
Reduction of Bromo- and Iodo-2,6-bis(diphenylphosphanylmethyl)benzene with Magnesium and Calcium



Abstract
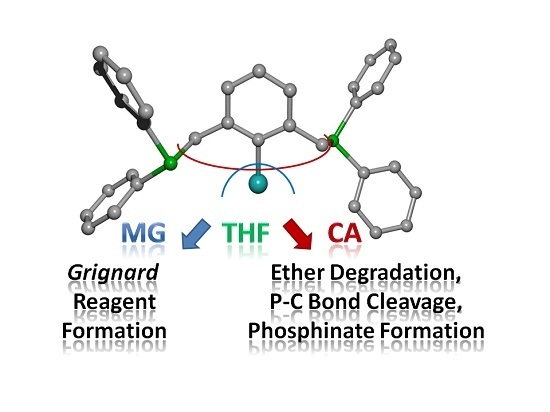
:
1. Introduction
2. Results
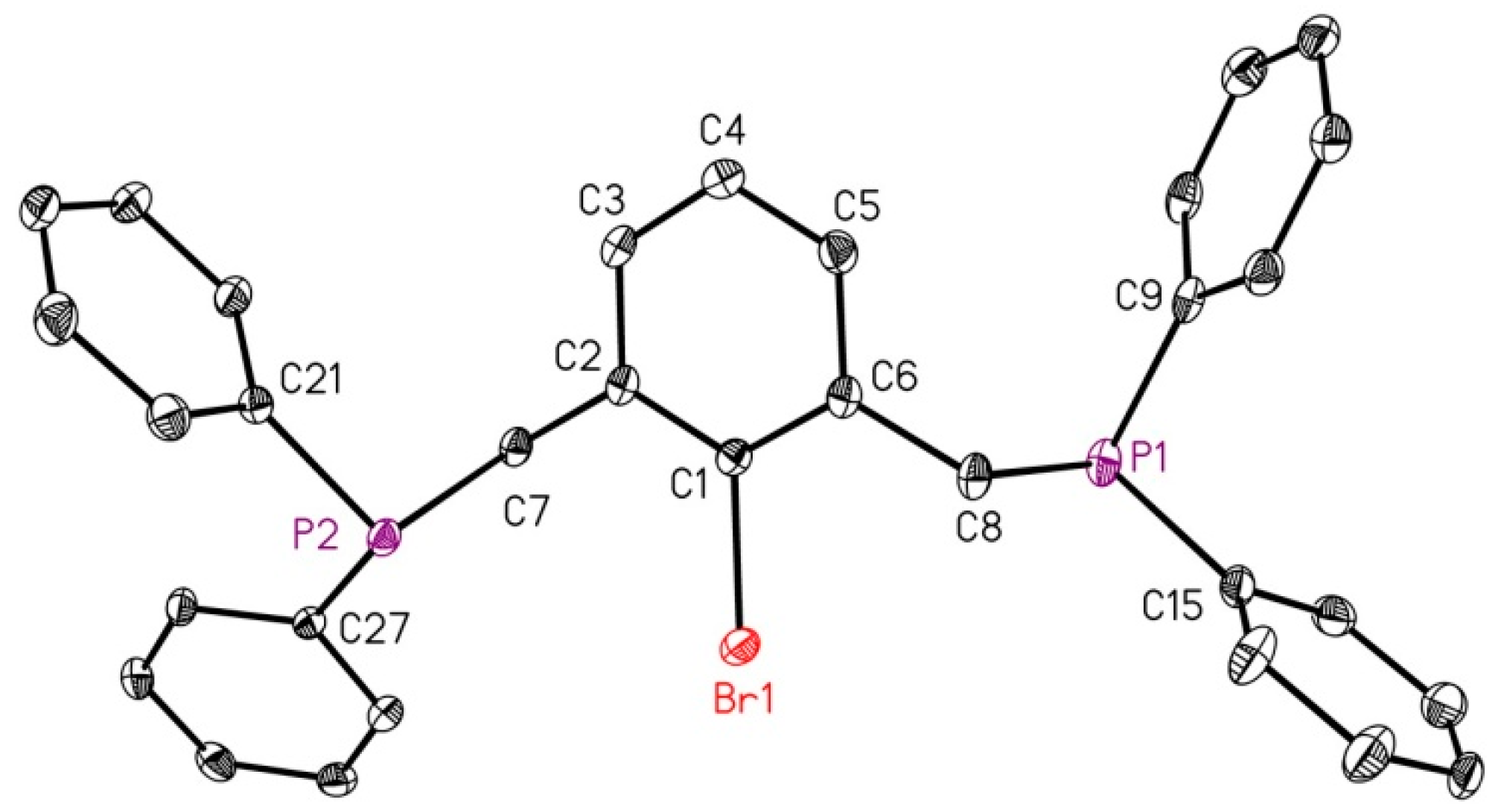
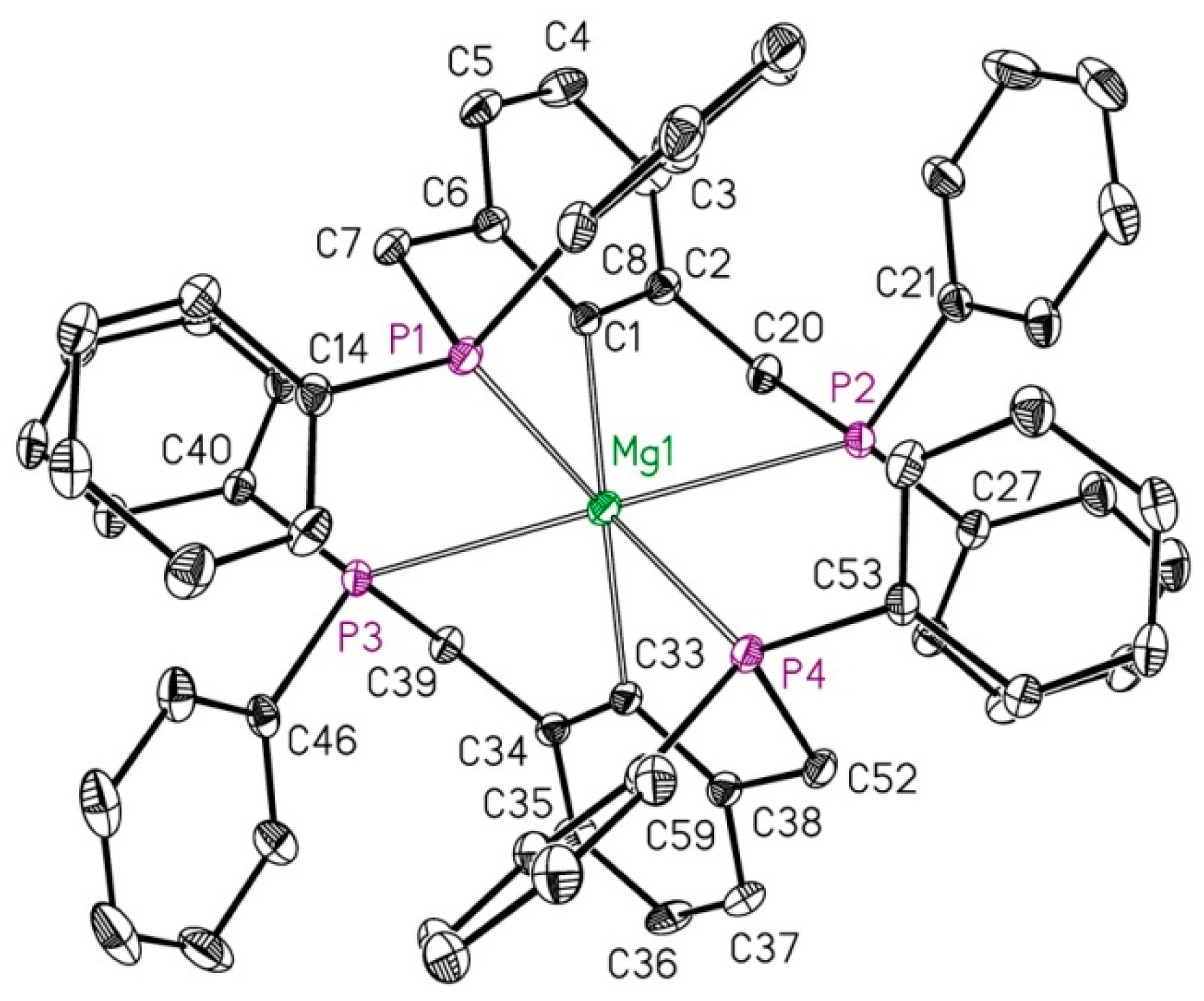
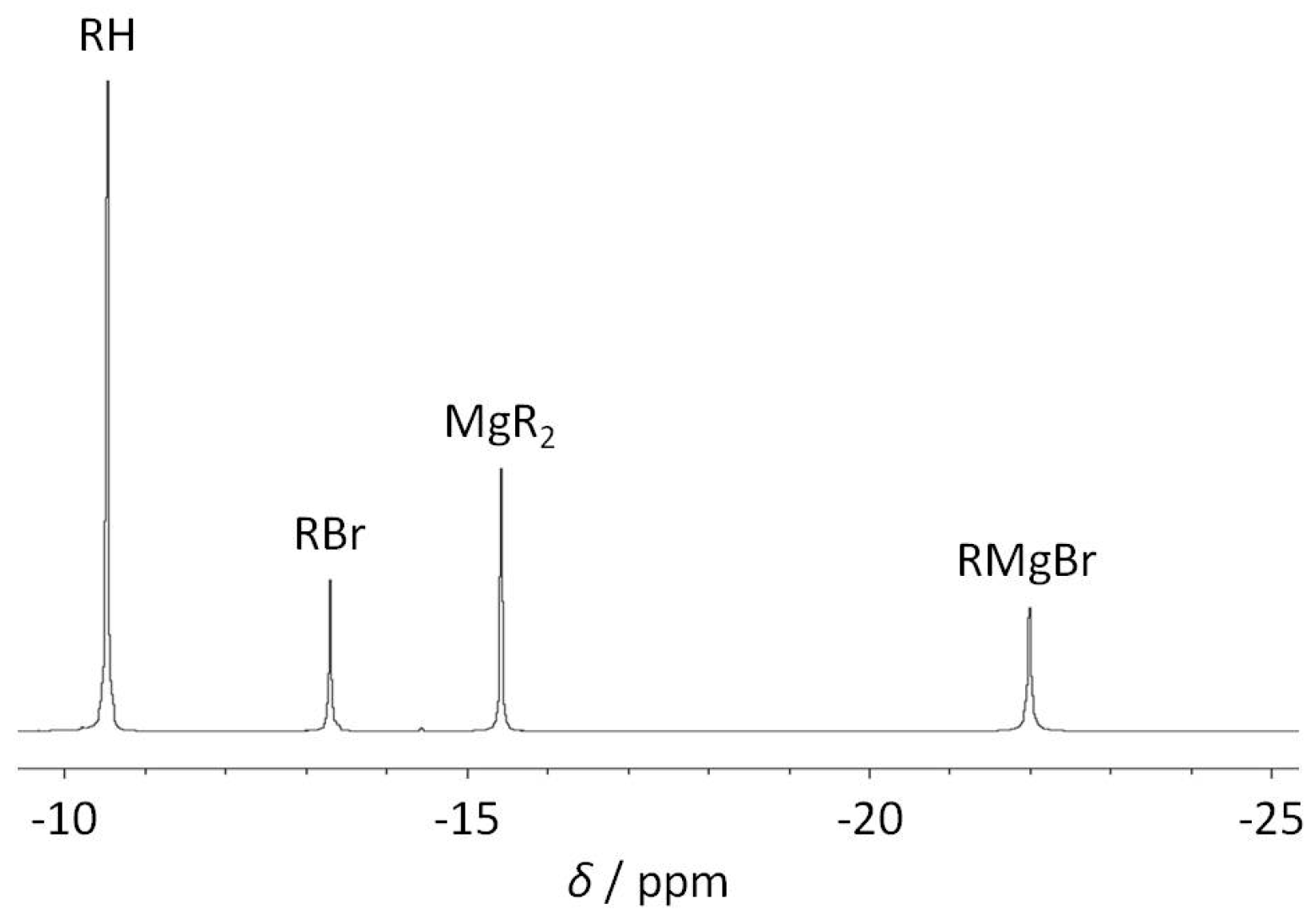
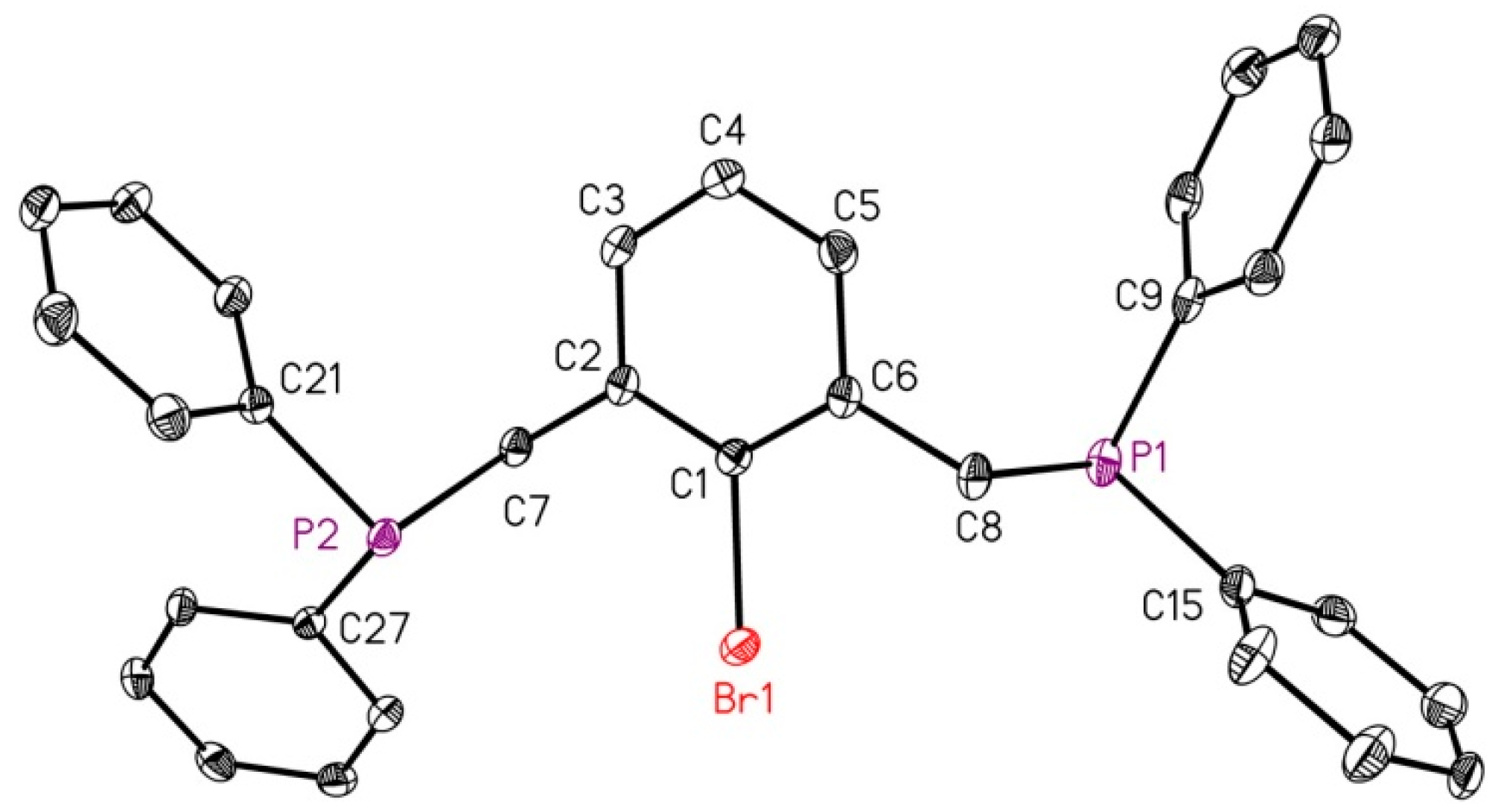
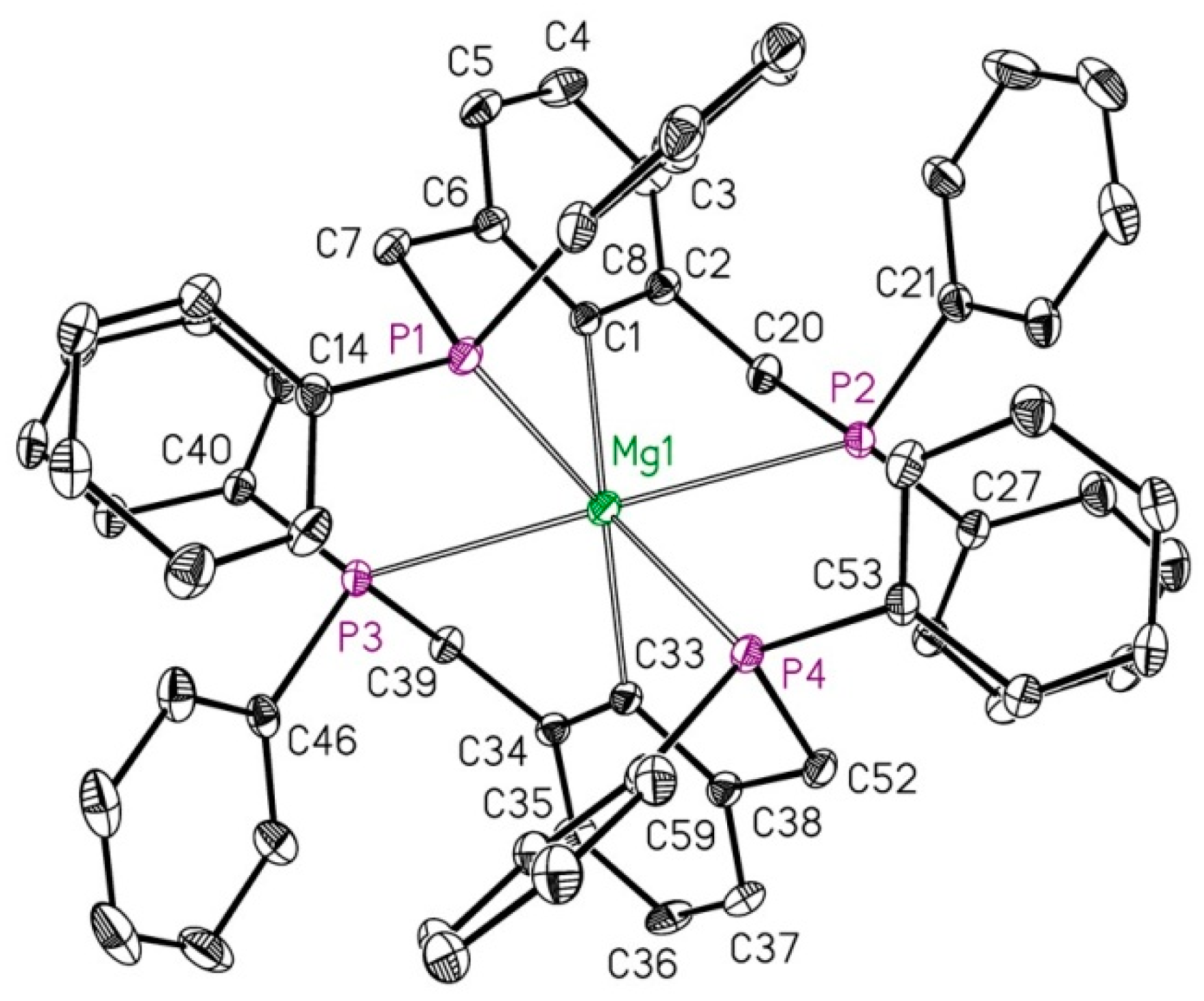
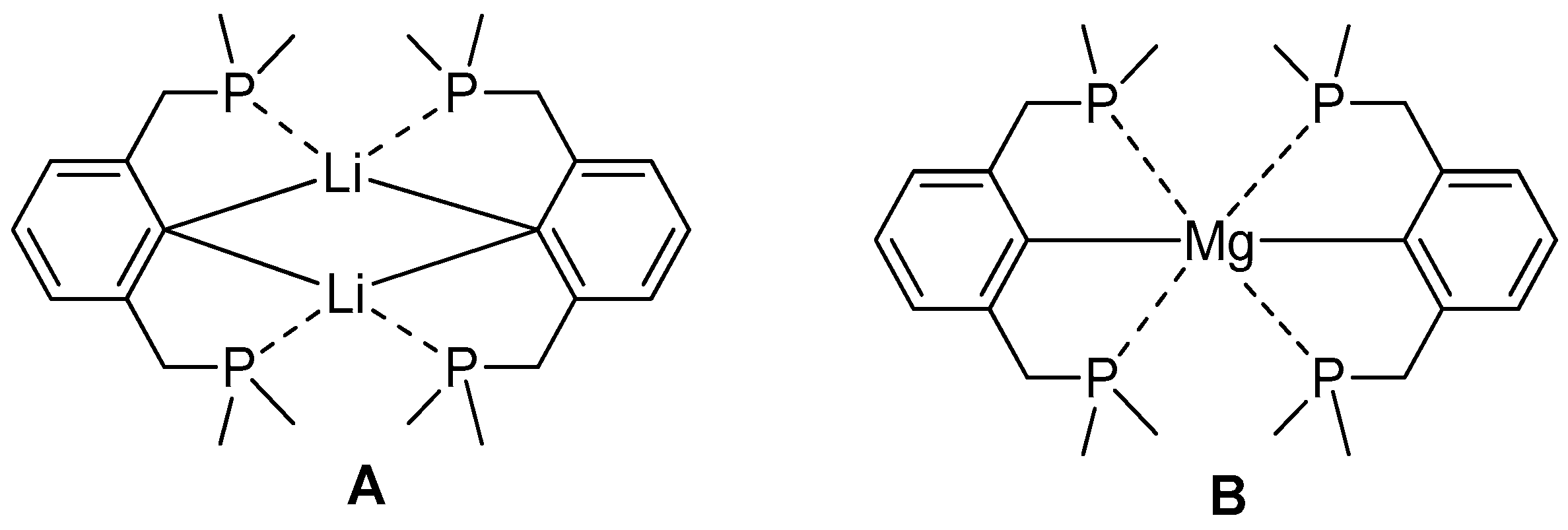
2.1. Reduction of Bromo-2,6-bis(diphosphanylmethyl)benzene with Magnesium

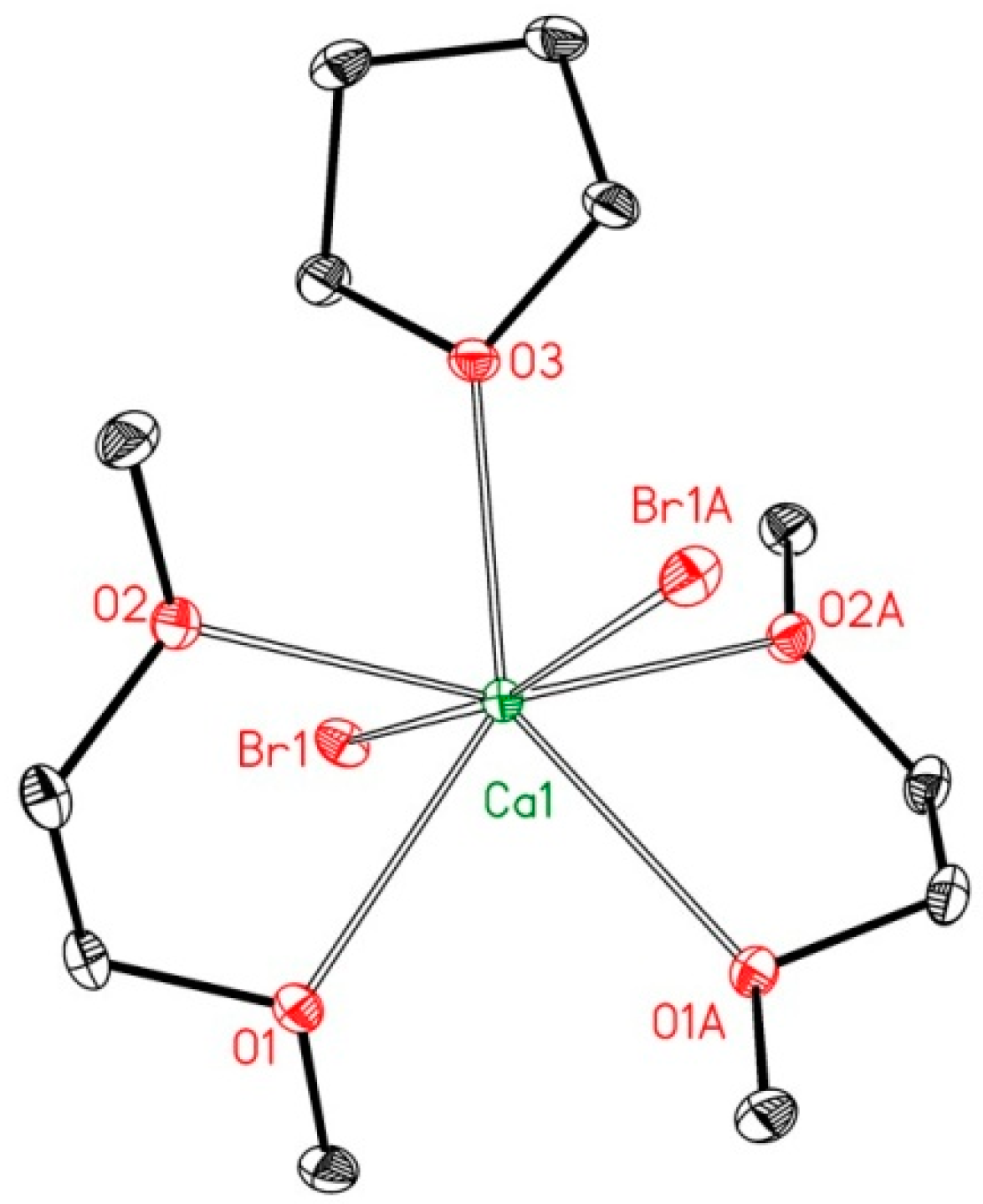
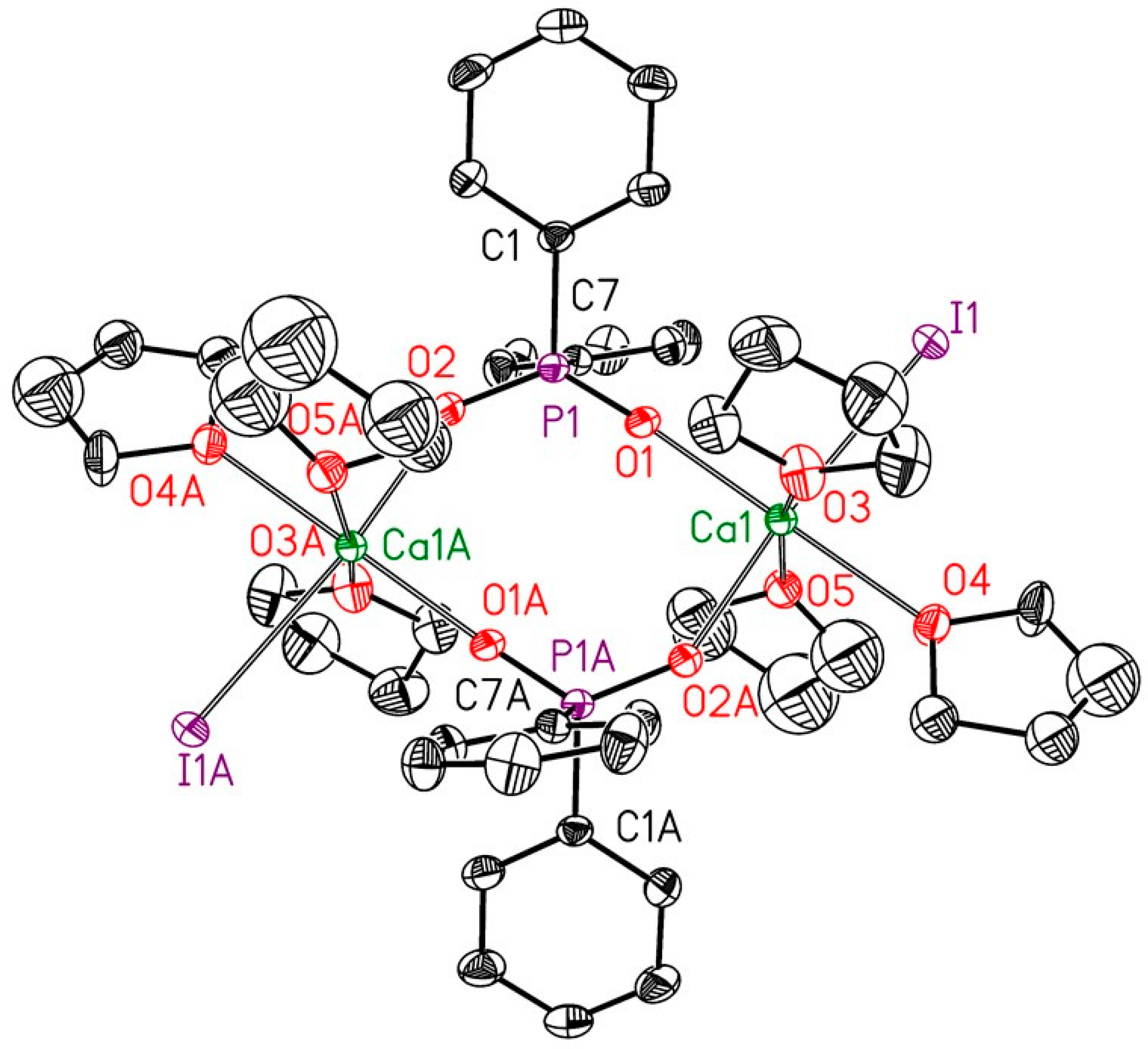
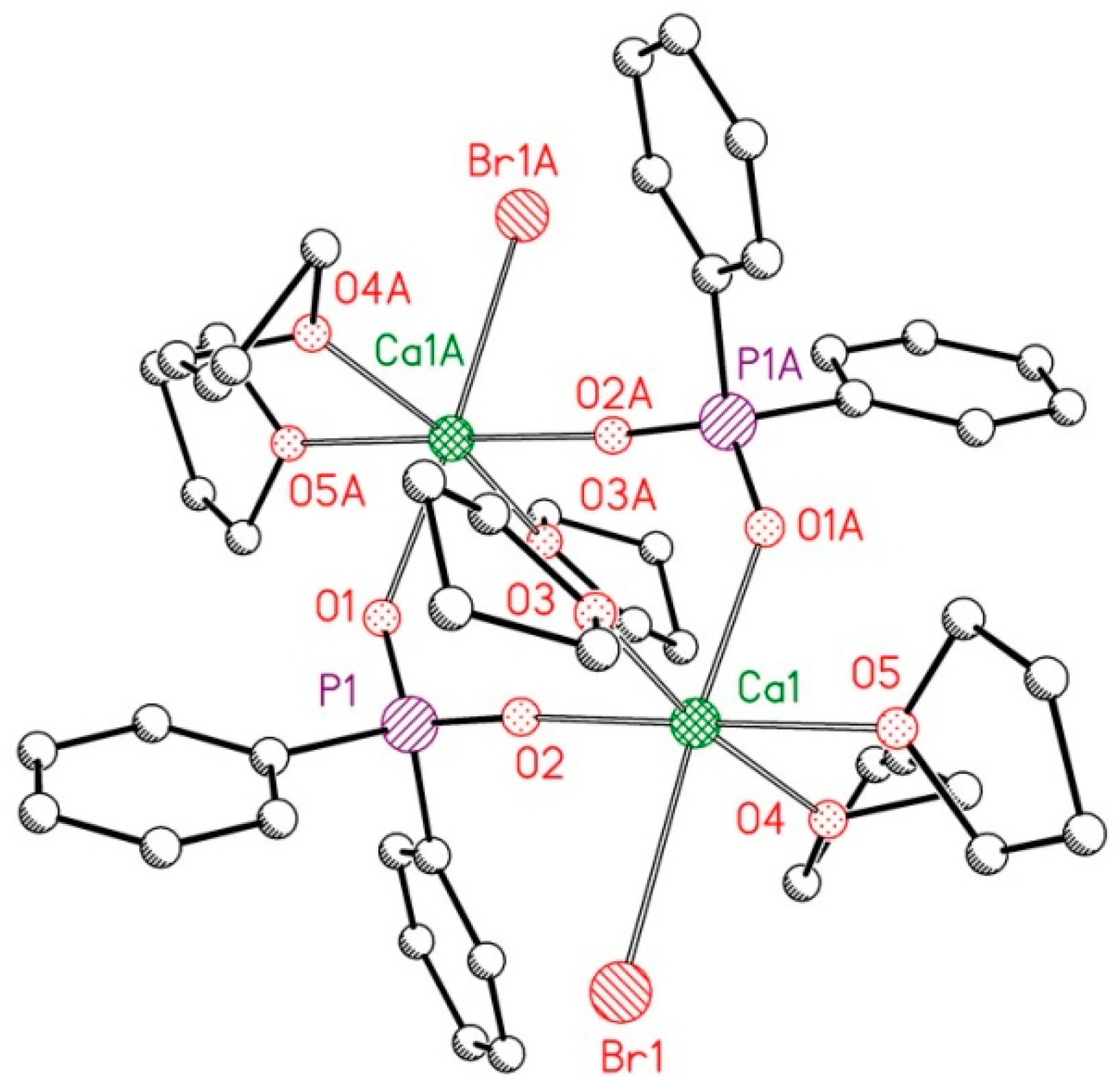
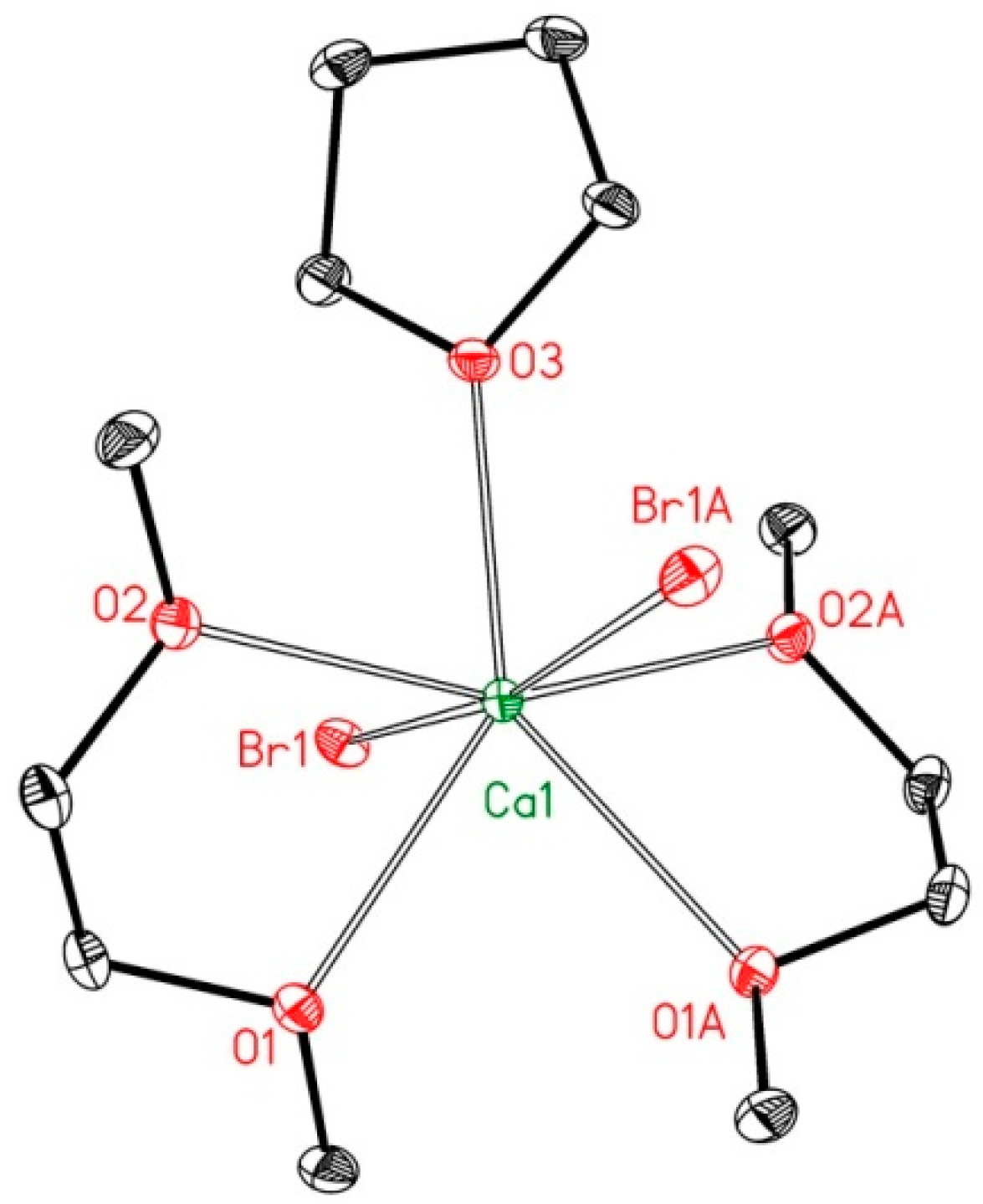
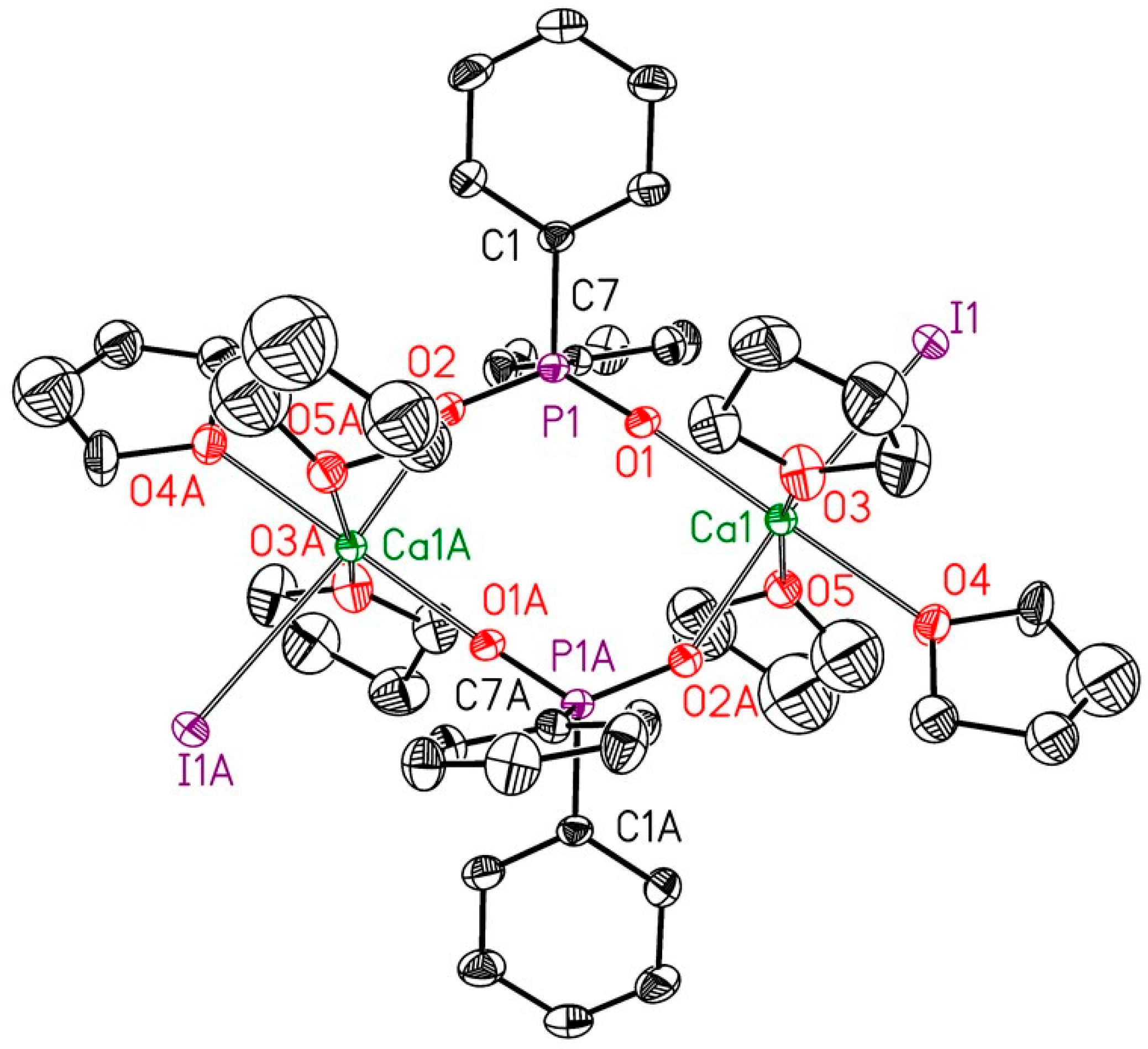
2.2. Reduction of 2,6-bis(diphosphanylmethyl)phenyl Halides with Calcium
2.3. Theoretical View on Ether and Phosphane Complexes of Magnesium and Calcium Ions
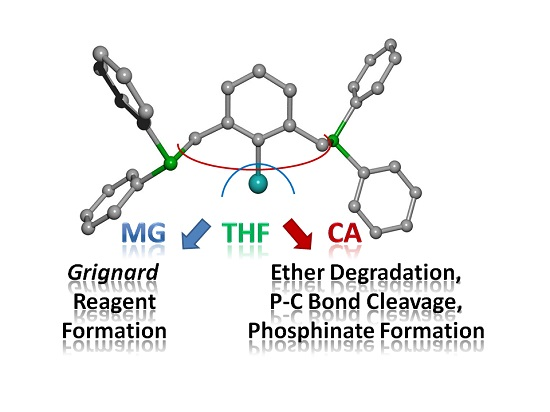
3. Discussion and Conclusions
4. Materials and Methods
4.1. General
4.2. Synthesis of Iodo-2,6-bis(diphenylphosphanylmethyl)benzene (1b)
4.3. Synthesis of Bis(2,6-bis(diphenylphosphanylmethyl)phenyl)Magnesium (2)
4.4. X-ray Structure Determinations
4.5. Quantum Chemical Calculations
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maercker, A. Ether Cleavage with Organo-Alkali-Metal Compounds and Alkali Metals. Angew. Chem. Int. Ed. Engl. 1987, 26, 972–989. [Google Scholar] [CrossRef]
- Langer, J.; Krieck, S.; Fischer, R.; Görls, H.; Walther, D.; Westerhausen, M. 1,4-Dioxane Adducts of Bis(2,4,6-trimethylphenyl)magnesium: Synthesis, Ether Cleavage Reactions, and Structural Diversity of Grignard Reagent/1,4-Dioxane Complexes. Organometallics 2009, 28, 5814–5820. [Google Scholar] [CrossRef]
- Krieck, S.; Westerhausen, M. Reimagining the Grignard Reaction. In The Lightest Metals: Science and Technology from Lithium to Calcium (Encyclopedia of Inorganic and Bioinorganic Chemistry); Hanusa, T.P., Ed.; Wiley: Chichester, UK, 2015; pp. 213–229. [Google Scholar]
- Westerhausen, M.; Langer, J.; Krieck, S.; Fischer, R.; Görls, H.; Köhler, M. Heavier Group 2 Grignard Reagents of the Type Aryl-Ae(L)n-X (Post-Grignard-Reagents). Top. Organomet. Chem. 2013, 45, 29–72. [Google Scholar] [CrossRef]
- Westerhausen, M.; Langer, J.; Krieck, S.; Glock, C. Calcium-based Organometallics and Superbases—Alkyl-, Aryl-, and Amidocalcium Compounds. Rev. Inorg. Chem. 2011, 31, 143–184. [Google Scholar]
- Buchanan, W.D.; Allis, D.G.; Ruhlandt-Senge, K. Synthesis and Stabilization—Advances in Organoalkaline Earth Metal Chemistry. Chem. Commun. 2010, 46, 4449–4465. [Google Scholar] [CrossRef] [PubMed]
- Westerhausen, M. Recent Developments in Organic Chemistry of Calcium—An Element with Unlimited Possibilities in Organometallic Chemistry? Z. Anorg. Allg. Chem. 2009, 635, 13–32. [Google Scholar] [CrossRef]
- Westerhausen, M. Heavy Grignard Reagents—Synthesis and Reactivity of Organocalcium Compounds. Coord. Chem. Rev. 2008, 252, 1516–1531. [Google Scholar] [CrossRef]
- Westerhausen, M.; Gärtner, M.; Fischer, R.; Langer, J. Arylcalcium Compounds: Syntheses, Structures, Physical Properties, and Chemical Behaviour. Angew. Chem. Int. Ed. 2007, 46, 1950–1956. [Google Scholar] [CrossRef] [PubMed]
- Westerhausen, M.; Gärtner, M.; Fischer, R.; Langer, J.; Yu, L.; Reiher, M. Heavy Grignard Reagents: Challenges and Possibilities of Aryl Alkaline Earth Metal Compounds. Chem. Eur. J. 2007, 13, 6292–6306. [Google Scholar] [CrossRef] [PubMed]
- Köhler, M.; Koch, A.; Görls, H.; Westerhausen, M. Trimethylsilylmethylcalcium Iodide, an Easily Accessible Grignard-Type Reagent of Calcium. Organometallics 2016, 35, 242–248. [Google Scholar] [CrossRef]
- Meinholz, M.M.; Pandey, S.K.; Deuerlein, S.M.; Stalke, D. Access to New Janus Head Ligands: Linking Sulfur Diimides and Phosphanes for Hemilabile Tripodal Scorpionates. Dalton Trans. 2011, 40, 1662–1671. [Google Scholar] [CrossRef] [PubMed]
- Peitz, S.; Peulecke, N.; Aluri, B.R.; Müller, B.H.; Spannenberg, A.; Rosenthal, U.; Al-Hazmi, M.H.; Mosa, F.M.; Wöhl, A.; Müller, W. Metalation and Transmetalation Studies on Ph2PN(iPr)P(Ph)N(iPr)H for Selective Ethene Trimerization to 1-Hexene. Organometallics 2010, 29, 5263–5268. [Google Scholar] [CrossRef]
- Pape, A.; Lutz, M.; Müller, G. Phosphane Coordination to Magnesium: Synthesis and Structure of Bis[ortho,ortho′-bis{(dimethylphosphino)methyl}phenyl]magnesium. Angew. Chem. Int. Ed. Engl. 1994, 33, 2281–2284. [Google Scholar] [CrossRef]
- Karsch, H.H.; Reisky, M. Phosphane Complexes of Alkaline Earth Metals. Eur. J. Inorg. Chem. 1998, 905–911. [Google Scholar] [CrossRef]
- Westerhausen, M.; Digeser, M.H.; Schwarz, W. 1,3-Bis(trimethylsilyl)-2-phenyl-1-aza-3-phosphapropenide Anions as Bidentate Ligands for the Alkaline Earth Metals Magnesium, Calcium, Strontium, and Barium. Inorg. Chem. 1997, 36, 521–527. [Google Scholar] [CrossRef]
- Meinholz, M.M.; Stalke, D. Monoanionic N,P,S-Janus Head Tripods in s-Block Metal Coordination. Eur. J. Inorg. Chem. 2011, 2011, 4578–4584. [Google Scholar] [CrossRef]
- Langer, J.; Wimmer, K.; Görls, H.; Westerhausen, M. Synthesis and Crystal Structures of Bis(diphenylphosphanyl)methanides of Lithium and Calcium as well as of their Borane Adducts. Dalton Trans. 2009, 28, 2951–2957. [Google Scholar] [CrossRef] [PubMed]
- Knapp, V.; Müller, G. Cyclopentadienyl-Free Calcium Alkyls with Heteroelement-Substituted Anionic Phosphane Ligands: Synthesis and Structure of a Trialkyl Calcate(II) and of an Organocalcium Heterocubane. Angew. Chem. Int. Ed. 2001, 40, 183–186. [Google Scholar] [CrossRef]
- Markies, P.R.; Akkerman, O.S.; Bickelhaupt, F.; Smeets, W.J.J.; Spek, A.L. X-ray Structural Analyses of Organomagnesium Compounds. Adv. Organomet. Chem. 1991, 32, 147–226. [Google Scholar]
- Holloway, C.E.; Melnik, M. Magnesium Compounds: Classification and Analysis of Crystallographic and Structural Data. J. Organomet. Chem. 1994, 465, 1–63. [Google Scholar] [CrossRef]
- Uhm, H.L. Crystal Structures of Grignard Reagents. In Handbook of Grignard Reagents; Silverman, G.S., Rakita, P.E., Eds.; CRC Press: Boca Raton, FL, USA, 1996; pp. 117–144. [Google Scholar]
- Bickelhaupt, F. Structures of Organomagnesium Compounds as Revealed by X-ray Diffraction Studies. In Grignard Reagents: New Developments; Richey, H.G., Ed.; Wiley: Chichester, UK, 2000; pp. 299–328. [Google Scholar]
- Jastrzebski, J.T.B.H.; Boersma, J.; van Koten, G. Structural Organomagnesium Chemistry. In The Chemistry of Organomagnesium Compounds (Patai Series: The Chemistry of Functional Groups); Wiley: Chichester, UK, 2008; pp. 1–99. [Google Scholar]
- Ruspic, C.; Harder, S. Synthesis and Structure of an Arylcalcium Compound with an Unusual Calcium Tetrahedron Containing an Encapsulated Oxide. Organometallics 2005, 24, 5506–5508. [Google Scholar] [CrossRef]
- Fischer, R.; Görls, H.; Westerhausen, M. Reinvestigation of the Synthesis of Phenylcalcium Iodide and the First Structural Characterization of a Heavy Grignard Reagent as [((thf)2CaPhI)3·(thf)CaO] with a Central Ca4 Tetrahedron. Inorg. Chem. Commun. 2005, 8, 1159–1161. [Google Scholar] [CrossRef]
- Gärtner, M.; Görls, H.; Westerhausen, M. Heteroleptic Phenylcalcium Derivatives via Metathesis Reaction of PhCa(thf)4I with Potassium Compounds. Organometallics 2007, 26, 1077–1083. [Google Scholar] [CrossRef]
- Krieck, S.; Görls, H.; Westerhausen, M. Reactivity Studies of Phenylcalcium Iodide Towards THF Yielding Phenyl-Free Cage Compounds—Crystal Structures of [{(thf)Ca(O–CH=CH2)2}4·CaO·CaI2] and [(CaO)4·4(thf)3CaI2]. J. Organomet. Chem. 2009, 694, 2204–2209. [Google Scholar] [CrossRef]
- Al-Shboul, T.M.A.; Volland, G.; Görls, H.; Krieck, S.; Westerhausen, M. Oxidation Products of Calcium and Strontium Bis(diphenylphosphanide). Inorg. Chem. 2012, 51, 7903–7912. [Google Scholar] [CrossRef] [PubMed]
- Westerhausen, M.; Krieck, S.; Langer, J.; Al-Shboul, T.M.A.; Görls, H. Phosphanides of Calcium and Their Oxidation Products. Coord. Chem. Rev. 2013, 257, 1049–1066. [Google Scholar] [CrossRef]
- Langer, J.; Krieck, S.; Fischer, R.; Görls, H.; Westerhausen, M. Post-Grignard Reagents: Influence of the Coligands L on the Molecular Structures of Phenylcalcium Iodides [(L)nCa(R)I] and Calcium Diiodides [(L)nCaI2]. Z. Anorg. Allg. Chem. 2010, 636, 1190–1198. [Google Scholar] [CrossRef]
- Burt, J.; Levason, W.; Reid, G. Coordination Chemistry of the Main Group Elements with Phosphine, Arsine and Stibine Ligands. Coord. Chem. Rev. 2014, 260, 65–115. [Google Scholar] [CrossRef]
- Michel, O.; Meermann, C.; Törnroos, K.W.; Anwander, R. Alkaline-Earth Metal Alkylaluminate Chemistry Revisited. Organometallics 2009, 28, 4783–4790. [Google Scholar] [CrossRef]
- Krieck, S.; Görls, H.; Westerhausen, M. Synthesis and Properties of Calcium Tetraorganylalanates with [Me4−nAlPhn]− Anions. Organometallics 2008, 27, 5052–5057. [Google Scholar] [CrossRef]
- Fischer, R.; Gärtner, M.; Görls, H.; Westerhausen, M. Synthesis of 2,4,6-Trimethylphenylcalcium Iodide and Degradation in THF Solution. Angew. Chem. Int. Ed. 2006, 45, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, J.R.; Zheng, Y.; Griffiths, D.V. Binuclear Complexes with Ligands Based on the 2,6-Bis(diphenylphosphinomethyl)benzene Framework. Synthesis and Crystal Structures of [Ir2Cl2(μ-CO){2,6-(Ph2PCH2)2C6H3S}2]·2CH2Cl2, [Ni2{2,6-(Ph2PCH2)2C6H3S}2]·Et2O·0.5CH2Cl2 and [Rh2Cl2(CO)2{1,3-(Ph2PCH2)2C6H4}2]. J. Chem. Soc. Dalton Trans. 1999, 1877–1881. [Google Scholar] [CrossRef]
- Rieke, R.D. Preparation of Highly Reactive Metal Powders and Their Use in Organic and Organometallic Synthesis. Acc. Chem. Res. 1977, 10, 301–306. [Google Scholar] [CrossRef]
- Rieke, R.D. Preparation of Organometallic Compounds from Highly Reactive Metal Powders. Science 1989, 246, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Hooft, R. COLLECT, Data Collection Software, Nonius B.V.: Rotterdam, The Netherlands, 1998.
- Otwinowski, Z.; Minor, W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In Methods in Enzymology, Macromolecular Crystallography; Carter, C.W., Sweet, R.M., Eds.; Academic Press: New York, NY, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- SADABS 2.10, Bruker-AXS Inc.: Madison, WI, USA, 2002.
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution of the Calculated Structure Factors. Acta Cryst. 2015, C71, 9–18. [Google Scholar]
- XP Molecular Graphics Software, Siemens Analytical X-ray Instruments Inc.: Karlsruhe, Germany, 1990; Madison, WI, USA, 1994.
- POV-Ray, Persistence of Vision Raytracer: Williamstown, Australia, 2007.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02, Gaussian, Inc.: Wallingford, UK, 2009.
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chem. Acc. 1973, 28, 213–222. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M, L | n = 0 | n = 1 | n = 2 | n = 3 | n = 4 | n = 5 |
|---|---|---|---|---|---|---|
| Mg, thf | −99 | −87 | −79 | −78 | −65 | −23 |
| Mg, PMe3 | −104 | −82 | −52 | −37 | +43 | - a |
| Ca, thf | −57 | −47 | −47 | −46 | −45 | −28 |
| Ca, PMe3 | −37 | −28 | −27 | −23 | −2 | −2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koch, A.; Krieck, S.; Görls, H.; Westerhausen, M. Reduction of Bromo- and Iodo-2,6-bis(diphenylphosphanylmethyl)benzene with Magnesium and Calcium. Inorganics 2016, 4, 39. https://doi.org/10.3390/inorganics4040039
Koch A, Krieck S, Görls H, Westerhausen M. Reduction of Bromo- and Iodo-2,6-bis(diphenylphosphanylmethyl)benzene with Magnesium and Calcium. Inorganics. 2016; 4(4):39. https://doi.org/10.3390/inorganics4040039
Chicago/Turabian StyleKoch, Alexander, Sven Krieck, Helmar Görls, and Matthias Westerhausen. 2016. "Reduction of Bromo- and Iodo-2,6-bis(diphenylphosphanylmethyl)benzene with Magnesium and Calcium" Inorganics 4, no. 4: 39. https://doi.org/10.3390/inorganics4040039
APA StyleKoch, A., Krieck, S., Görls, H., & Westerhausen, M. (2016). Reduction of Bromo- and Iodo-2,6-bis(diphenylphosphanylmethyl)benzene with Magnesium and Calcium. Inorganics, 4(4), 39. https://doi.org/10.3390/inorganics4040039