
Symmetric Assembly of a Sterically Encumbered Allyl Complex: Mechanochemical and Solution Synthesis of the Tris(allyl)beryllate, K[BeA′3] (A′ = 1,3-(SiMe3)2C3H3)


Abstract
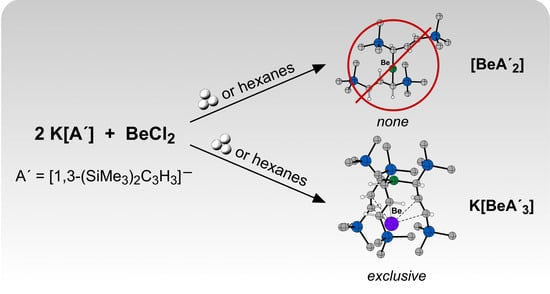
:
1. Introduction
2. Results and Discussion
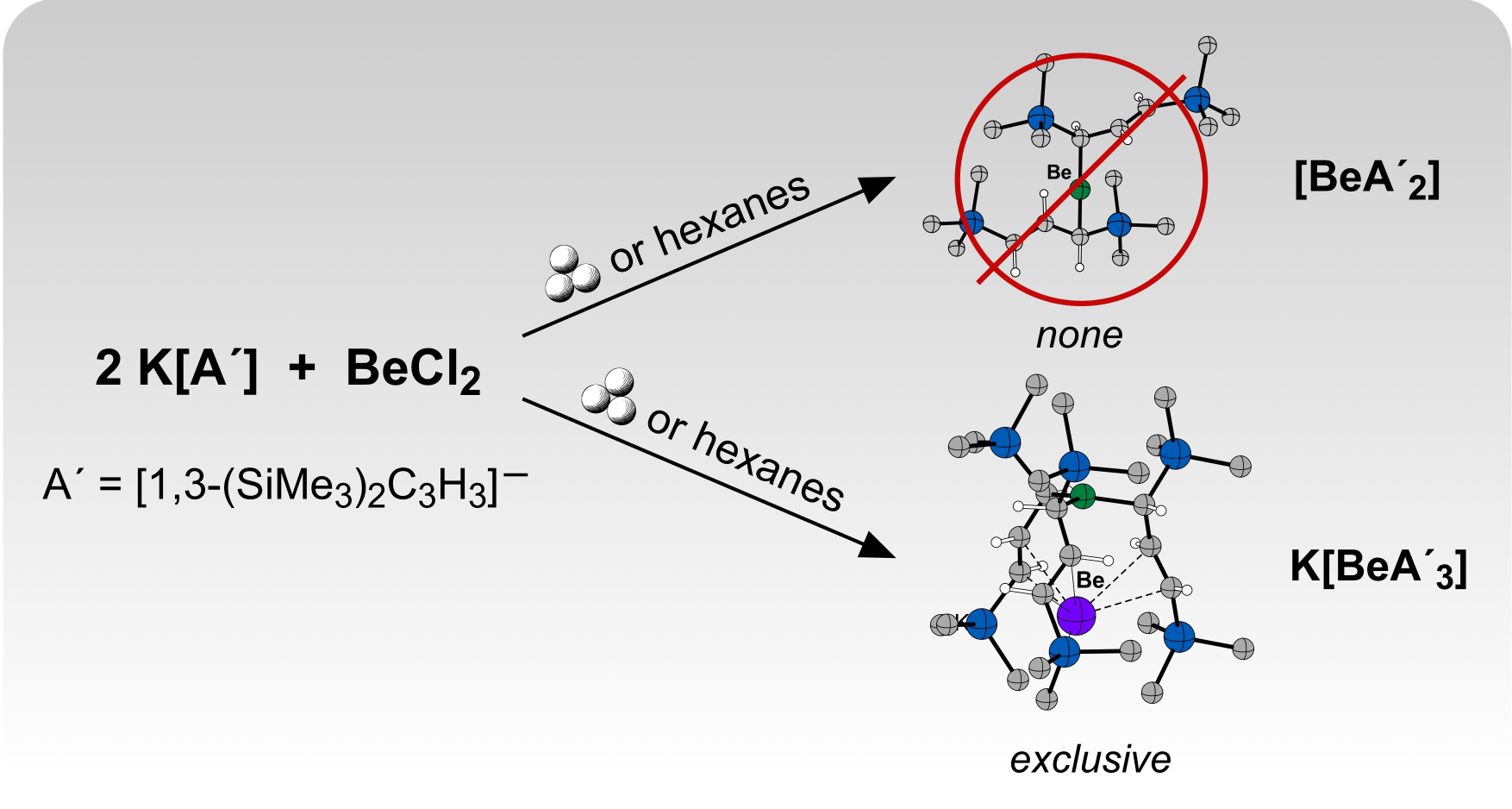
2.1. Solid-State Synthesis
2.2. Synthesis in Solution
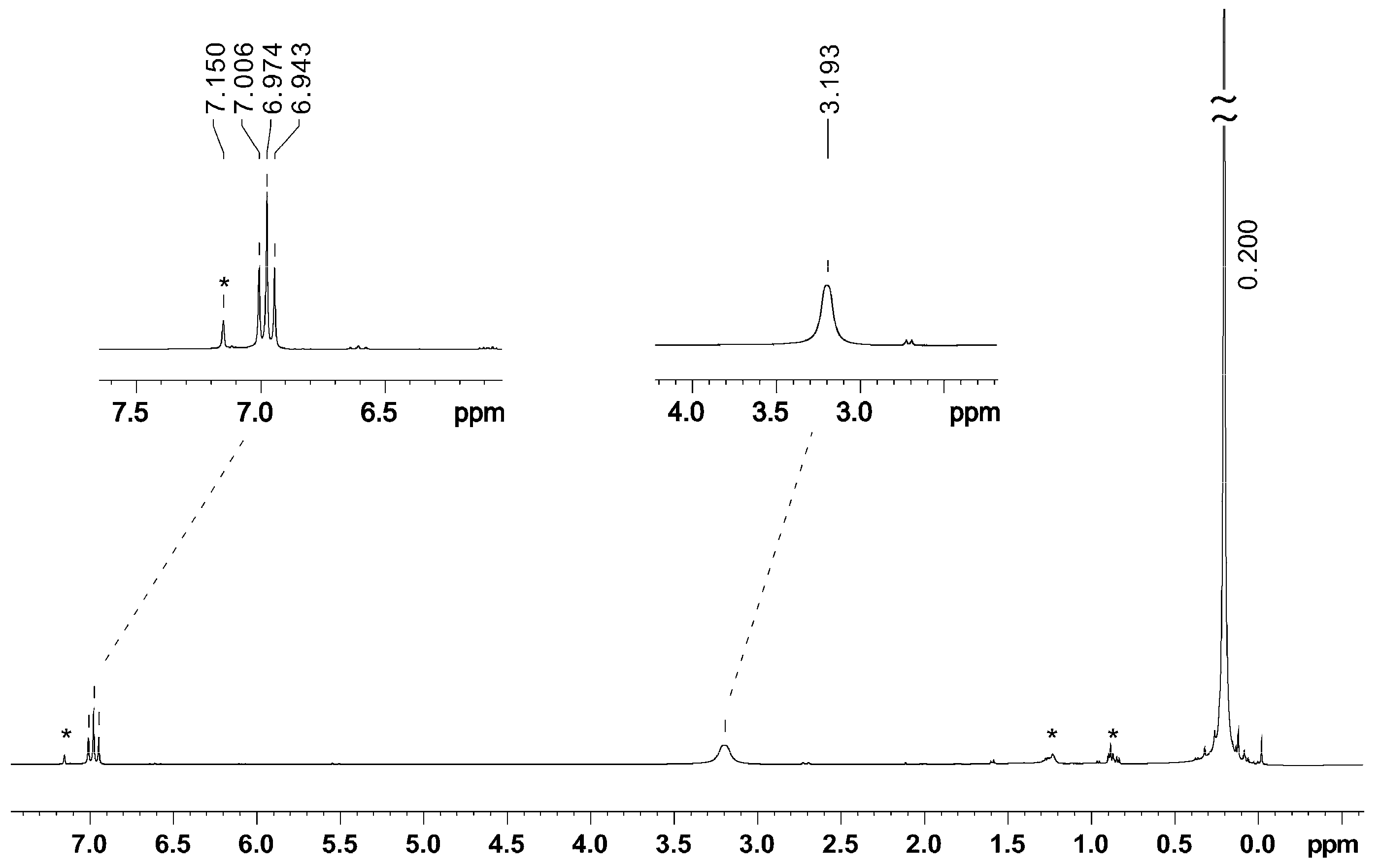
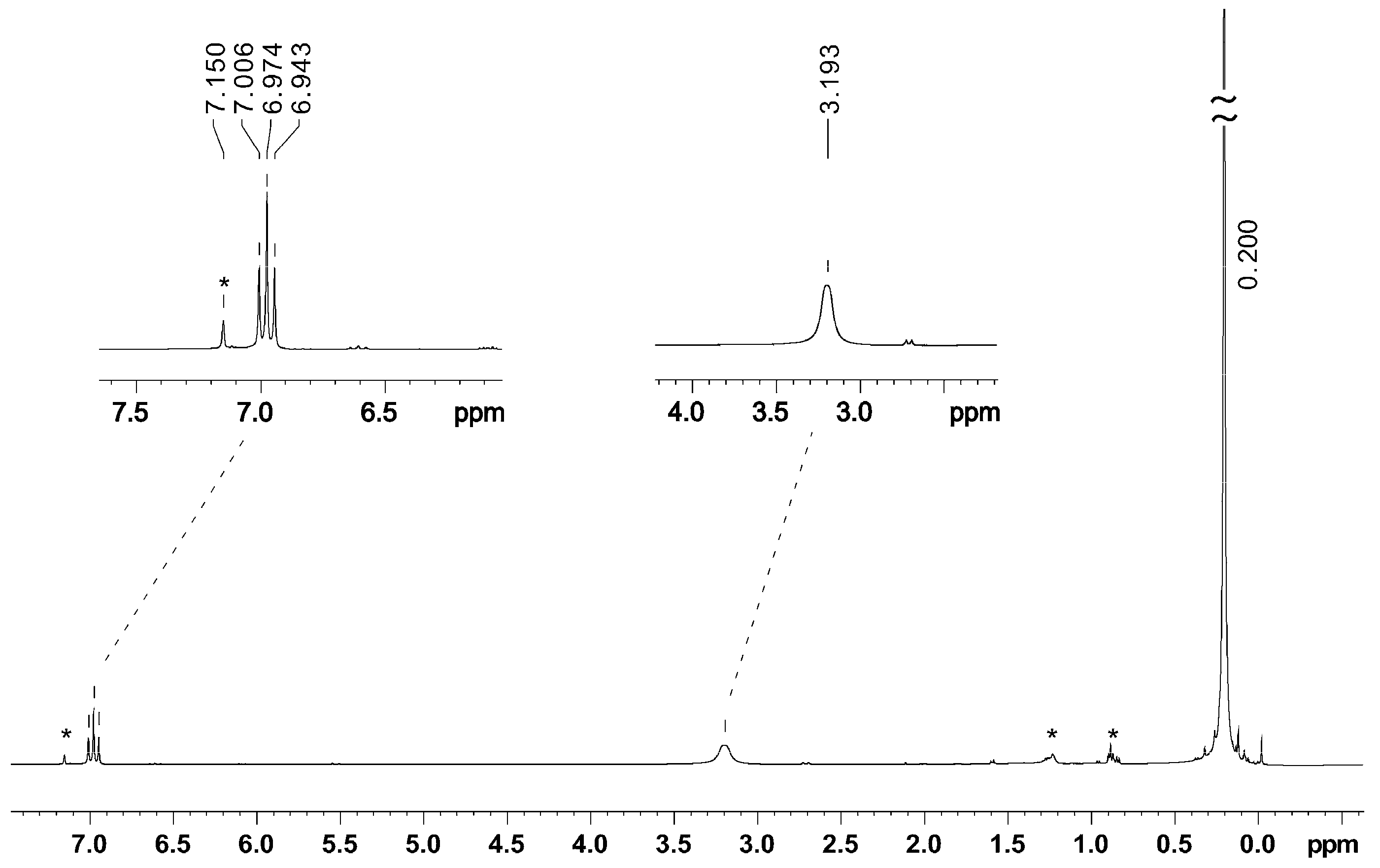
2.3. NMR Spectroscopy
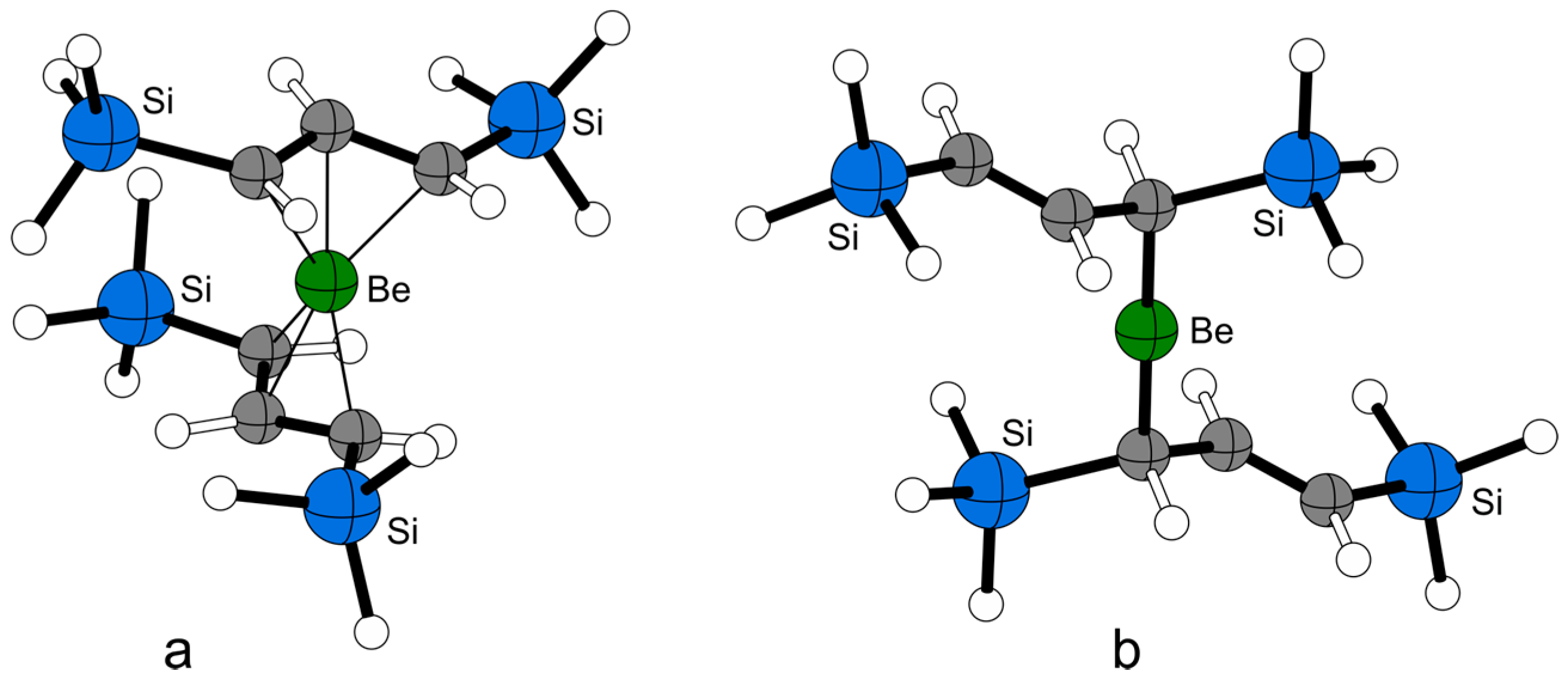
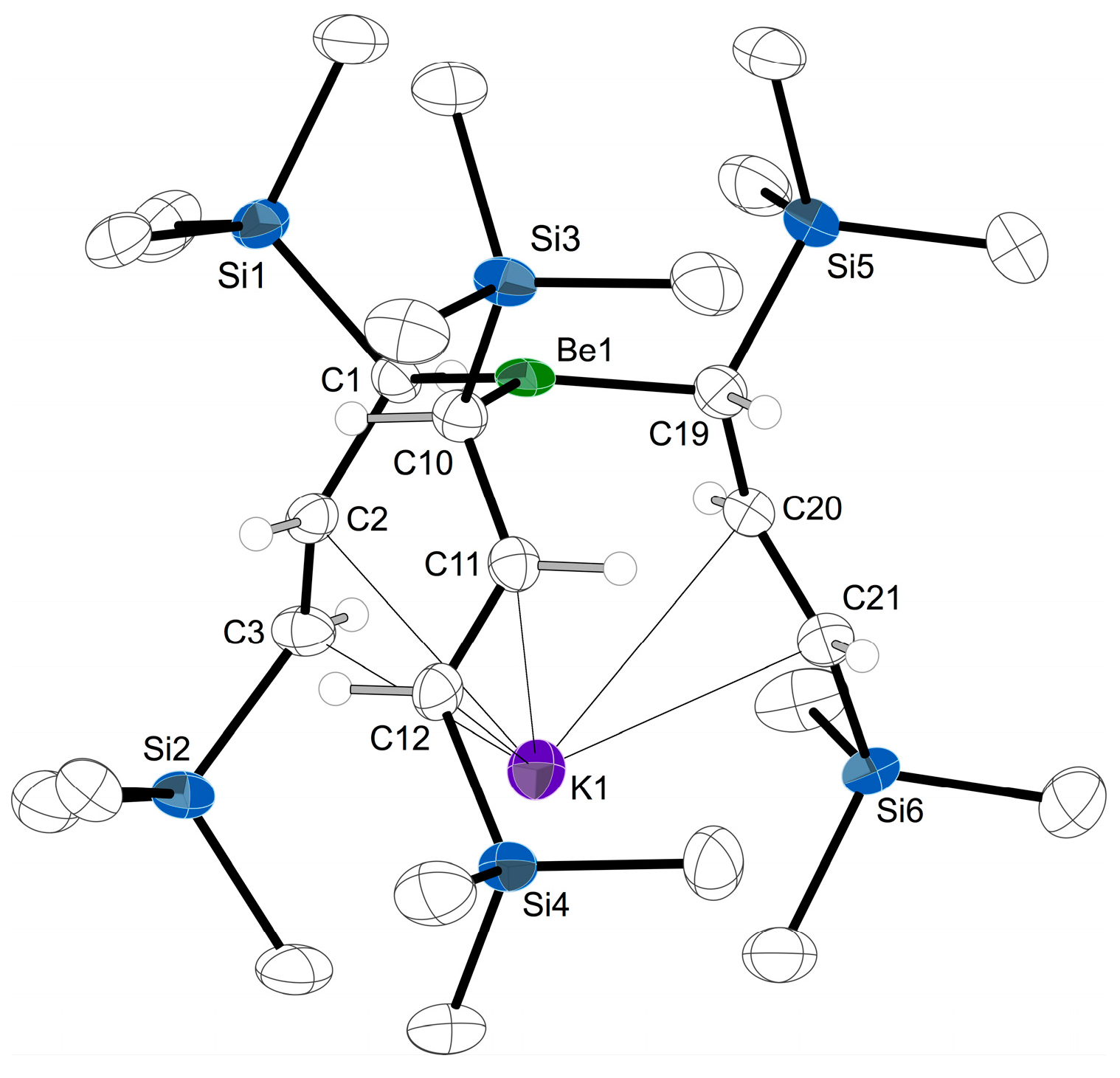
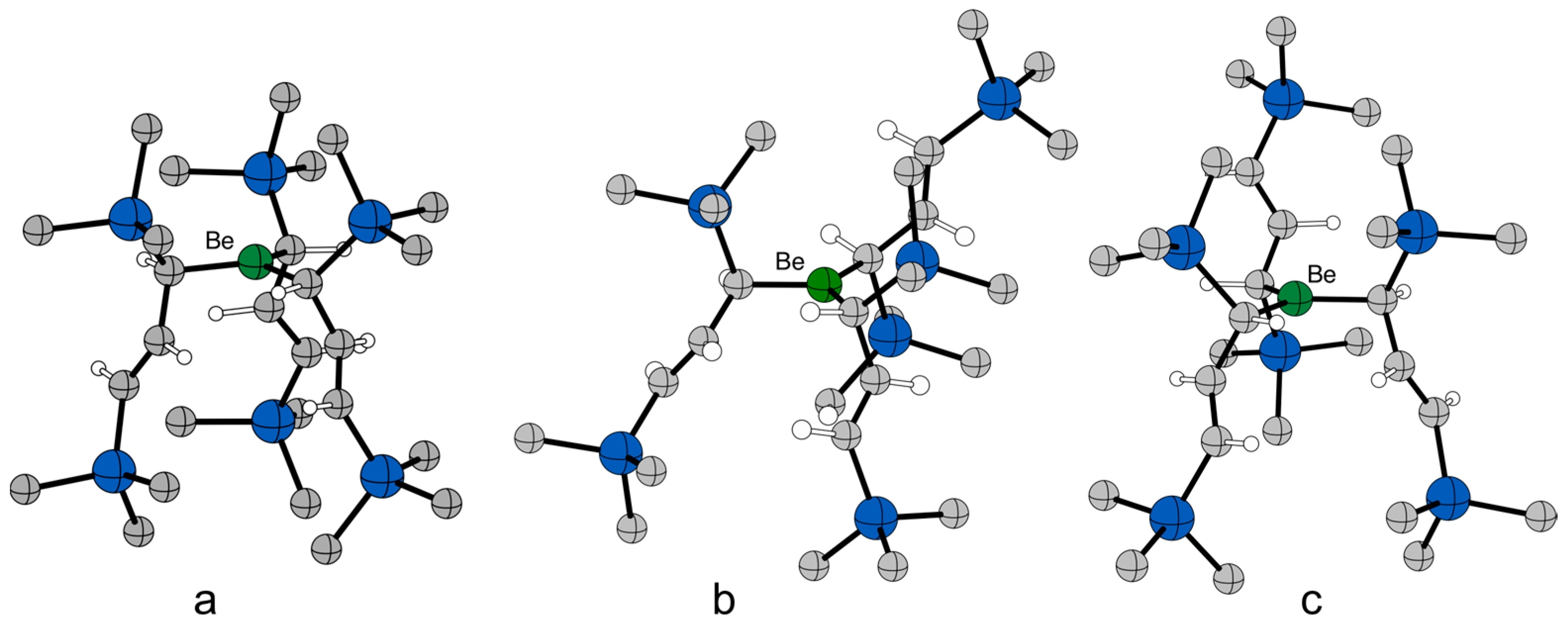
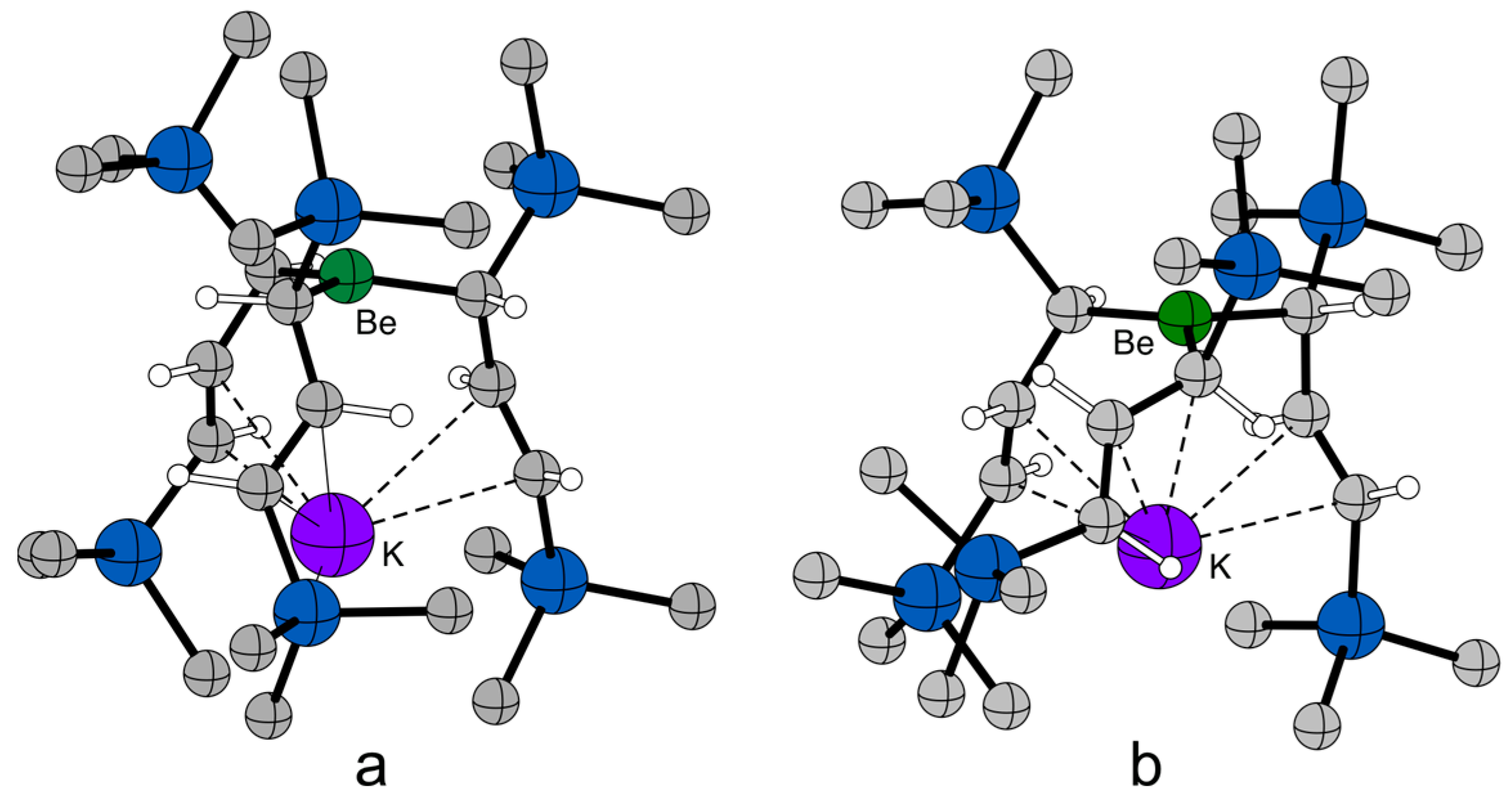
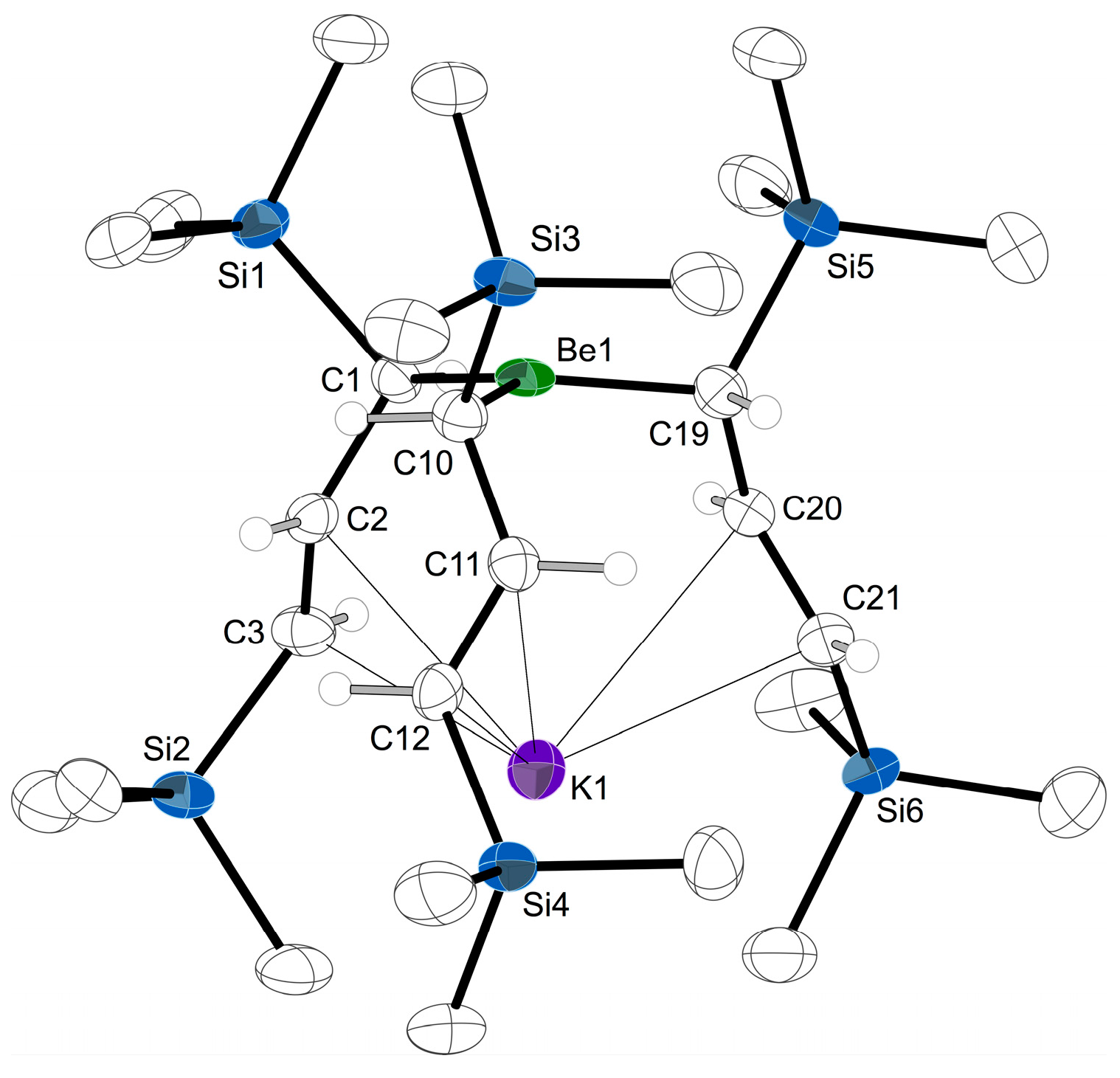
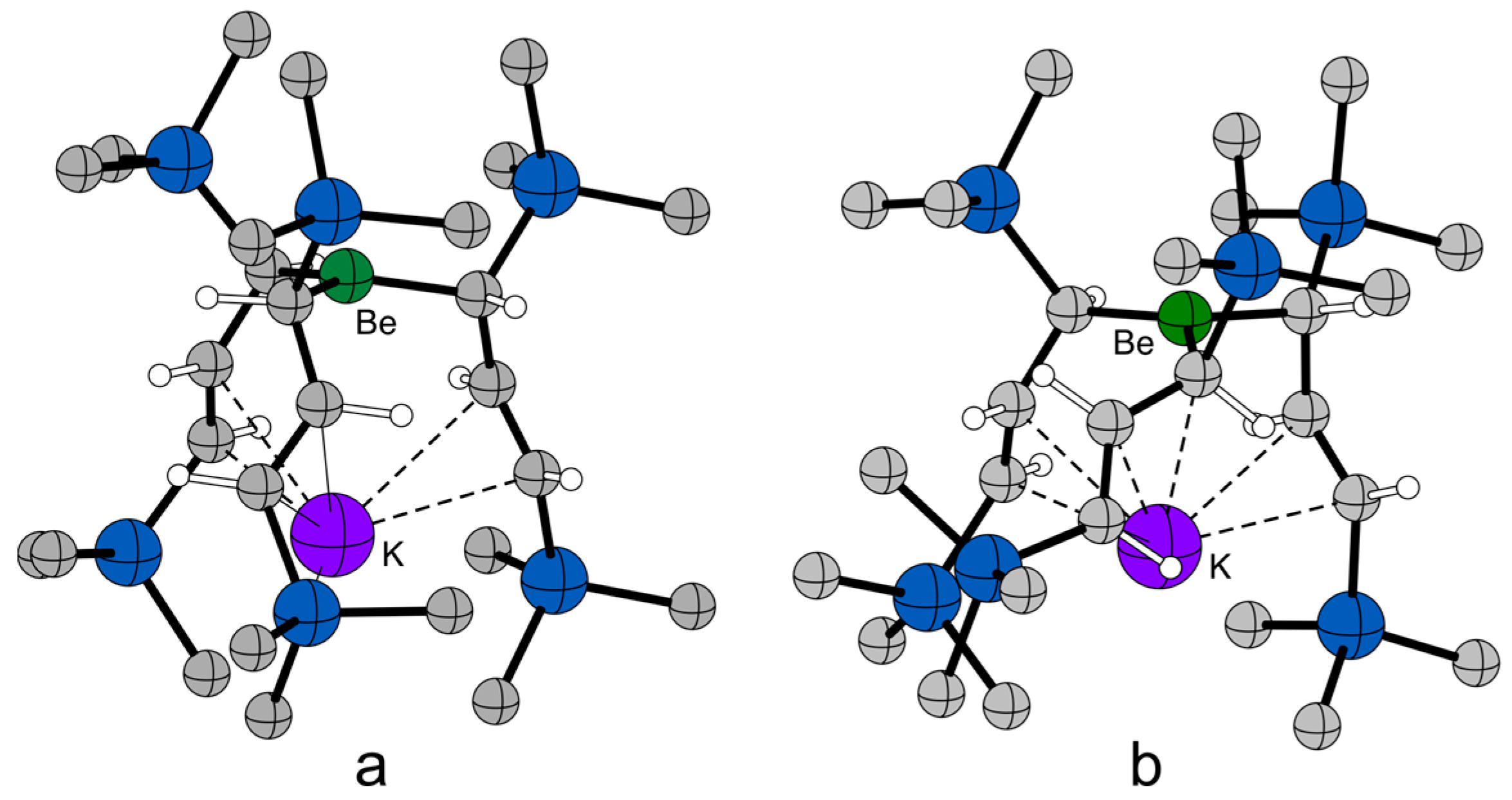
2.4. Solid State Structure
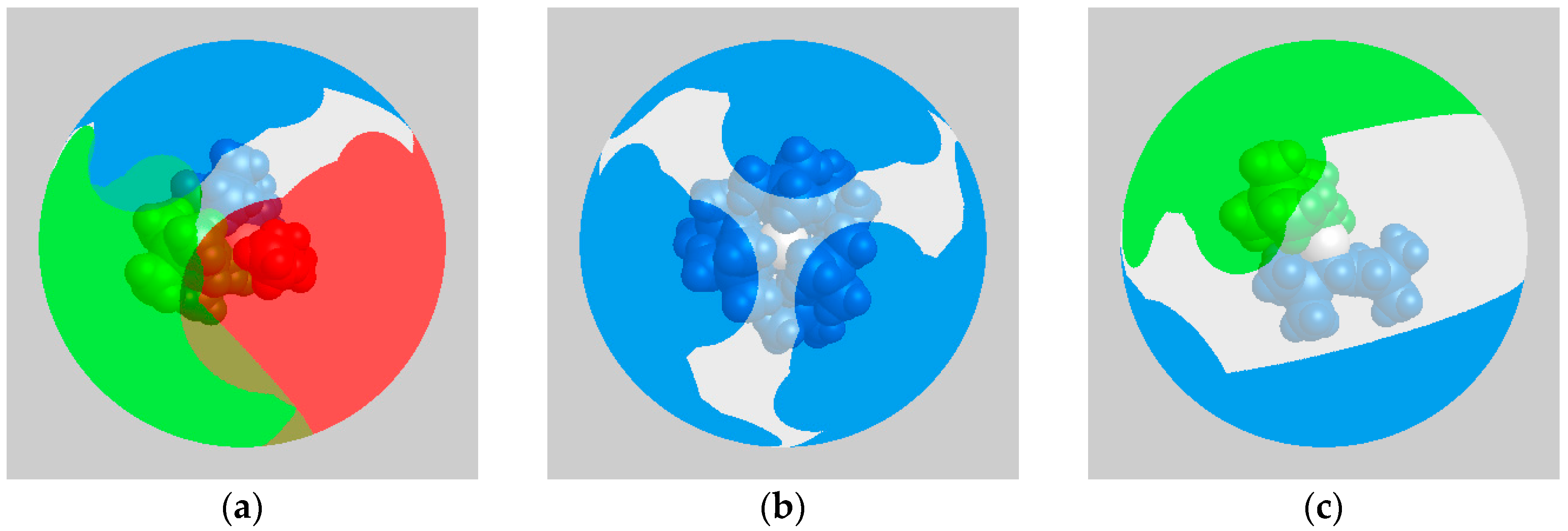
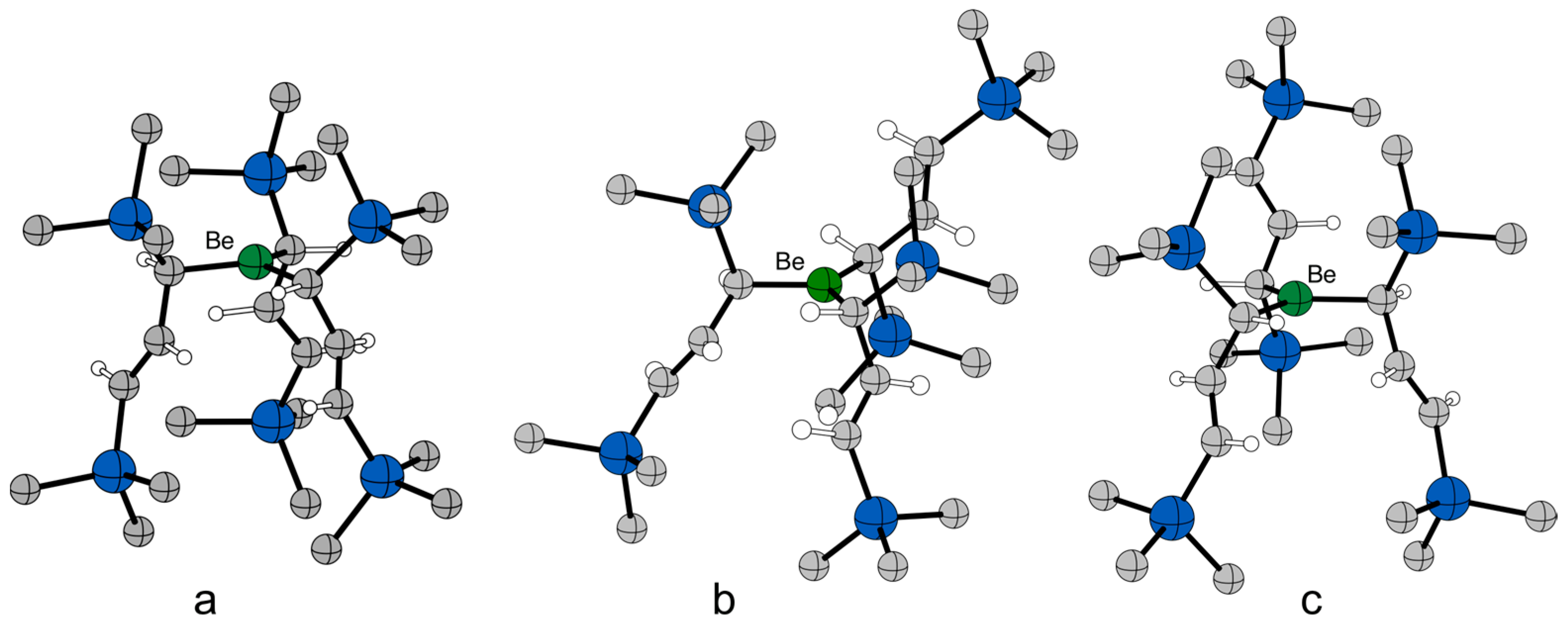
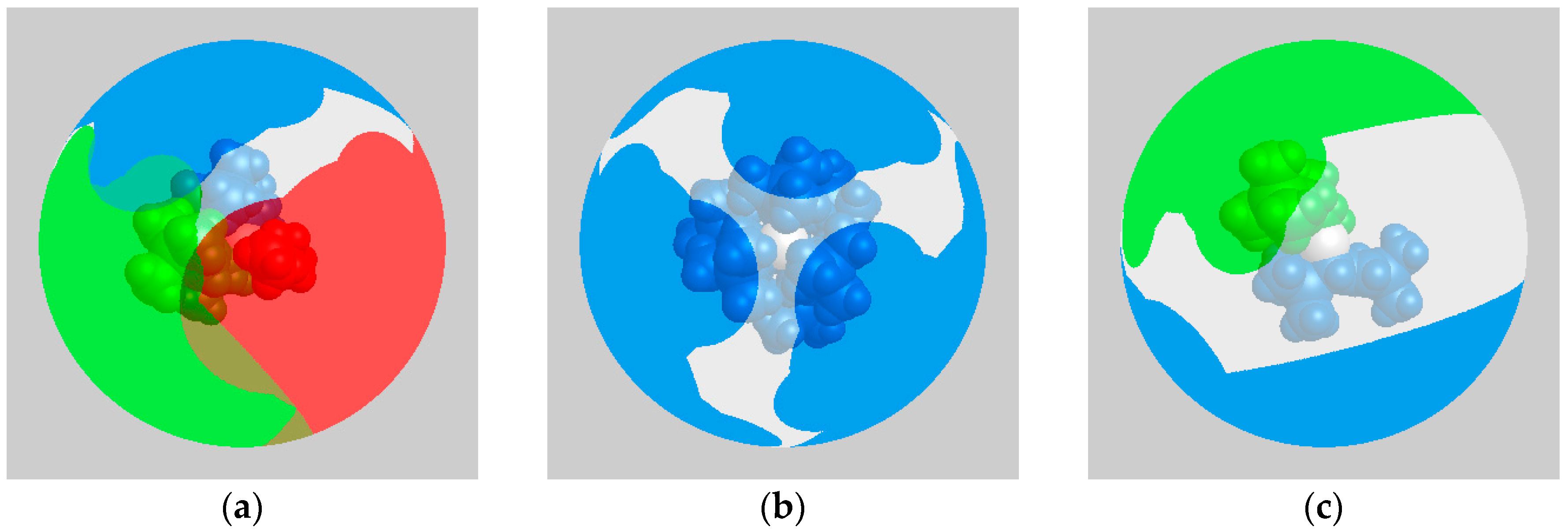
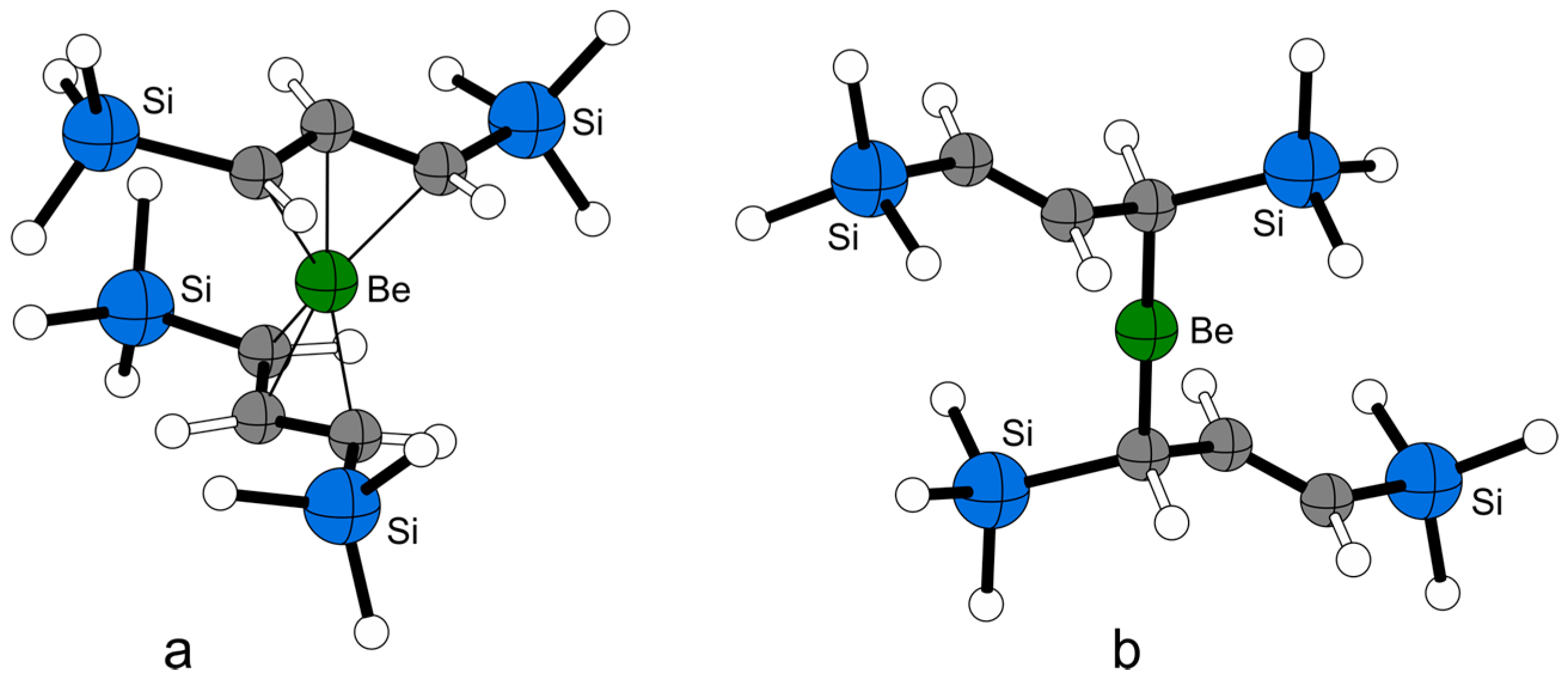
2.5. Computational Investigations
3. Materials and Methods
3.1. Mechanochemical Synthesis of K[BeA’3] (1)
3.2. General Procedures for Synthesis of K[BeA’3] (1) with Solvents
3.3. Procedures for X-ray Crystallography
3.4. General Procedures for Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arrowsmith, M.; Hill, M.S.; Kociok-Köhn, G. Activation of N-Heterocyclic Carbenes by {BeH2} and {Be(H)(Me)} Fragments. Organometallics 2015, 34, 653–662. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Westerhausen, M. Synthesis and Spectroscopic Properties of Bis(trimethylsilyl)amides of the Alkaline-earth Metals Magnesium, Calcium, Strontium, and Barium. Inorg. Chem. 1991, 30, 96–101. [Google Scholar] [CrossRef]
- Naglav, D.; Neumann, A.; Blaser, D.; Wolper, C.; Haack, R.; Jansen, G.; Schulz, S. Bonding situation in Be[N(SiMe3)2]2—An experimental and computational study. Chem. Commun. 2015, 51, 3889–3891. [Google Scholar] [CrossRef] [PubMed]
- Nugent, K.W.; Beattie, J.K.; Hambley, T.W.; Snow, M.R. A precise Low-temperature crystal structure of bis(cyclopentadienyl)beryllium. Aust. J. Chem. 1984, 37, 1601–1606. [Google Scholar] [CrossRef]
- Burkey, D.J.; Hanusa, T.P. Structural Lessons from Main-Group Metallocenes. Comments Inorg. Chem. 1995, 17, 41–77. [Google Scholar] [CrossRef]
- Civic, T.M. Beryllium Metal Toxicology: A Current Perspective. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Hanusa, T.P.; Bierschenk, E.J.; Engerer, L.K.; Martin, K.A.; Rightmire, N.R. 1.37—Alkaline Earth Chemistry: Synthesis and Structures. In Comprehensive Inorganic Chemistry II, 2nd ed.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1133–1187. [Google Scholar]
- Iversen, K.J.; Couchman, S.A.; Wilson, D.J.D.; Dutton, J.L. Modern organometallic and coordination chemistry of beryllium. Coord. Chem. Rev. 2015, 297–298, 40–48. [Google Scholar] [CrossRef]
- Naglav, D.; Buchner, M.R.; Bendt, G.; Kraus, F.; Schulz, S. Off the Beaten Track—A Hitchhiker's Guide to Beryllium Chemistry. Angew. Chem. Int. Ed. 2016, 55, 10562–10576. [Google Scholar] [CrossRef] [PubMed]
- Chmely, S.C.; Hanusa, T.P.; Brennessel, W.W. Bis(1,3-trimethylsilylallyl)beryllium. Angew. Chem. Int. Ed. 2010, 49, 5870–5874. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.A.; Muryn, C.A.; Layfield, R.A. s-Block metal complexes of a bulky, donor-functionalized allyl ligand. Chem. Commun. 2008, 3142–3144. [Google Scholar] [CrossRef] [PubMed]
- Chmely, S.C.; Carlson, C.N.; Hanusa, T.P.; Rheingold, A.L. Classical versus Bridged Allyl Ligands in Magnesium Complexes: The Role of Solvent. J. Am. Chem. Soc. 2009, 131, 6344–6345. [Google Scholar] [CrossRef] [PubMed]
- Rightmire, N.R.; Hanusa, T.P. Advances in Organometallic Synthesis with Mechanochemical Methods. Dalton Trans. 2016, 45, 2352–2362. [Google Scholar] [CrossRef] [PubMed]
- Gren, C.K.; Hanusa, T.P.; Rheingold, A.L. Threefold Cation–π Bonding in Trimethylsilylated Allyl Complexes. Organometallics 2007, 26, 1643–1649. [Google Scholar] [CrossRef]
- Layfield, R.A.; Garcia, F.; Hannauer, J.; Humphrey, S.M. Ansa-tris(allyl) complexes of alkali metals: Tripodal analogues of cyclopentadienyl and ansa-metallocene ligands. Chem. Commun. 2007, 5081–5083. [Google Scholar] [CrossRef] [PubMed]
- Neumüller, B.; Weller, F.; Dehnicke, K. Synthese, Schwingungsspektren und Kristallstrukturen der Chloroberyllate (Ph4P)2[BeCl4] und (Ph4P)2[Be2Cl6]. Z. Anorg. Allg. Chem. 2003, 629, 2195–2199. [Google Scholar] [CrossRef]
- Allred, A.L. Electronegativity values from thermochemical data. J. Inorg. Nucl. Chem. 1961, 17, 215–221. [Google Scholar] [CrossRef]
- Plieger, P.G.; John, K.D.; Keizer, T.S.; McCleskey, T.M.; Burrell, A.K.; Martin, R.L. Predicting 9Be Nuclear Magnetic Resonance Chemical Shielding Tensors Utilizing Density Functional Theory. J. Am. Chem. Soc. 2004, 126, 14651–14658. [Google Scholar] [CrossRef] [PubMed]
- Arnold, T.; Braunschweig, H.; Ewing, W.C.; Kramer, T.; Mies, J.; Schuster, J.K. Beryllium bis(diazaborolyl): Old neighbors finally shake hands. Chem. Commun. 2015, 51, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Wermer, J.R.; Gaines, D.F.; Harris, H.A. Synthesis and molecular structure of lithium tri-tert-butylberyllate, Li[Be(tert-C4H9)3]. Organometallics 1988, 7, 2421–2422. [Google Scholar] [CrossRef]
- Gottfriedsen, J.; Blaurock, S. The First Carbene Complex of a Diorganoberyllium: Synthesis and Structural Characterization of Ph2Be(i-Pr-carbene) and Ph2Be(n-Bu2O). Organometallics 2006, 25, 3784–3786. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hill, M.S.; Kociok-Köhn, G.; MacDougall, D.J.; Mahon, M.F. Beryllium-Induced C–N Bond Activation and Ring Opening of an N-Heterocyclic Carbene. Angew. Chem. Inter. Ed. 2012, 51, 2098–2100. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.; Wolfrum, R. Über metall–alkyl-verbindungen VIII. Die kristallstruktur des lithium-tetramethylberyllats. J. Organomet. Chem. 1968, 12, 257–262. [Google Scholar] [CrossRef]
- Rightmire, N.R.; Bruns, D.L.; Hanusa, T.P.; Brennessel, W.W. Mechanochemical Influence on the Stereoselectivity of Halide Metathesis: Synthesis of Group 15 Tris(allyl) Complexes. Organometallics 2016, 35, 1698–1706. [Google Scholar] [CrossRef]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssell, K. A Density Functional with Spherical Atom Dispersion Terms. J. Chem. Theory Comp. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Rightmire, N.R.; Hanusa, T.P.; Rheingold, A.L. Mechanochemical Synthesis of [1,3-(SiMe3)2C3H3]3(Al,Sc), a Base-Free Tris(allyl)aluminum Complex and Its Scandium Analogue. Organometallics 2014, 33, 5952–5955. [Google Scholar] [CrossRef]
- Guzei, I.A.; Wendt, M. An improved method for the computation of ligand steric effects based on solid angles. Dalton Trans. 2006, 3991–3999. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, G.; Chow, A.; Winchester, W.R. Dynamics of solvated lithium(+) within exo,exo-[1,3-bis(trimethylsilyl)allyl]lithium N,N,N′,N′-tetramethylethylenediamine complex. J. Am. Chem. Soc. 1990, 112, 1382–1386. [Google Scholar] [CrossRef]
- Harvey, M.J.; Hanusa, T.P.; Young, V.G., Jr. Synthesis and Crystal Structure of a Bis(allyl) Complex of Calcium, [Ca{C3(SiMe3)2H3}2·(thf)2]. Angew. Chem. Int. Ed. 1999, 38, 217–219. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Pergamon: Oxford, UK, 1988. [Google Scholar]
- APEX2, Version 2014.11–0; Bruker AXS: Madison, WI, USA, 2013.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G.; Spagna, R. SIR2011: A New Package for Crystal Structure Determination and Refinement, Version 1.0; Istituto di Cristallografia: Bari, Italy, 2012.
- Sheldrick, G.M. SHELXL-2014/7, University of Göttingen: Göttingen, Germany, 2014.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09W, Revision D.01; Gaussian, Inc.: Wallingford CT, USA, 2009.
- Glock, C.; Görls, H.; Westerhausen, M. N,N,N′,N′-Tetramethylethylendiamine adducts of amido calcium bases—Synthesis of monomeric [(tmeda)Ca{N(SiMe3)2}2], [(tmeda)Ca{NiPr2}2], and dimeric Hauser base-type [(tmeda)Ca(tmp)(μ-I)]2 (tmp = 2,2,6,6-tetramethylpiperidide). Inorg. Chim. Acta 2011, 374, 429–434. [Google Scholar] [CrossRef]
- Fitts, L.S.; Bierschenk, E.J.; Hanusa, T.P.; Rheingold, A.L.; Pink, M.; Young, V.G. Selective modification of the metal coordination environment in heavy alkaline-earth iodide complexes. New J. Chem. 2016, 40, 8229–8238. [Google Scholar] [CrossRef]
- Hernández, J.G.; Friščić, T. Metal-catalyzed organic reactions using mechanochemistry. Tetrahedron Lett. 2015, 56, 4253–4265. [Google Scholar] [CrossRef]
- Boldyreva, E. Mechanochemistry of inorganic and organic systems: What is similar, what is different? Chem. Soc. Rev. 2013, 42, 7719–7738. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef] [PubMed]
- Waddell, D.C.; Thiel, I.; Clark, T.D.; Marcum, S.T.; Mack, J. Making kinetic and thermodynamic enolates via solvent-free high speed ball milling. Green Chem. 2010, 12, 209–211. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | K[A’]:BeCl2 | Medium a | Time | Organoberyllium Product(s) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | 1:1 |  | 15 min | K[BeA’3] | 97 |
| 2 | 2:1 | | 15 min | K[BeA’3] | 21 |
| 3 | 3:1 | | 15 min | K[BeA’3] | 25 |
| 4 | 1:1 | Et2O | 1 h | 2A’BeCl ⇄ BeA’2 + BeCl | n/a c,d |
| 5 | 2:1 | Et2O | 2 h | BeA’2∙OEt2 | 77 c |
| 6 | 2:1 | Et2O | 16 h | K[BeA’3] | 98 |
| 7 | 1:1 | hexanes | 1 h | K[BeA’3] | 24 |
| Complex | δ H(α)/H(γ) | δ H(β) | δ SiMe3 | M–C (σ) Å | M’···C(olefin) Å | Ref. |
|---|---|---|---|---|---|---|
| Li[ZnA’3] | 6.46 | 3.50 | 0.15 | 2.117(3) b | 2.745(4), 2.268(3) b | [15] |
| Na[ZnA’3] | 7.59 | 4.00 | 0.16 | 2.103(3) | 2.857(3), 2.567(3) | [15] |
| K[BeA’3] | 6.97 | 3.19 | 0.20 | 1.805(10) | 3.153(7), 2.940(7) | this work |
| K[ZnA’3] | 7.05 | 3.42 | 0.23 | 2.068(4) | 3.205(3), 2.945(3) | [15] |
| K(thf)[SnA’3] | 6.43 | 4.42 | 0.42, 0.23 a | 2.344(7) | 3.201(7), 3.164(8), 3.065(8) | [16] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyde, N.C.; Rightmire, N.R.; Hanusa, T.P.; Brennessel, W.W. Symmetric Assembly of a Sterically Encumbered Allyl Complex: Mechanochemical and Solution Synthesis of the Tris(allyl)beryllate, K[BeA′3] (A′ = 1,3-(SiMe3)2C3H3). Inorganics 2017, 5, 36. https://doi.org/10.3390/inorganics5020036
Boyde NC, Rightmire NR, Hanusa TP, Brennessel WW. Symmetric Assembly of a Sterically Encumbered Allyl Complex: Mechanochemical and Solution Synthesis of the Tris(allyl)beryllate, K[BeA′3] (A′ = 1,3-(SiMe3)2C3H3). Inorganics. 2017; 5(2):36. https://doi.org/10.3390/inorganics5020036
Chicago/Turabian StyleBoyde, Nicholas C., Nicholas R. Rightmire, Timothy P. Hanusa, and William W. Brennessel. 2017. "Symmetric Assembly of a Sterically Encumbered Allyl Complex: Mechanochemical and Solution Synthesis of the Tris(allyl)beryllate, K[BeA′3] (A′ = 1,3-(SiMe3)2C3H3)" Inorganics 5, no. 2: 36. https://doi.org/10.3390/inorganics5020036
APA StyleBoyde, N. C., Rightmire, N. R., Hanusa, T. P., & Brennessel, W. W. (2017). Symmetric Assembly of a Sterically Encumbered Allyl Complex: Mechanochemical and Solution Synthesis of the Tris(allyl)beryllate, K[BeA′3] (A′ = 1,3-(SiMe3)2C3H3). Inorganics, 5(2), 36. https://doi.org/10.3390/inorganics5020036