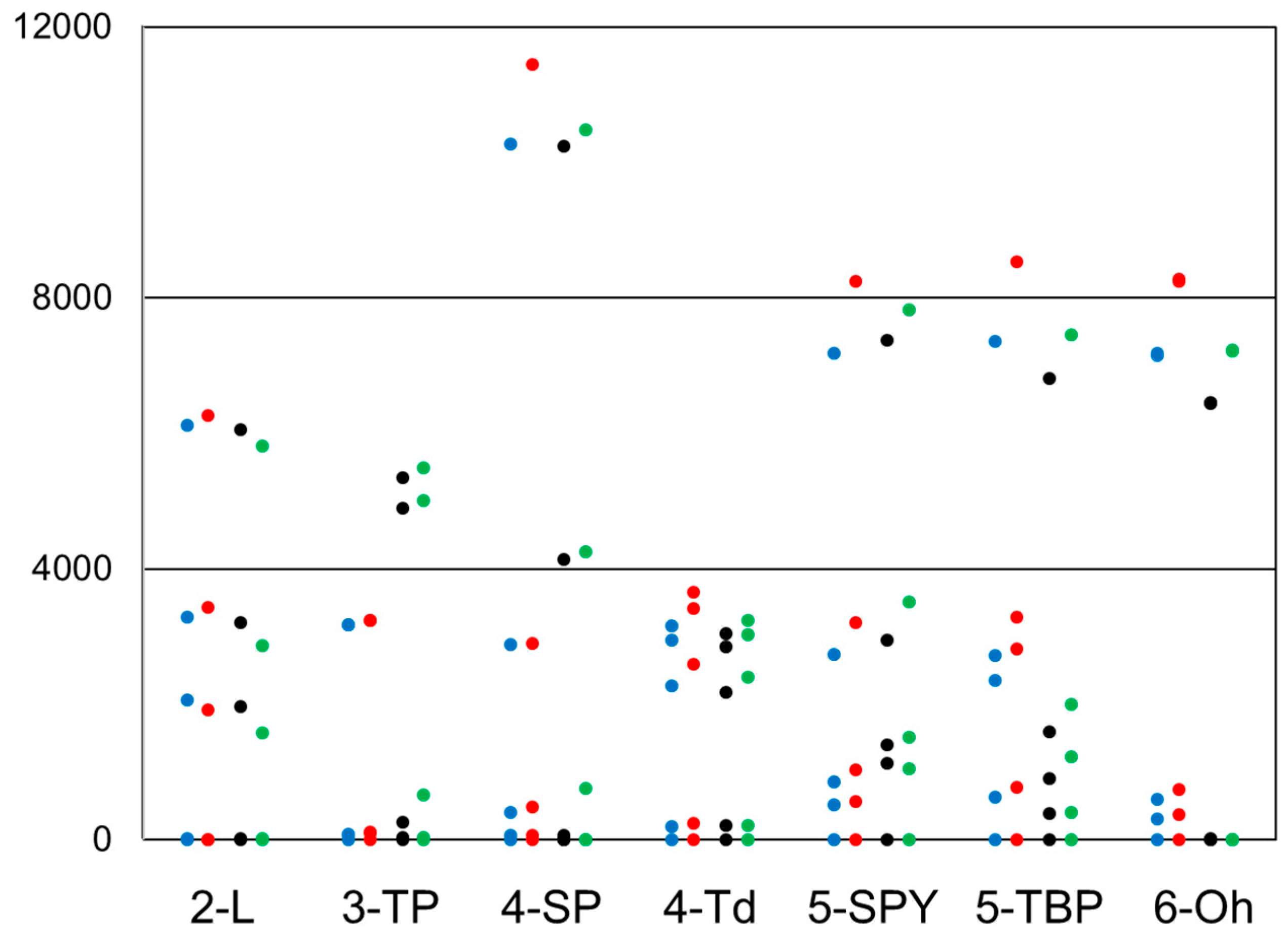{kind=link}
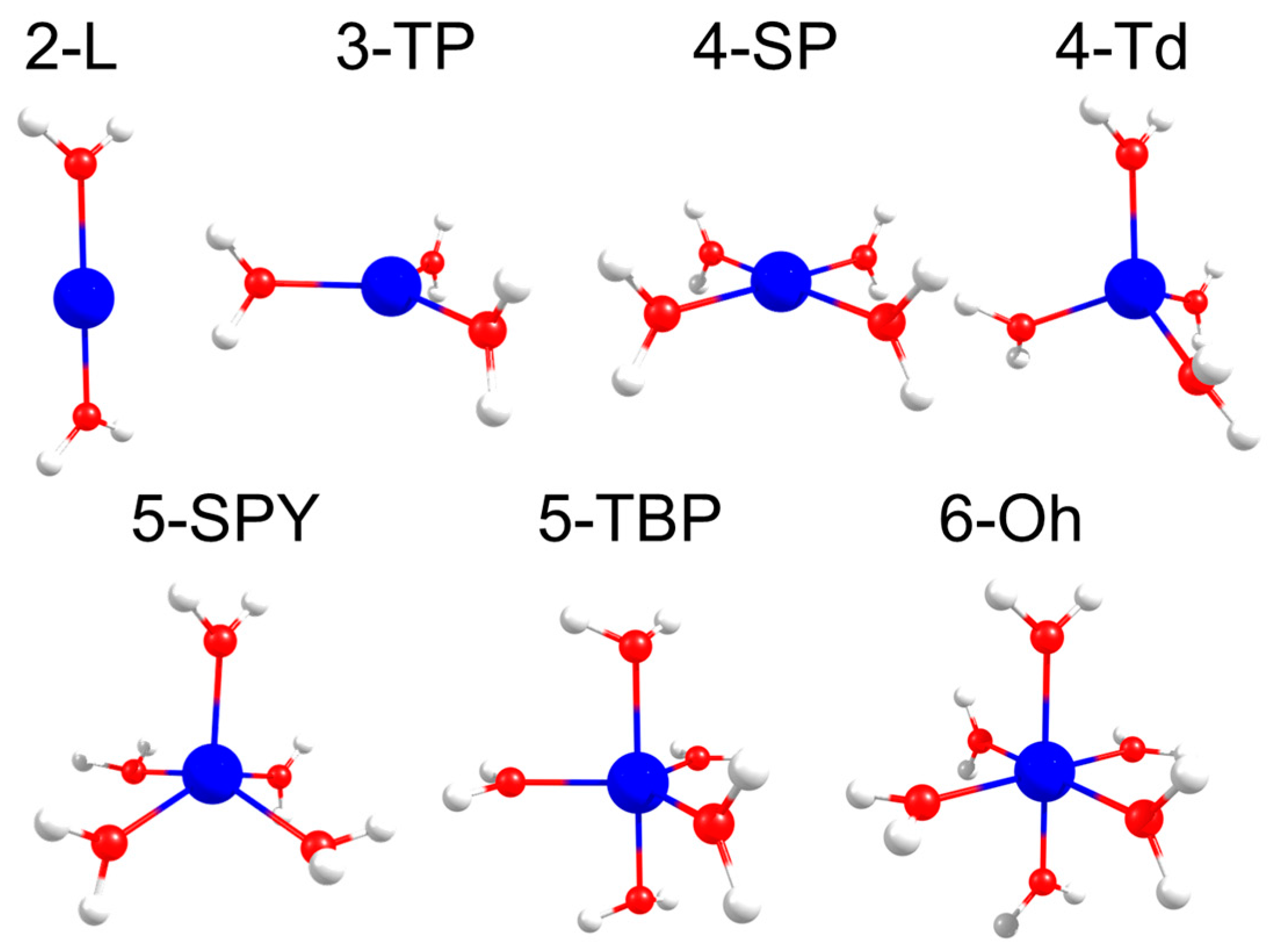{kind=link}
| Geometry | Fe(II) | Co(II) | ||
|---|---|---|---|---|
| noJT | JT | noJT | JT | |
| 2-L | 8.9 | 26.3 | 7.3 | 3.8 |
| 3-TP | 63.7 | 609 | 1408.2 | 2217.8 |
| 4-SP | 68.8 | 565.1 | 1000.5 | 1195.9 |
| 4-Td | 203.4 | 304.3 | 2268.5 | 3016.5 |
| 5-SPY | 526.2 | 970.9 | 664.5 | 1155.2 |
| 5-TBP | 638 | 913.8 | 588.4 | 2196.3 |
| 6-Oh | 299.3 | 461.6 | 4.2 | 394.7 |
| Fe(II) | Co(II) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| D2PT | DHeff | E/D2PT | E/DHeff | D2PT | DHeff | E/D2PT | E/DHeff | ||
| noJT | 2-L | −4799.7 | −83.1 | 0.00 | 0.01 | −36459.3 | −239.8 | 0.00 | 0.00 |
| 3-TP | 475.9 | 21.4 | 0.05 | 0.02 | 133.8 | 79.1 | 0.02 | 0.02 | |
| 4-SP | −438.4 | −37.7 | 0.03 | 0.26 | 160.2 | 87.0 | 0.00 | 0.00 | |
| 4-Td | −10.1 | −4.9 | 0.10 | 0.06 | −18.6 | −14.6 | 0.25 | 0.24 | |
| 5-SPY | −22.1 | 18.0 | 0.28 | 0.31 | −185.3 | −117.9 | 0.17 | 0.21 | |
| 5-TBP | −11.1 | −10.1 | 0.24 | 0.15 | 83.7 | 62.7 | 0.02 | 0.15 | |
| 6-Oh | −33.0 | 25.8 | 0.23 | 0.24 | 15175.6 | 175.0 | 0.03 | 0.02 | |
| JT | 2-L | −1602.8 | −82.5 | 0.00 | 0.00 | −64329.6 | −236.0 | 0.00 | 0.00 |
| 3-TP | −49.0 | −33.4 | 0.09 | 0.00 | 86.6 | 64.5 | 0.00 | 0.00 | |
| 4-SP | −54.6 | −38.0 | 0.02 | 0.04 | 134.5 | 82.9 | 0.00 | 0.00 | |
| 4-Td | −8.1 | −5.0 | 0.02 | 0.09 | −15.7 | −13.4 | 0.24 | 0.24 | |
| 5-SPY | −7.5 | −6.1 | 0.30 | 0.27 | −82.2 | −66.6 | 0.26 | 0.26 | |
| 5-TBP | 7.8 | −6.7 | 0.29 | 0.32 | 50.7 | 42.0 | 0.02 | 0.02 | |
| 6-Oh | 22.1 | 21.9 | 0.28 | 0.16 | 143.5 | 116.4 | 0.01 | 0.00 | |
| CASSCF | NEVPT2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DAll | E/DAll | DHS | E/DHS | DAll | E/DAll | DHS | E/DHS | ||||
| Fe(II) | noJT | 5-SPY | −22.1 | 0.28 | −21.0 | 0.29 | −19.9 | 0.25 | −19.0 | 0.27 | |
| noJT | 5-TBP | −11.1 | 0.24 | −11.5 | 0.14 | −8.8 | 0.27 | −9.4 | 0.15 | ||
| JT | 3-TP | −49.0 | 0.09 | −48.1 | 0.09 | −44.6 | 0.07 | −32.5 | 0.01 | ||
| JT | 4-SP | −54.6 | 0.02 | −53.1 | 0.02 | −54.2 | 0.00 | −36.5 | 0.02 | ||
| JT | 4-Td | −8.1 | 0.02 | −6.2 | 0.15 | −7.7 | 0.04 | −3.8 | 0.06 | ||
| JT | 5-SPY | −7.5 | 0.30 | −6.2 | 0.29 | 7.0 | 0.32 | −5.6 | 0.22 | ||
| JT | 5-TBP | 7.8 | 0.29 | −7.3 | 0.25 | 6.7 | 0.26 | −7.1 | 0.19 | ||
| JT | 6-Oh | 22.1 | 0.28 | 21.6 | 0.23 | 18.1 | 0.32 | 21.8 | 0.13 | ||
| Co(II) | noJT | 3-TP | 133.8 | 0.02 | 125.9 | 0.02 | 108.3 | 0.03 | 102.9 | 0.03 | |
| noJT | 4-SP | 160.2 | 0.00 | 151.4 | 0.00 | 135.5 | 0.00 | 127.3 | 0.00 | ||
| noJT | 4-Td | −18.6 | 0.25 | −17.4 | 0.24 | −14.8 | 0.27 | −13.9 | 0.27 | ||
| noJT | 5-SPY | −185.3 | 0.17 | −177.8 | 0.15 | −147.7 | 0.17 | −145.9 | 0.15 | ||
| noJT | 5-TBP | 83.7 | 0.02 | 76.4 | 0.02 | 69.4 | 0.01 | 63.4 | 0.01 | ||
| JT | 3-TP | 86.6 | 0.00 | 78.4 | 0.00 | 73.3 | 0.00 | 58.2 | 0.00 | ||
| JT | 4-SP | 134.5 | 0.00 | 124.7 | 0.00 | 109.3 | 0.00 | 77.1 | 0.00 | ||
| JT | 4-Td | −15.7 | 0.24 | −14.4 | 0.23 | −13.1 | 0.25 | −12.3 | 0.23 | ||
| JT | 5-SPY | −82.2 | 0.26 | −78.9 | 0.23 | −62.0 | 0.28 | −64.4 | 0.24 | ||
| JT | 5-TBP | 50.7 | 0.02 | 43.5 | 0.02 | 42.3 | 0.02 | 36.2 | 0.02 | ||
| CASSCF | NEVPT2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DAll | DHigh | DLow | DHS | DAll | DHigh | DLow | DHS | |||
| Fe(II) | noJT | 5-SPY | −22.1 | −20.3 | −1.8 | −21.0 | −19.9 | −18.0 | −1.9 | −19.0 |
| noJT | 5-TBP | −11.1 | −11.1 | 0.0 | −11.5 | −8.8 | −8.6 | −0.2 | −9.4 | |
| JT | 3-TP | −49.0 | −46.9 | −2.1 | −48.1 | −44.6 | −42.4 | −2.2 | −32.5 | |
| JT | 4-SP | −54.6 | −52.1 | −2.6 | −53.1 | −54.2 | −51.4 | −2.8 | −36.5 | |
| JT | 4-Td | −8.1 | −6.2 | −1.9 | −6.2 | −7.7 | −5.8 | −1.9 | −3.8 | |
| JT | 5-SPY | −7.5 | −5.8 | −1.8 | −6.2 | 7.0 | 5.3 | 1.7 | −5.6 | |
| JT | 5-TBP | 7.8 | 6.3 | 1.5 | −7.3 | 6.7 | 5.1 | 1.6 | −7.1 | |
| JT | 6-Oh | 22.1 | 21.2 | 1.0 | 21.6 | 18.1 | 17.1 | 1.0 | 21.8 | |
| Co(II) | noJT | 3-TP | 133.8 | 125.2 | 8.6 | 125.9 | 108.3 | 99.5 | 8.8 | 102.9 |
| noJT | 4-SP | 160.2 | 149.9 | 10.3 | 151.4 | 135.5 | 123.8 | 11.7 | 127.3 | |
| noJT | 4-Td | −18.6 | −17.5 | −1.2 | −17.4 | −14.8 | −13.6 | −1.2 | −13.9 | |
| noJT | 5-SPY | −185.3 | −180.0 | −5.4 | −177.8 | −147.7 | −142.1 | −5.7 | −145.9 | |
| noJT | 5-TBP | 83.7 | 76.1 | 7.6 | 76.4 | 69.4 | 61.5 | 7.9 | 63.4 | |
| JT | 3-TP | 86.6 | 77.8 | 8.8 | 78.4 | 73.3 | 64.3 | 9.0 | 58.2 | |
| JT | 4-SP | 134.5 | 123.1 | 11.4 | 124.7 | 109.3 | 95.6 | 13.7 | 77.1 | |
| JT | 4-Td | −15.7 | −14.4 | −1.3 | −14.4 | −13.1 | −11.7 | −1.3 | −12.3 | |
| JT | 5-SPY | −82.2 | −79.2 | −3.1 | −78.9 | −62.0 | −58.6 | −3.4 | −64.4 | |
| JT | 5-TBP | 50.7 | 43.2 | 7.5 | 43.5 | 42.3 | 34.5 | 7.8 | 36.2 | |
| All | HS | All | HS | |||
|---|---|---|---|---|---|---|
| Fe(II) | 2-L (noJT) | 0.0 | 0.0 | 4-Td (noJT) | 0.0 | 0.0 |
| 0.0 | 0.0 | 1.2 | 0.7 | |||
| 193.8 | 190.9 | 14.8 | 10.2 | |||
| 193.8 | 190.9 | 15.7 | 14.0 | |||
| 388.0 | 385.1 | 20.5 | 16.0 | |||
| 3-TP (noJT) | 0.0 | 0.0 | 6-Oh (noJT) | 0.0 | 0.0 | |
| 19.0 | 18.7 | 5.9 | 6.3 | |||
| 21.0 | 21.2 | 39.8 | 37.7 | |||
| 84.8 | 84.7 | 101.9 | 101.2 | |||
| 84.8 | 84.8 | 110.6 | 108.9 | |||
| 4-SP (noJT) | 0.0 | 0.0 | 2-L (JT) | 0.0 | 0.0 | |
| 0.6 | 0.6 | 0.0 | 0.0 | |||
| 104.5 | 102.3 | 195.8 | 192.9 | |||
| 113.7 | 111.5 | 196.0 | 193.0 | |||
| 195.5 | 194.1 | 381.5 | 379.3 | |||
| Co(II) | 2-L (noJT) | 0.0 | 0.0 | 2-L (JT) | 0.0 | 0.0 |
| 479.6 | 470.9 | 472.0 | 464.2 | |||
| 960.9 | 954.7 | 941.7 | 937.5 | |||
| 1444.5 | 1451.2 | 1423.3 | 1433.3 | |||
| 6-Oh (noJT) | 0.0 | 0.0 | 6-Oh (JT) | 0.0 | 0.0 | |
| 350.3 | 350.1 | 232.9 | 234.4 | |||
| 353.0 | 352.8 | 512.5 | 512.2 | |||
| 933.8 | 947.7 | 960.8 | 969.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llanos, L.; Aravena, D. Effect of Low Spin Excited States for Magnetic Anisotropy of Transition Metal Mononuclear Single Molecule Magnets. Inorganics 2018, 6, 24. https://doi.org/10.3390/inorganics6010024
Llanos L, Aravena D. Effect of Low Spin Excited States for Magnetic Anisotropy of Transition Metal Mononuclear Single Molecule Magnets. Inorganics. 2018; 6(1):24. https://doi.org/10.3390/inorganics6010024
Chicago/Turabian StyleLlanos, Leonel, and Daniel Aravena. 2018. "Effect of Low Spin Excited States for Magnetic Anisotropy of Transition Metal Mononuclear Single Molecule Magnets" Inorganics 6, no. 1: 24. https://doi.org/10.3390/inorganics6010024
APA StyleLlanos, L., & Aravena, D. (2018). Effect of Low Spin Excited States for Magnetic Anisotropy of Transition Metal Mononuclear Single Molecule Magnets. Inorganics, 6(1), 24. https://doi.org/10.3390/inorganics6010024