
Catalytic Organic Transformations Mediated by Actinide Complexes


Abstract
:
1. Introduction
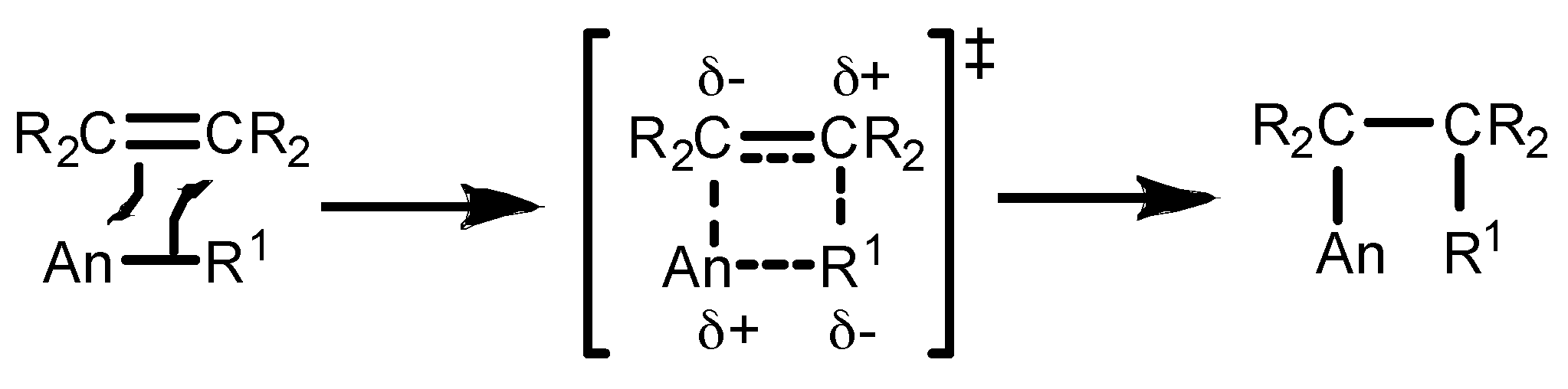
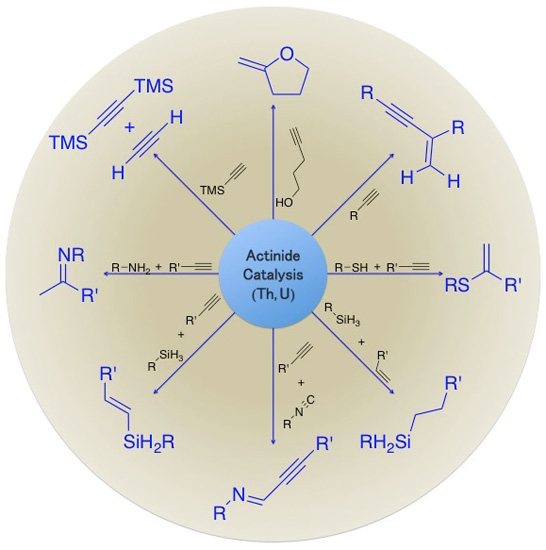
2. Application of Actinide Complexes in Catalytic Transformations
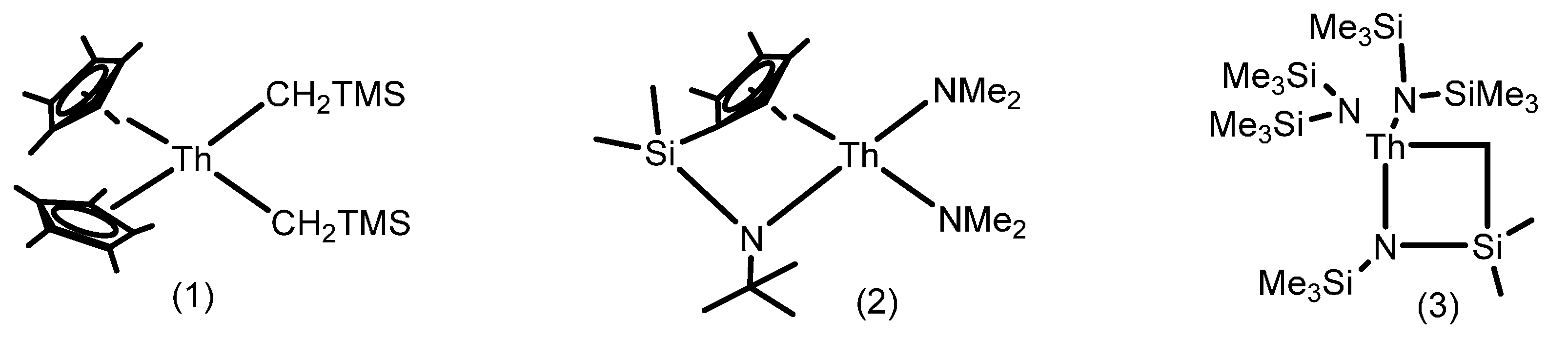
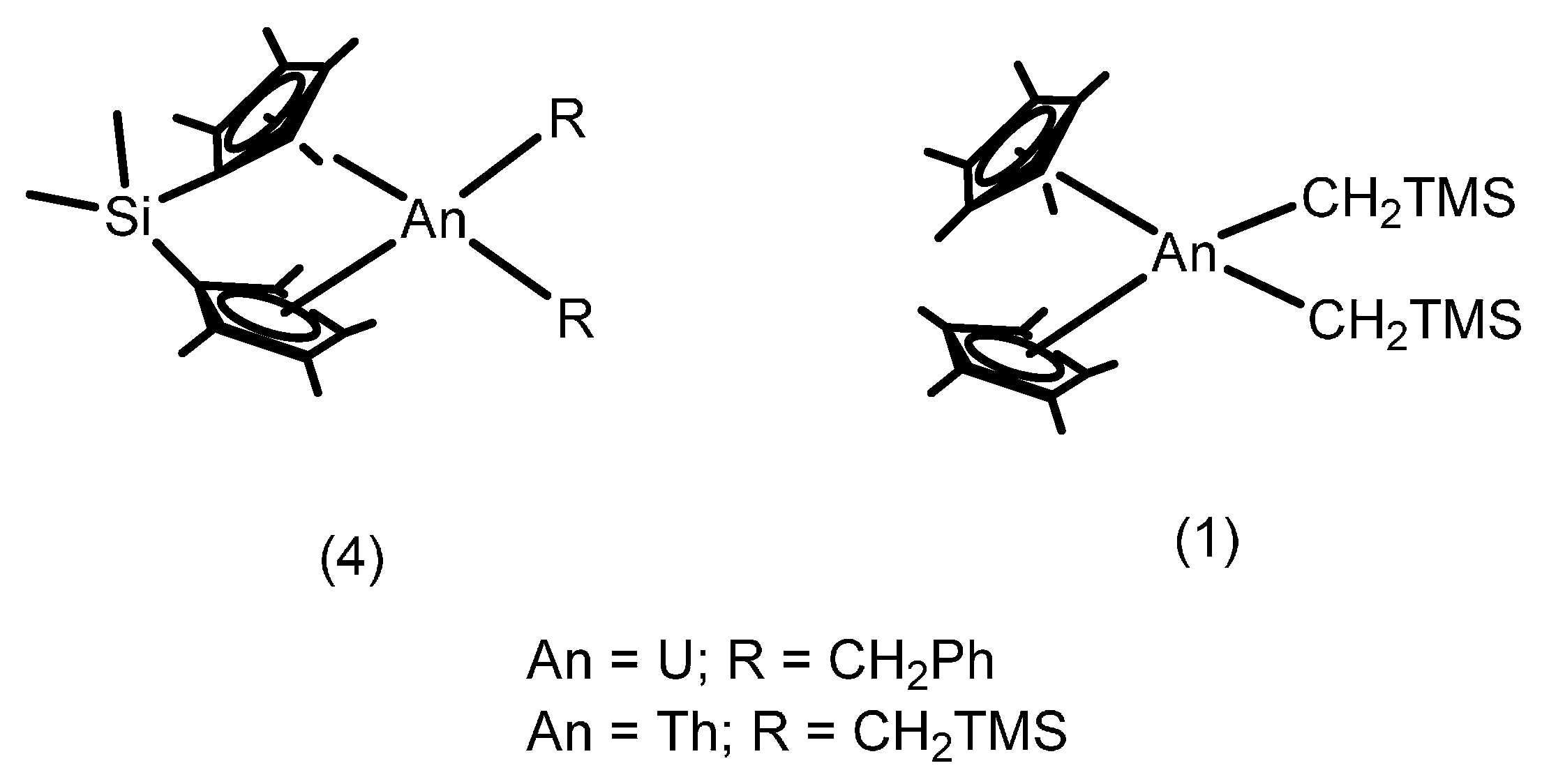
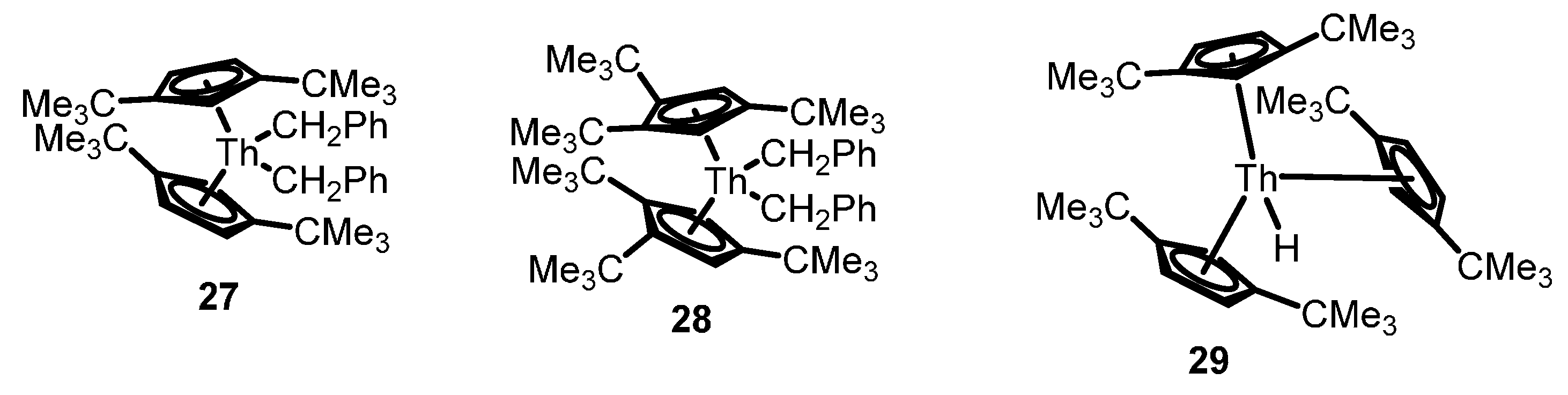
2.1. Hydroelementation Reactions
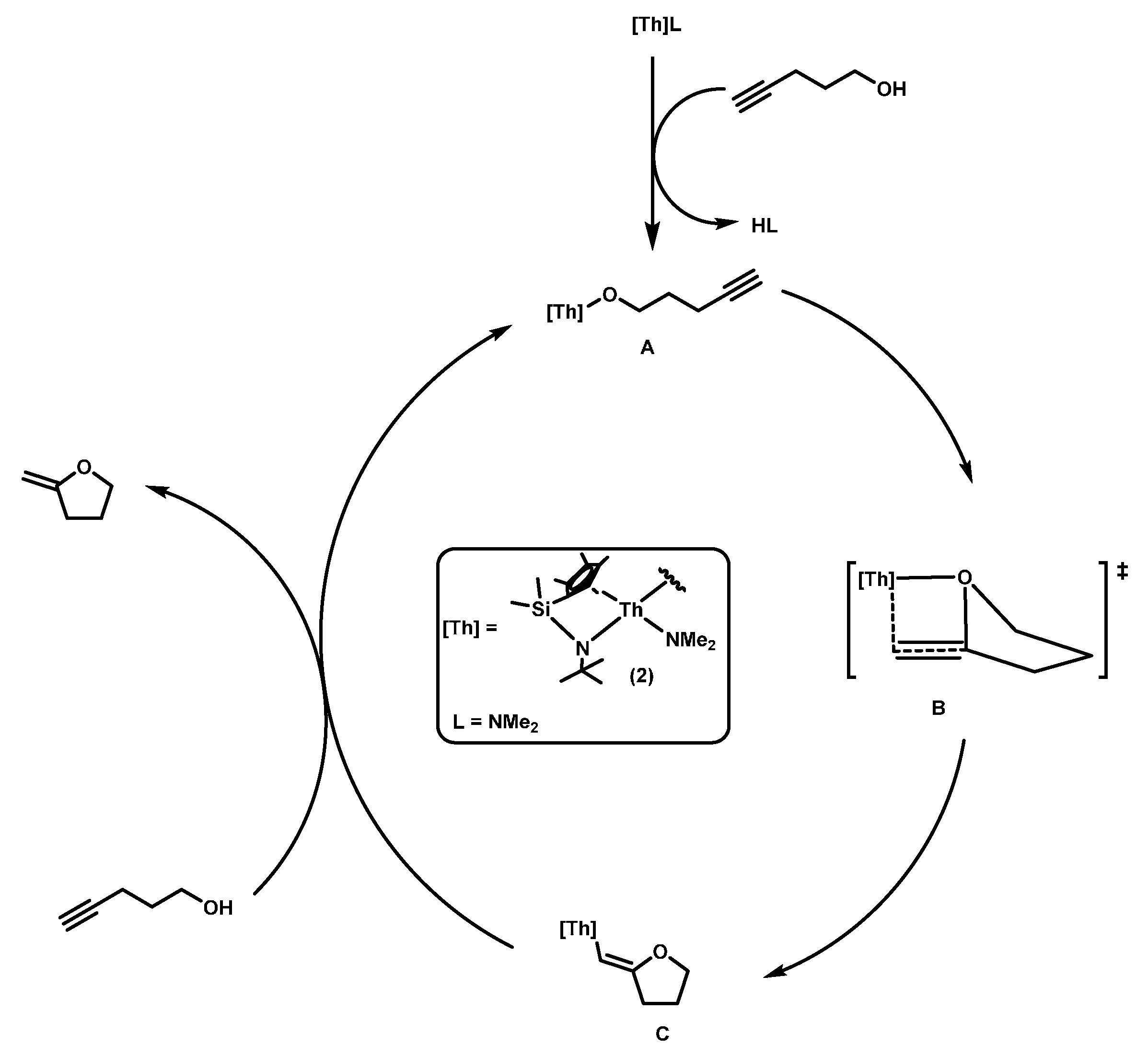
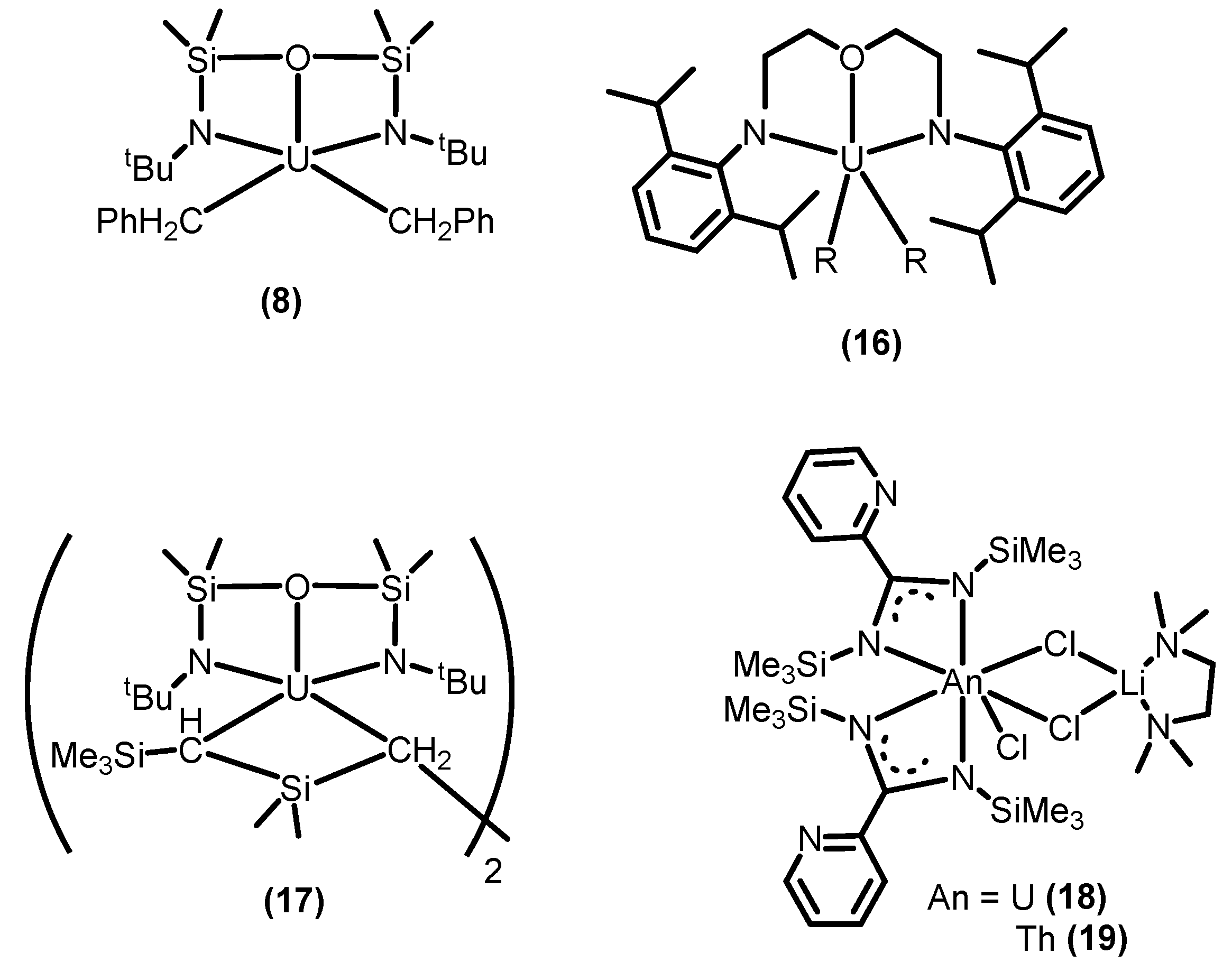
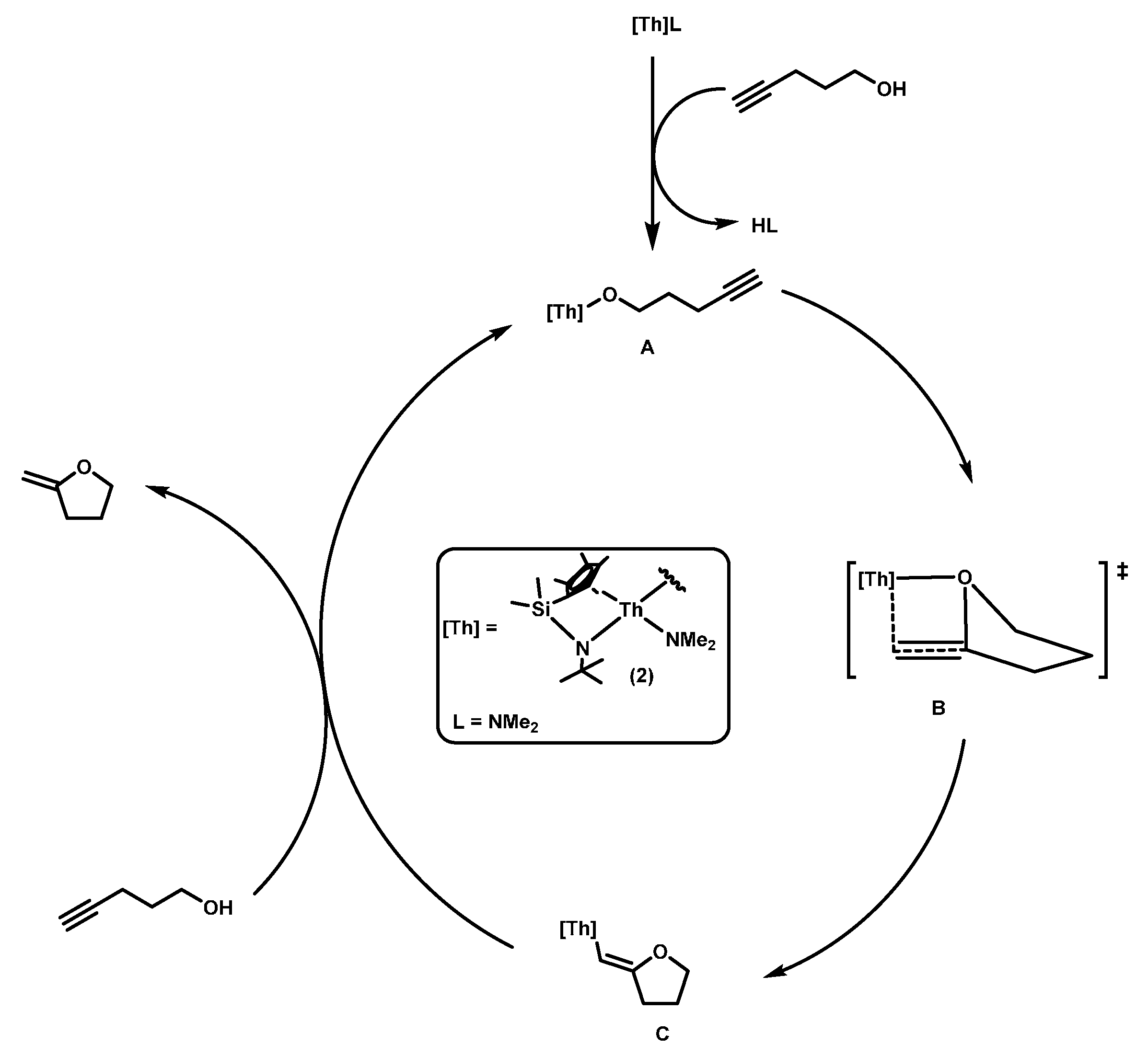
2.1.1. Hydroalkoxylation/Cyclization of Alkynyl Alcohols
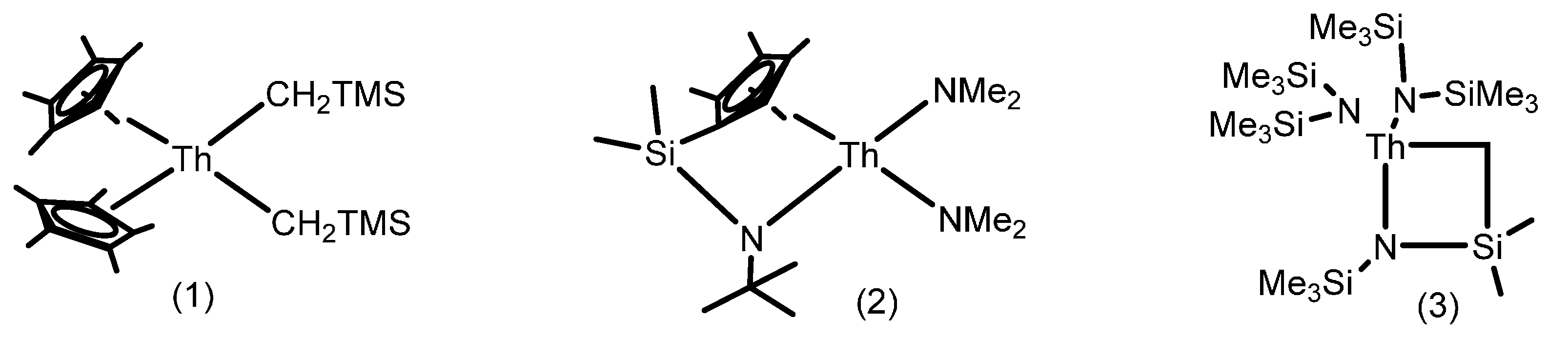
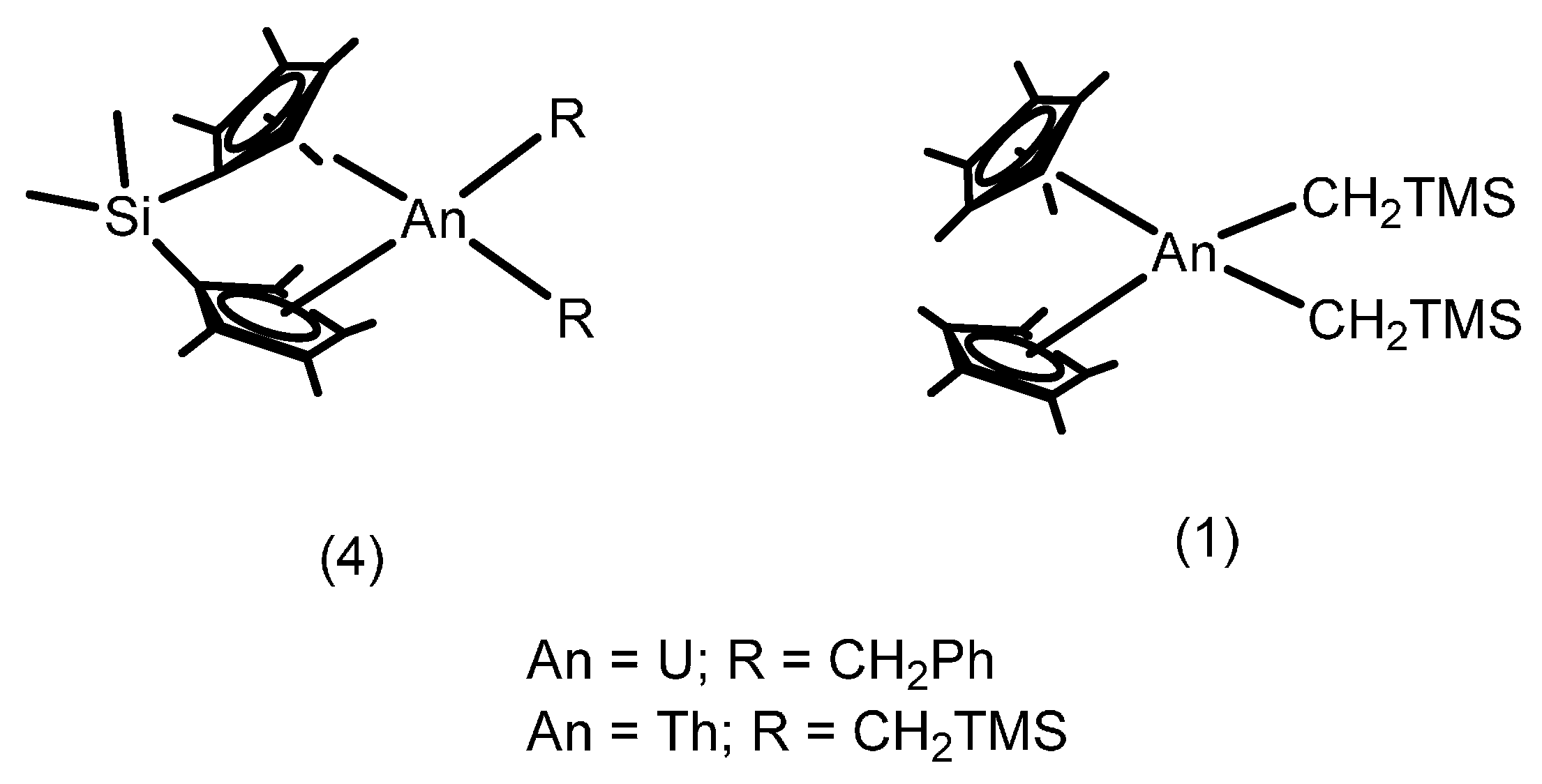
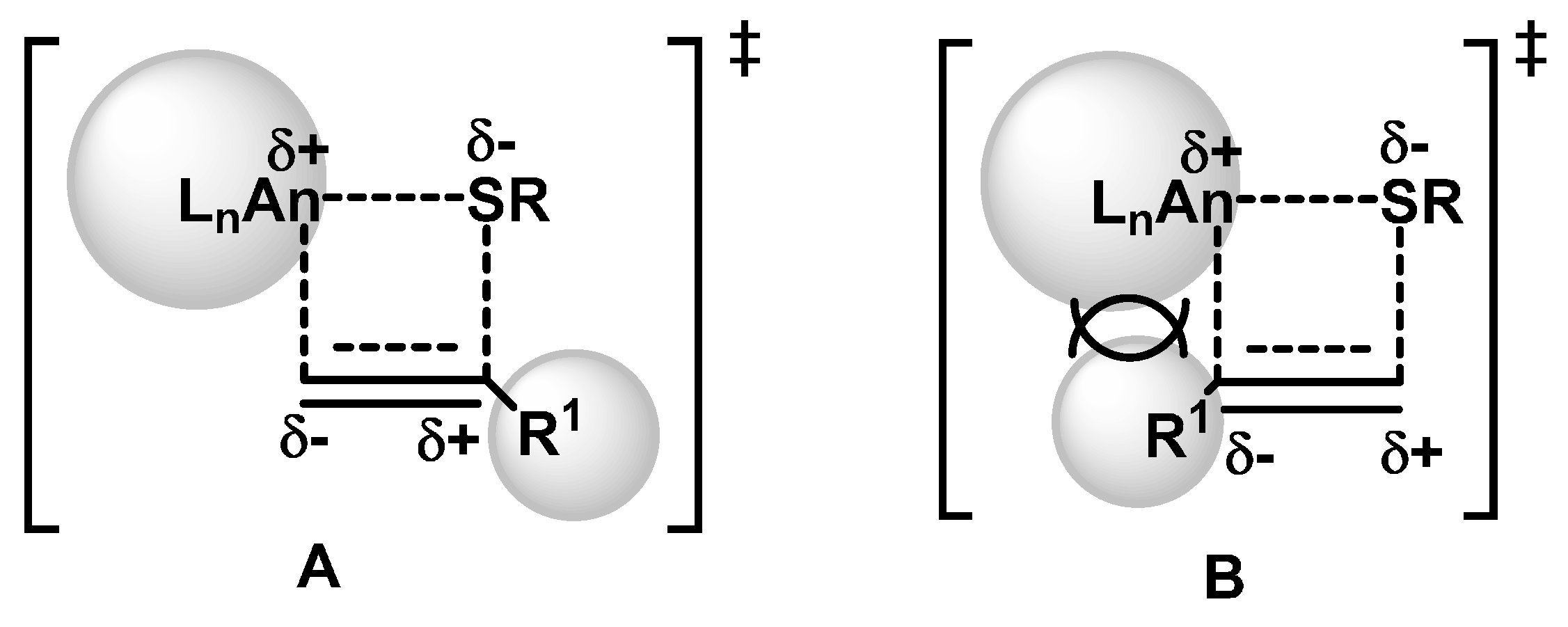
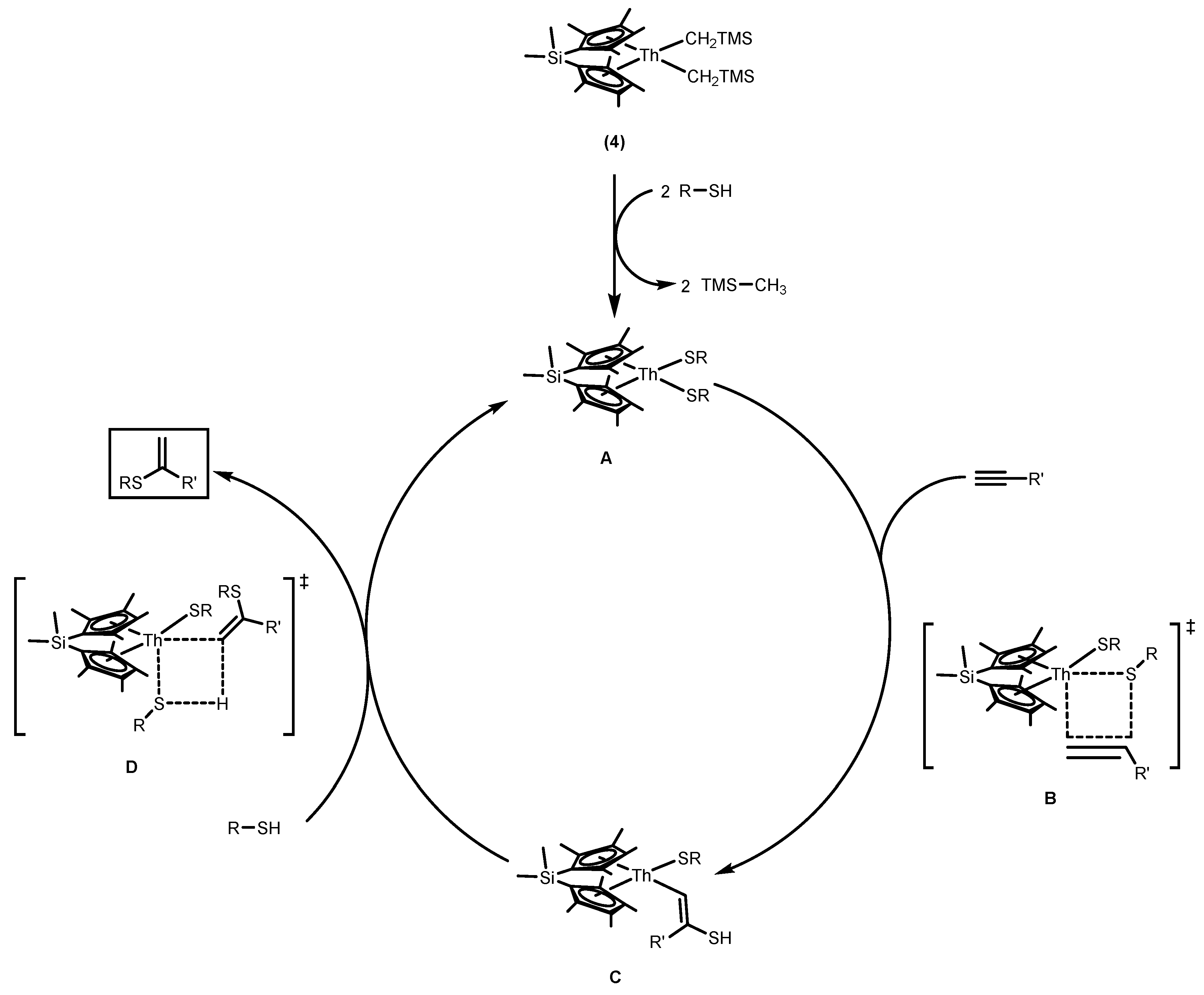
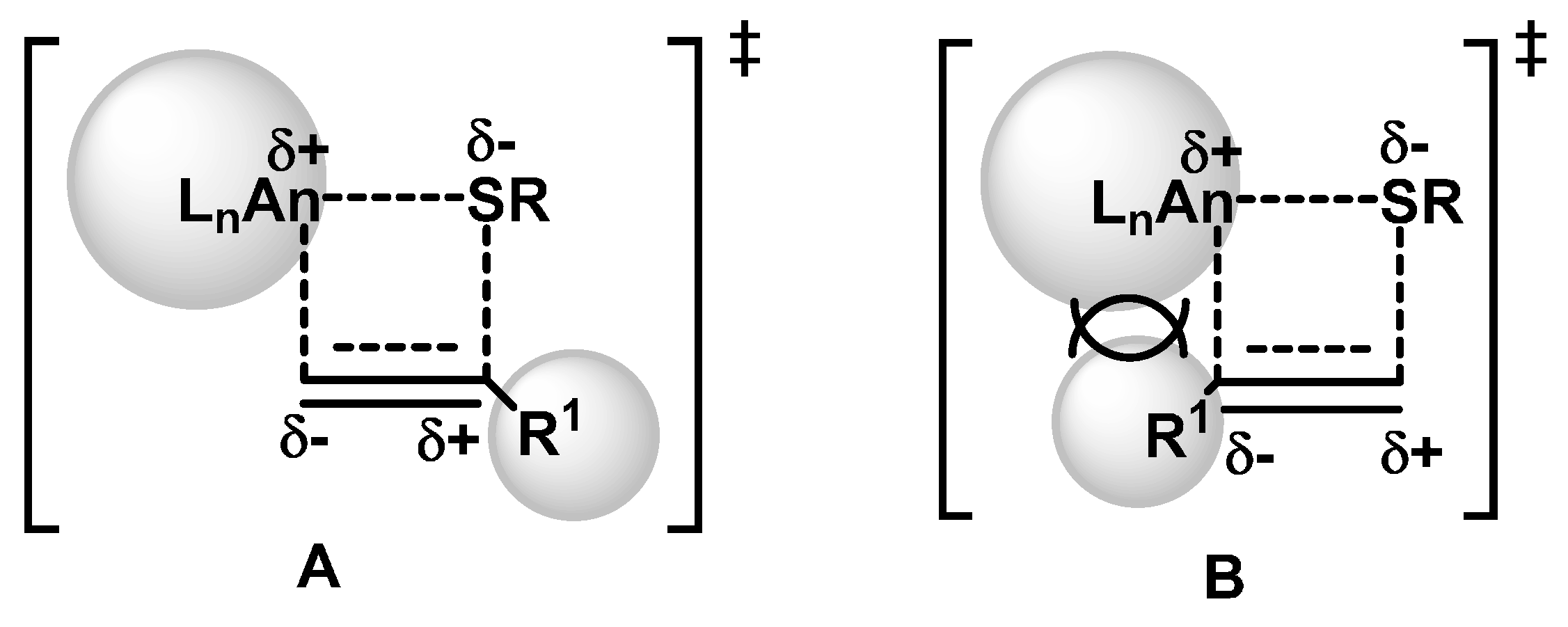
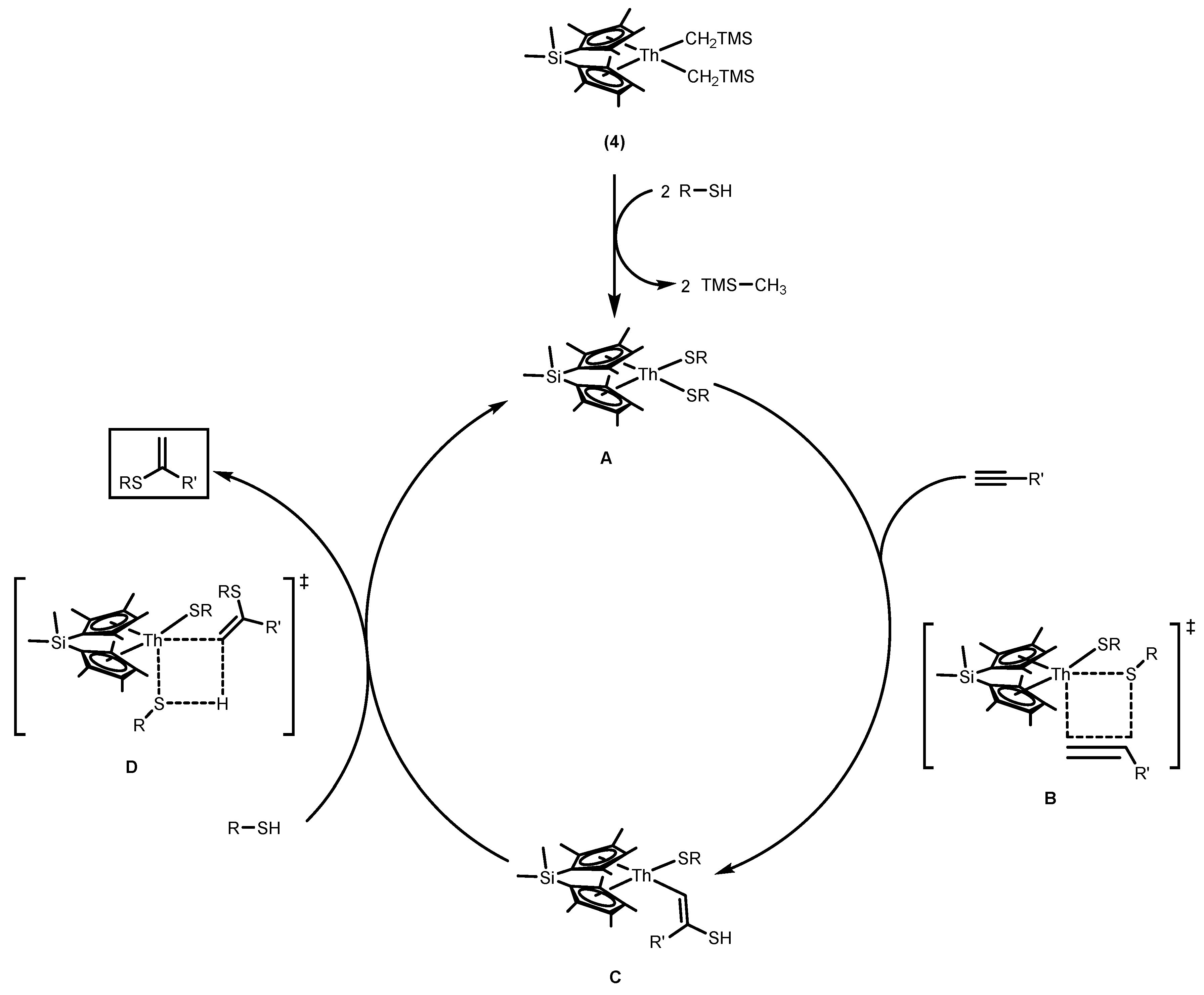
2.1.2. Hydrothiolation of Terminal Alkynes
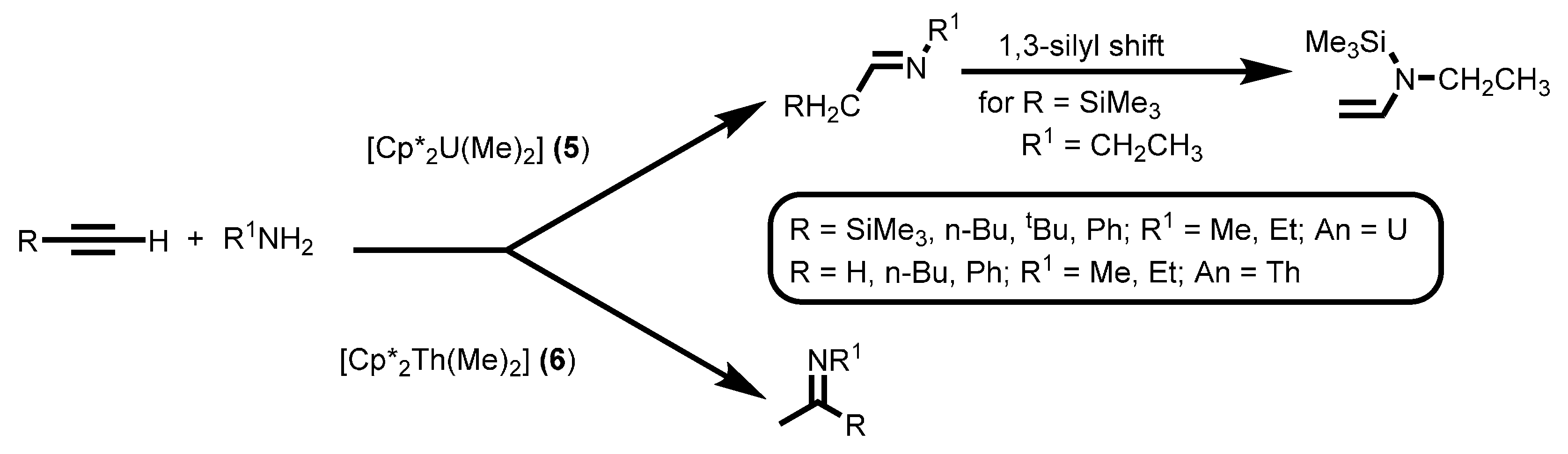
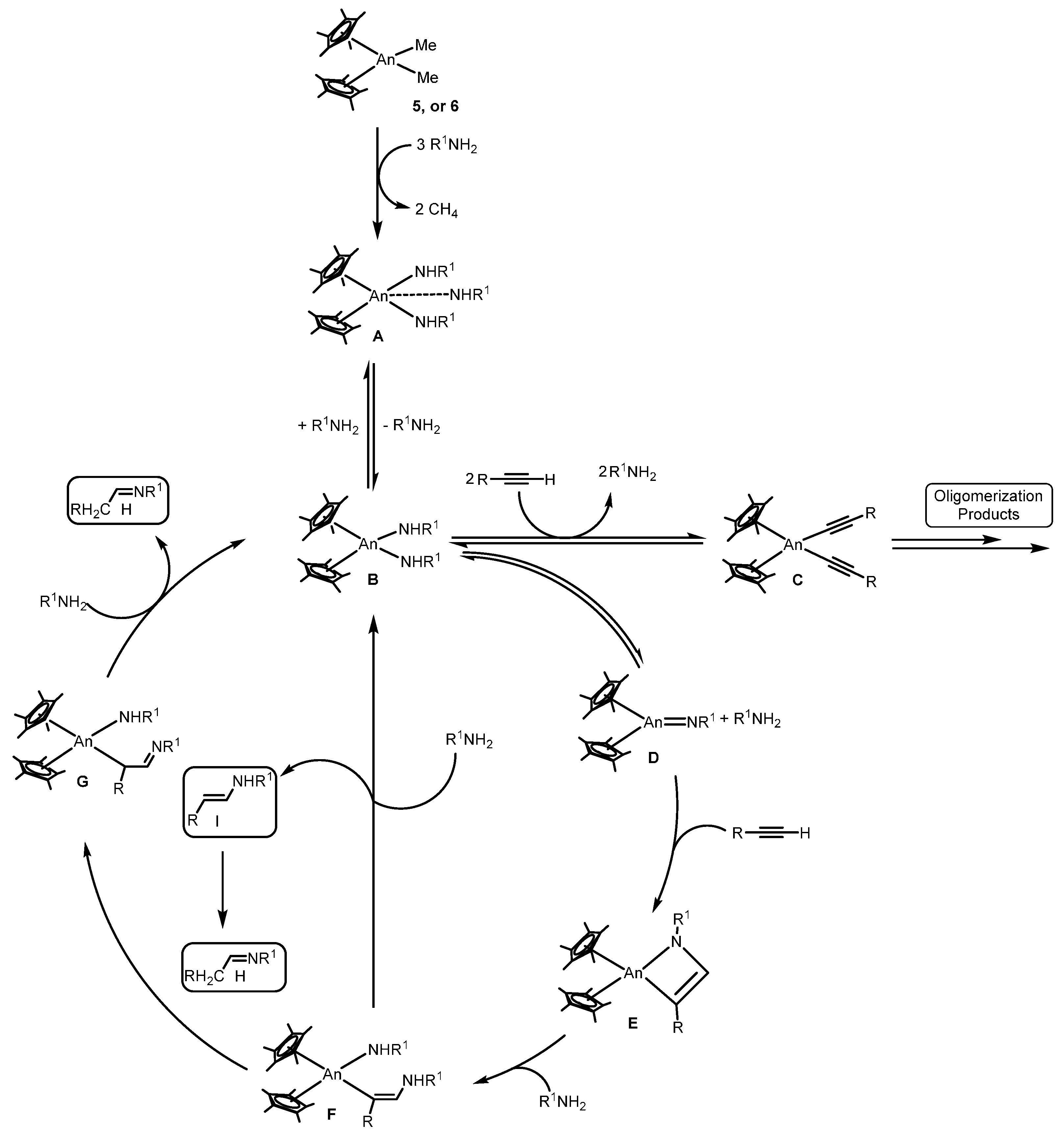
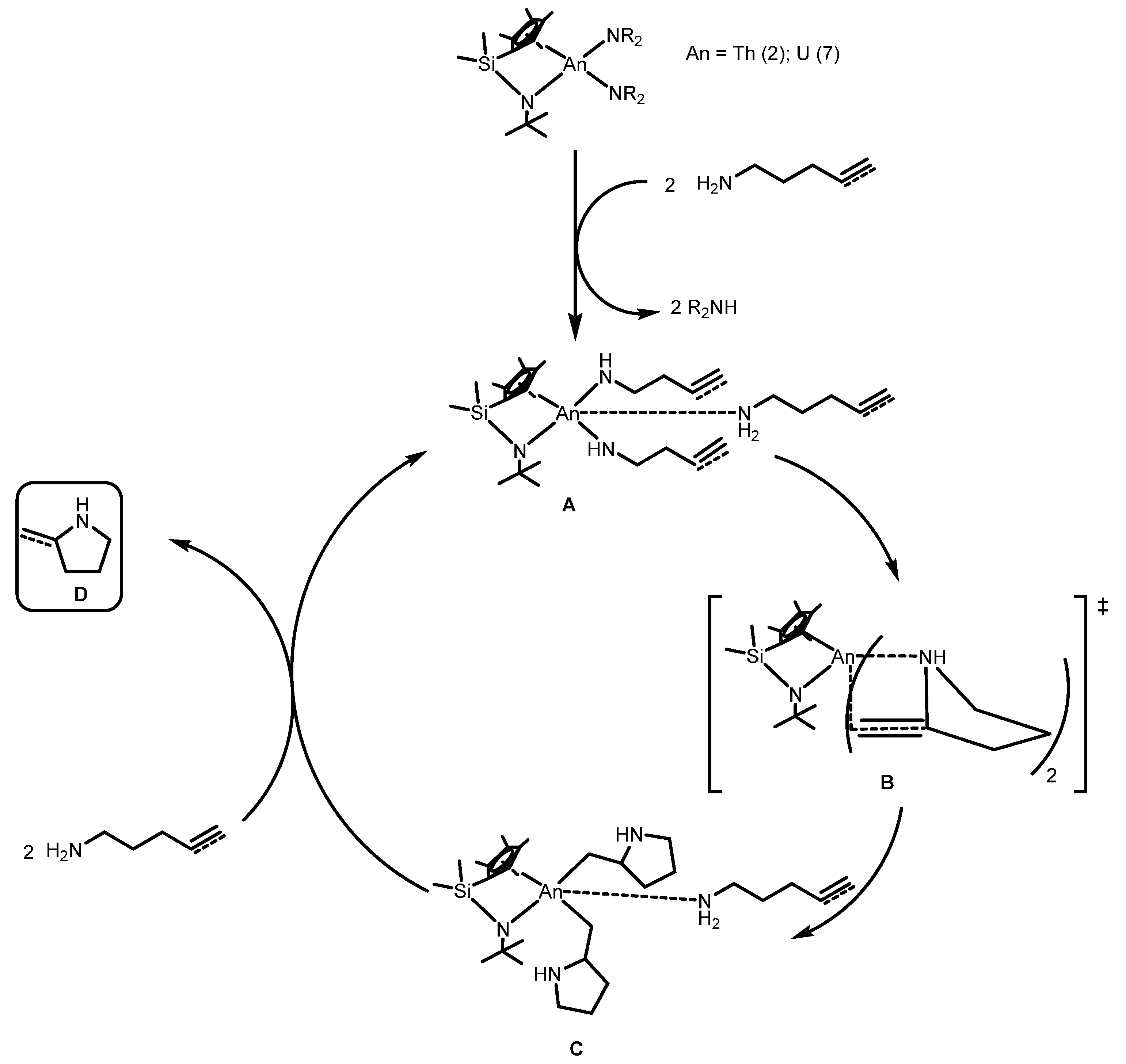
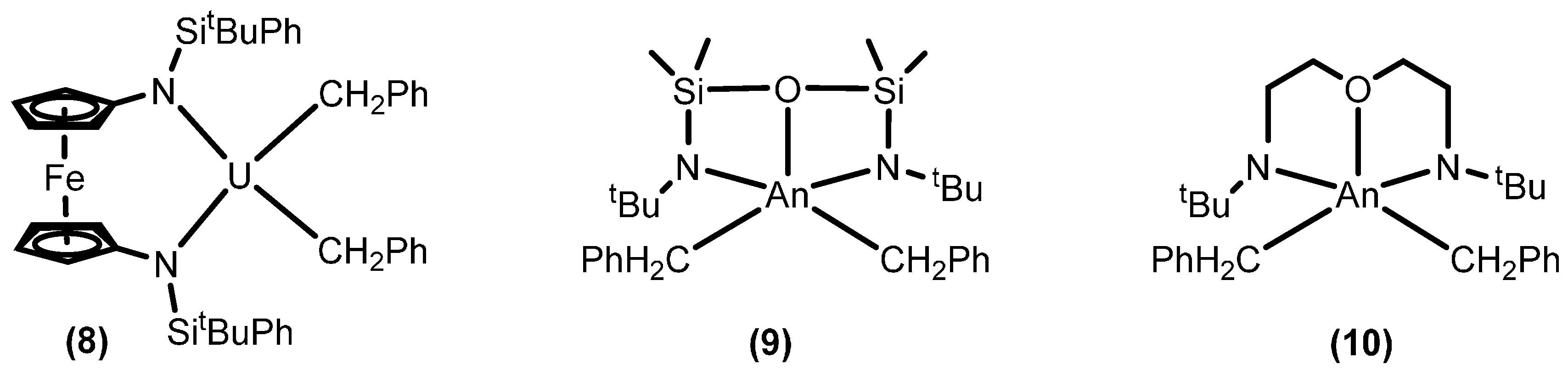
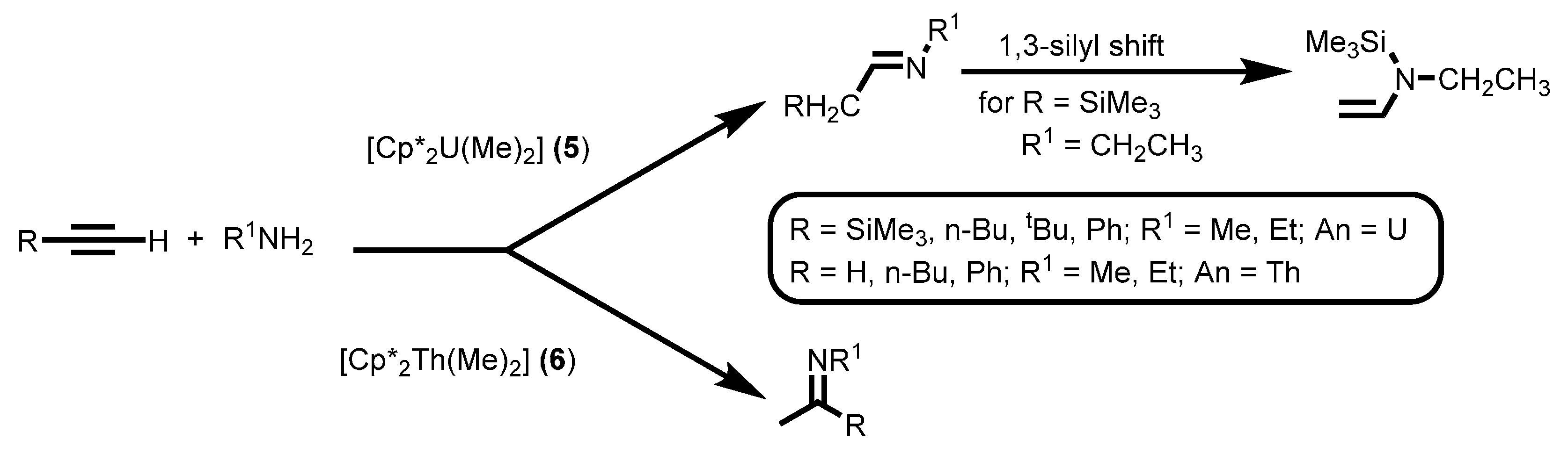
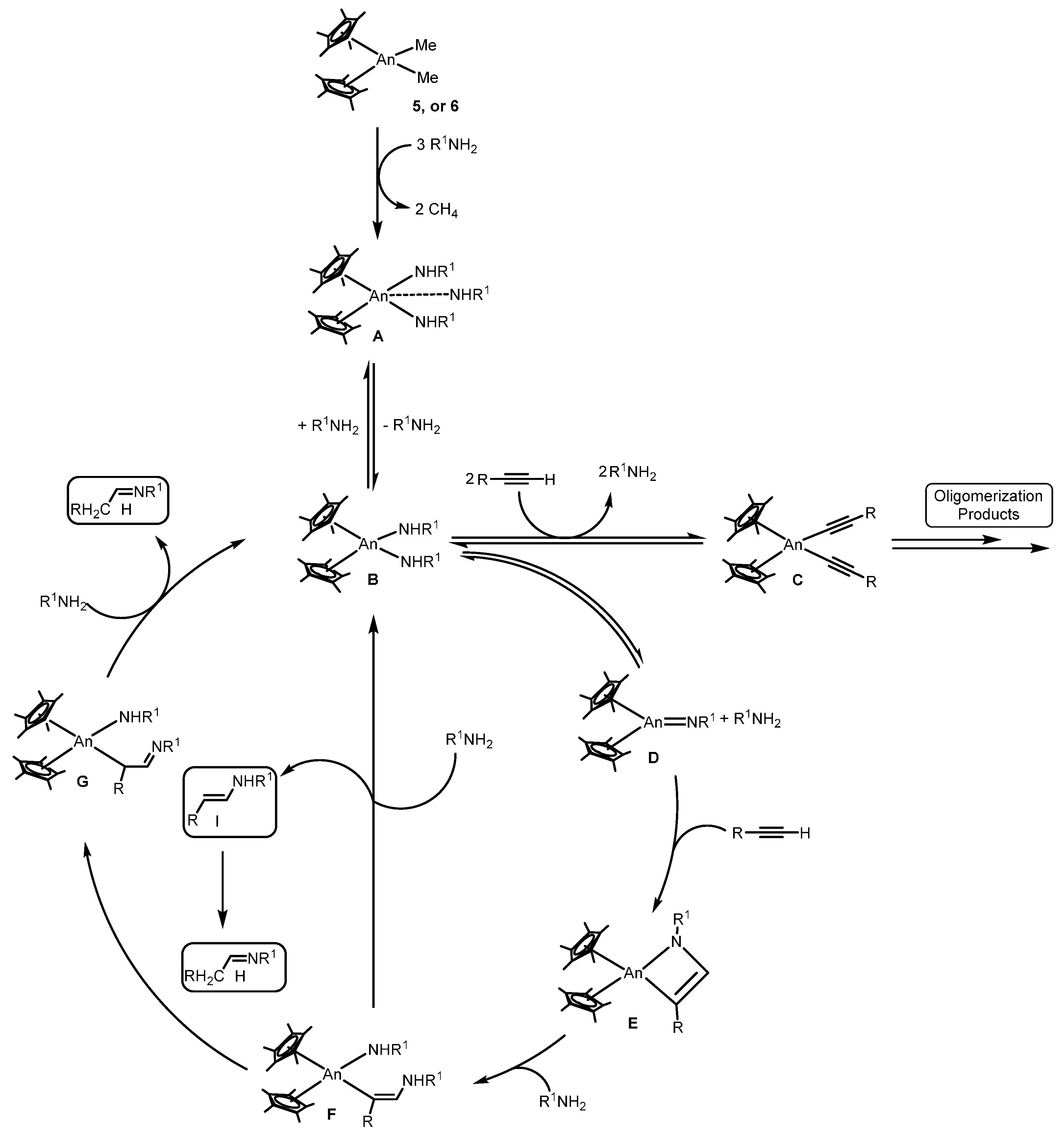
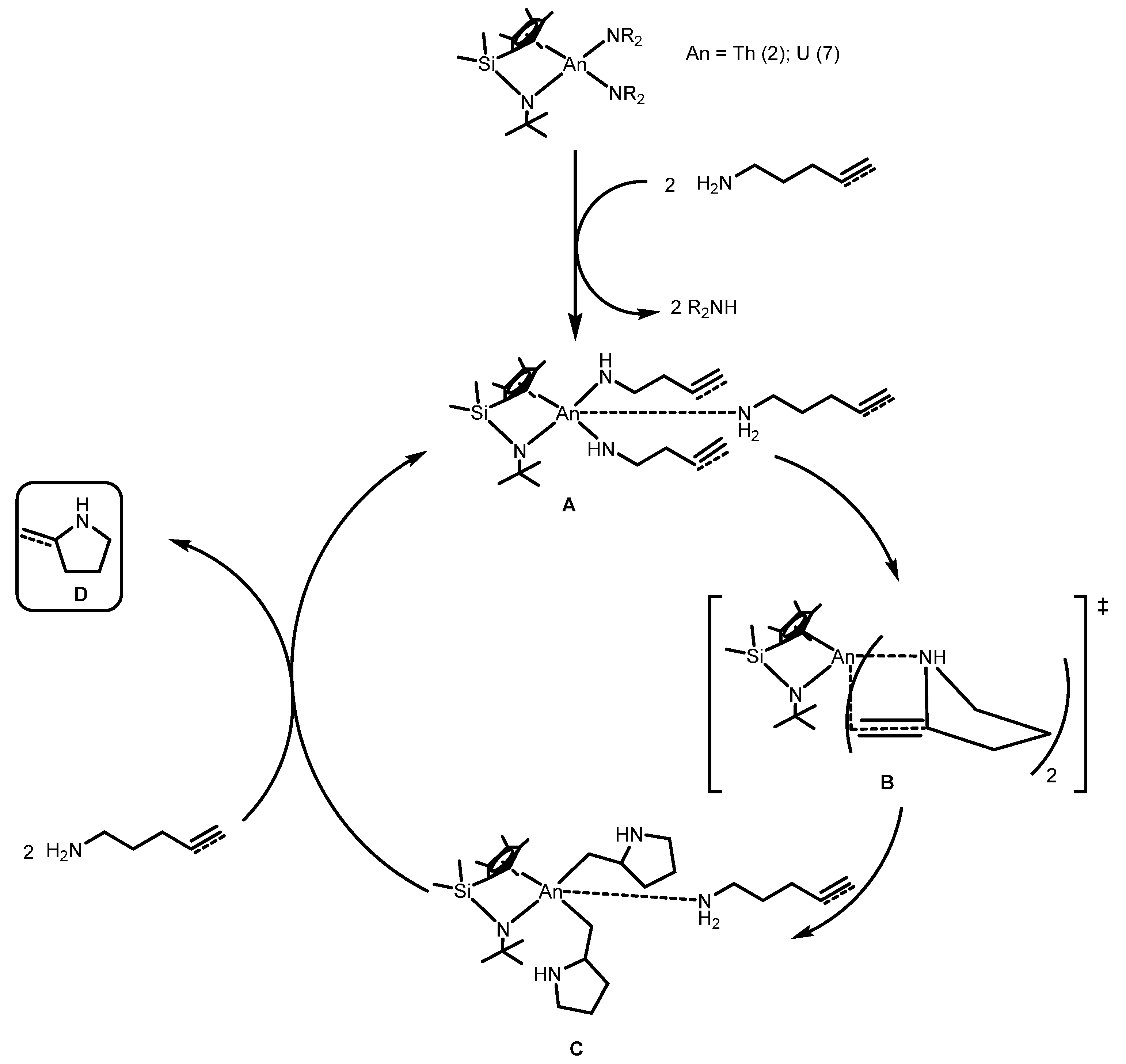
2.1.3. Inter- and Intra-Molecular Hydroamination
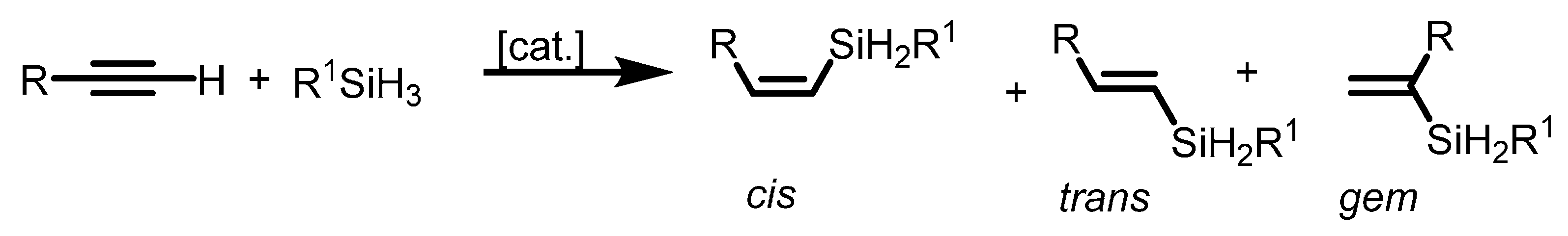
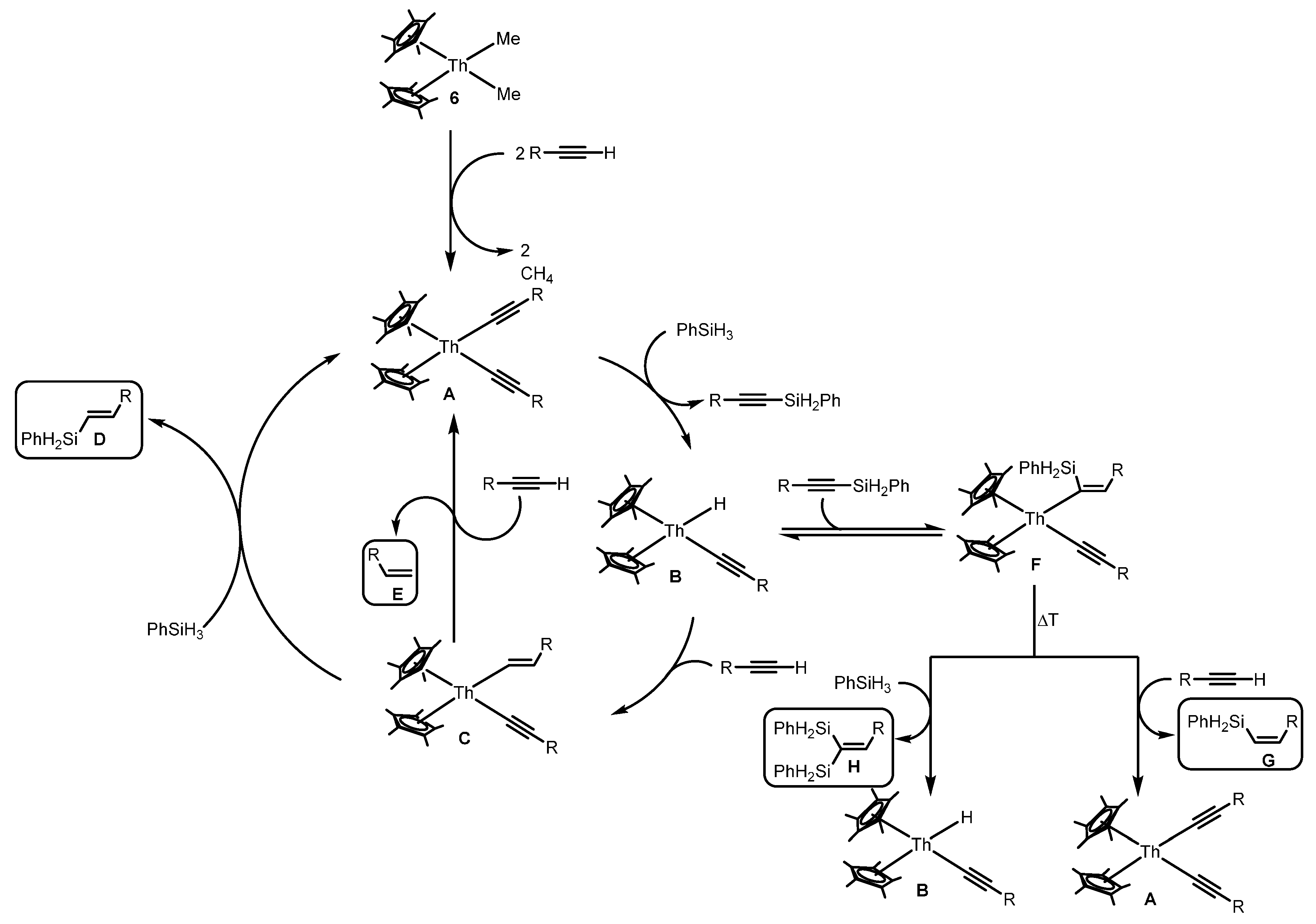
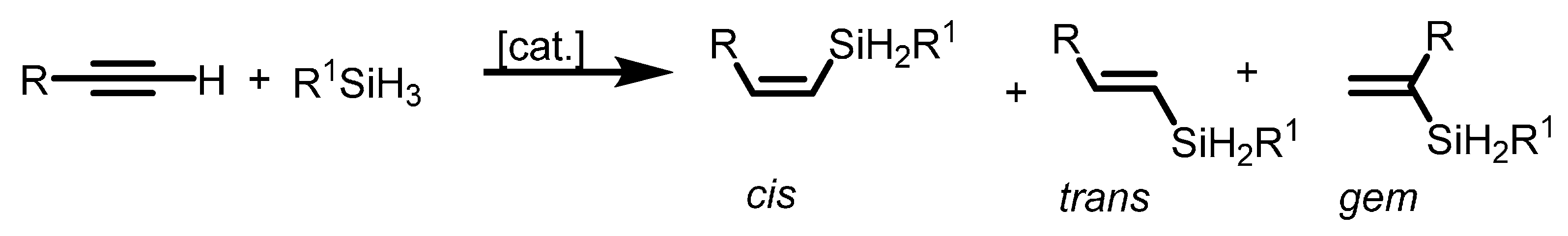
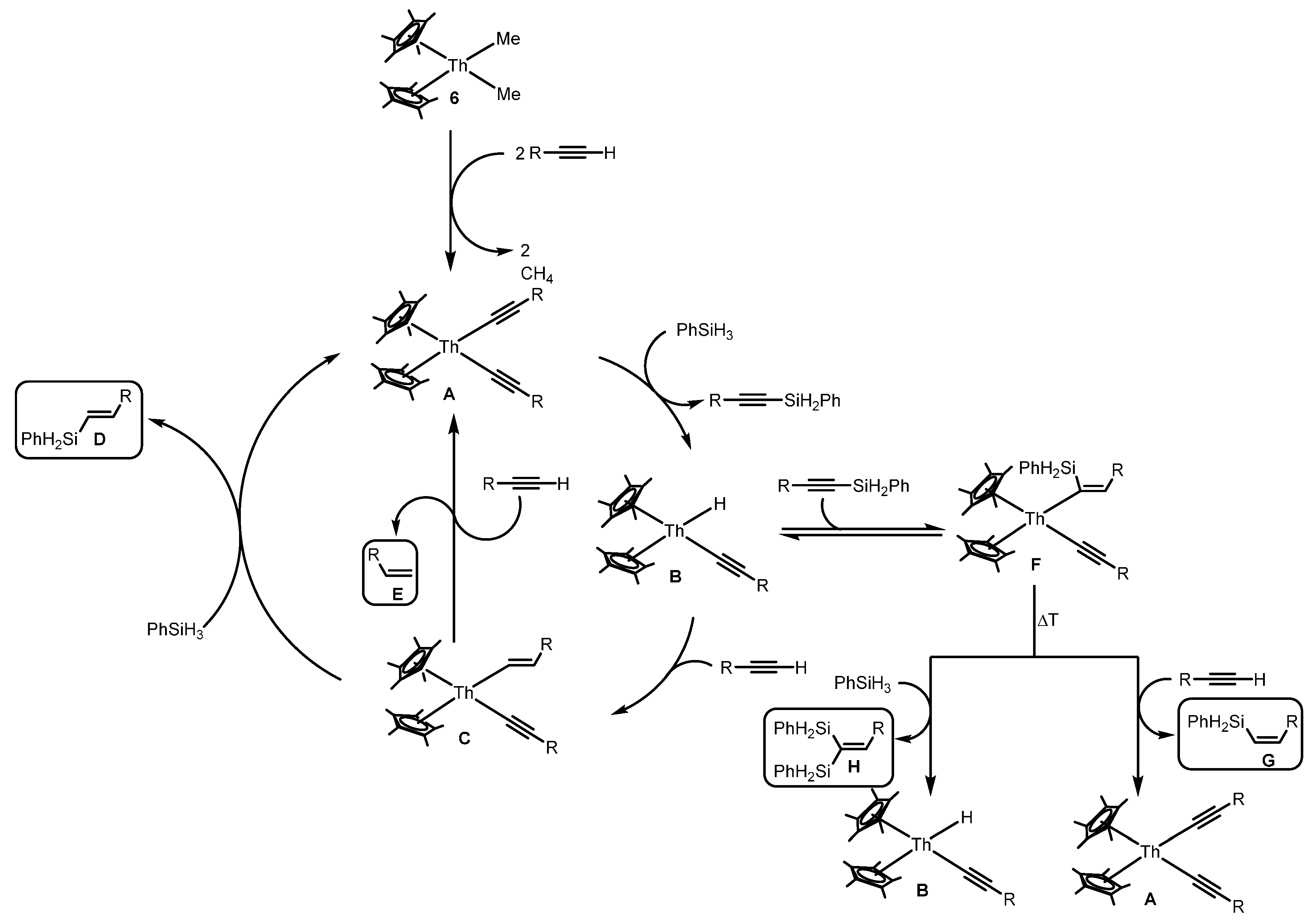
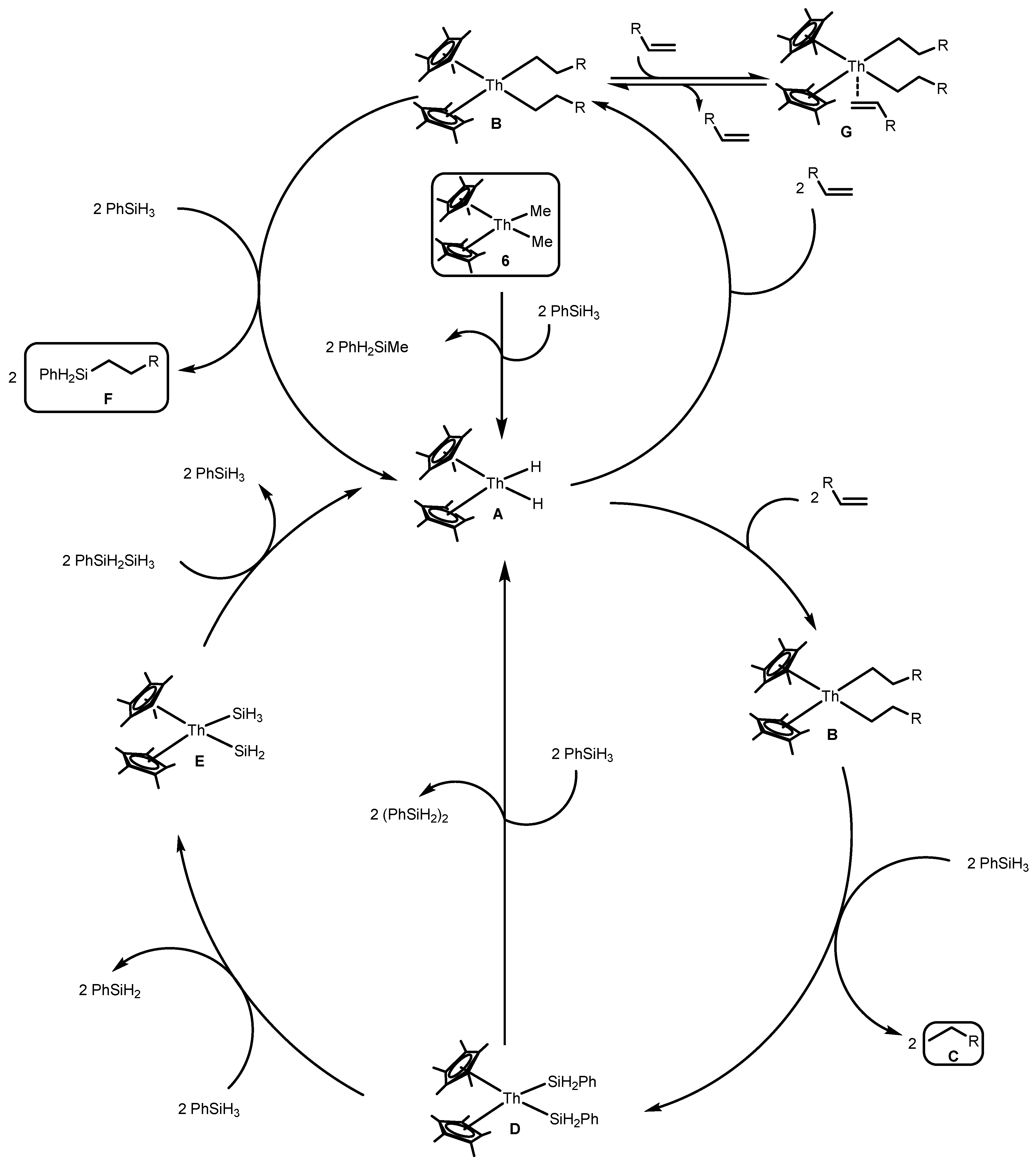
2.1.4. Hydrosilylation of Terminal Alkynes
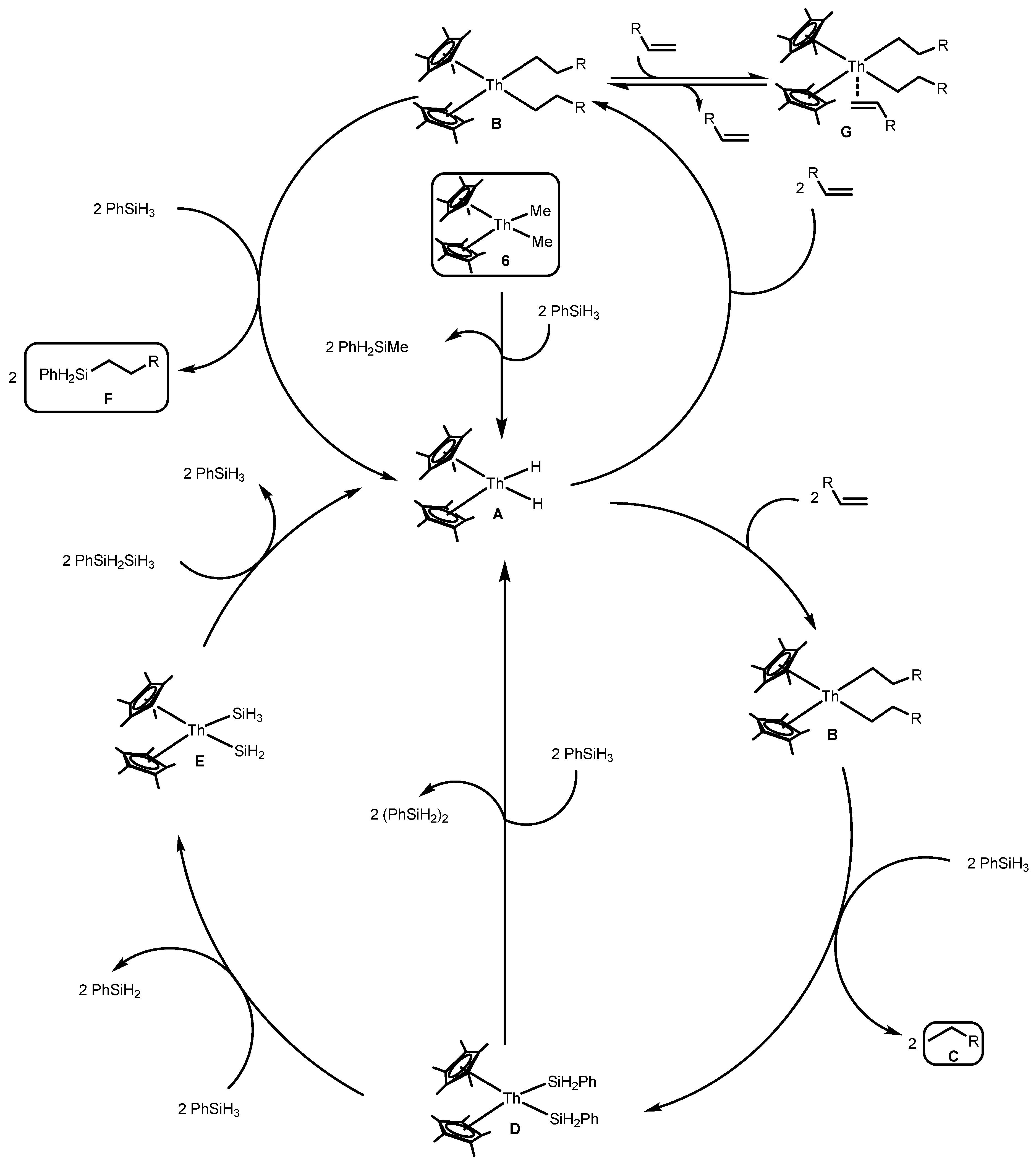
2.2. Coupling Reactions
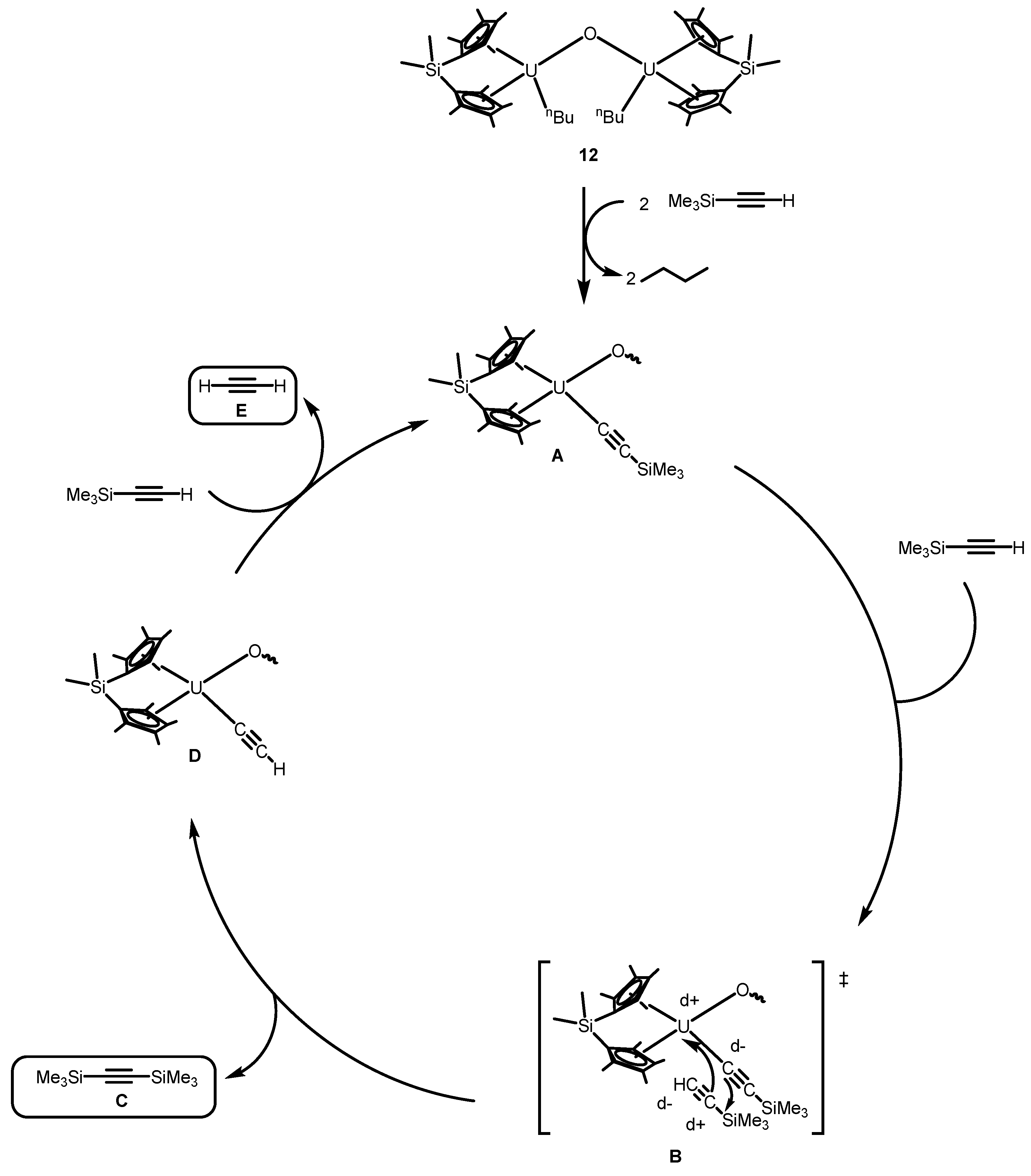
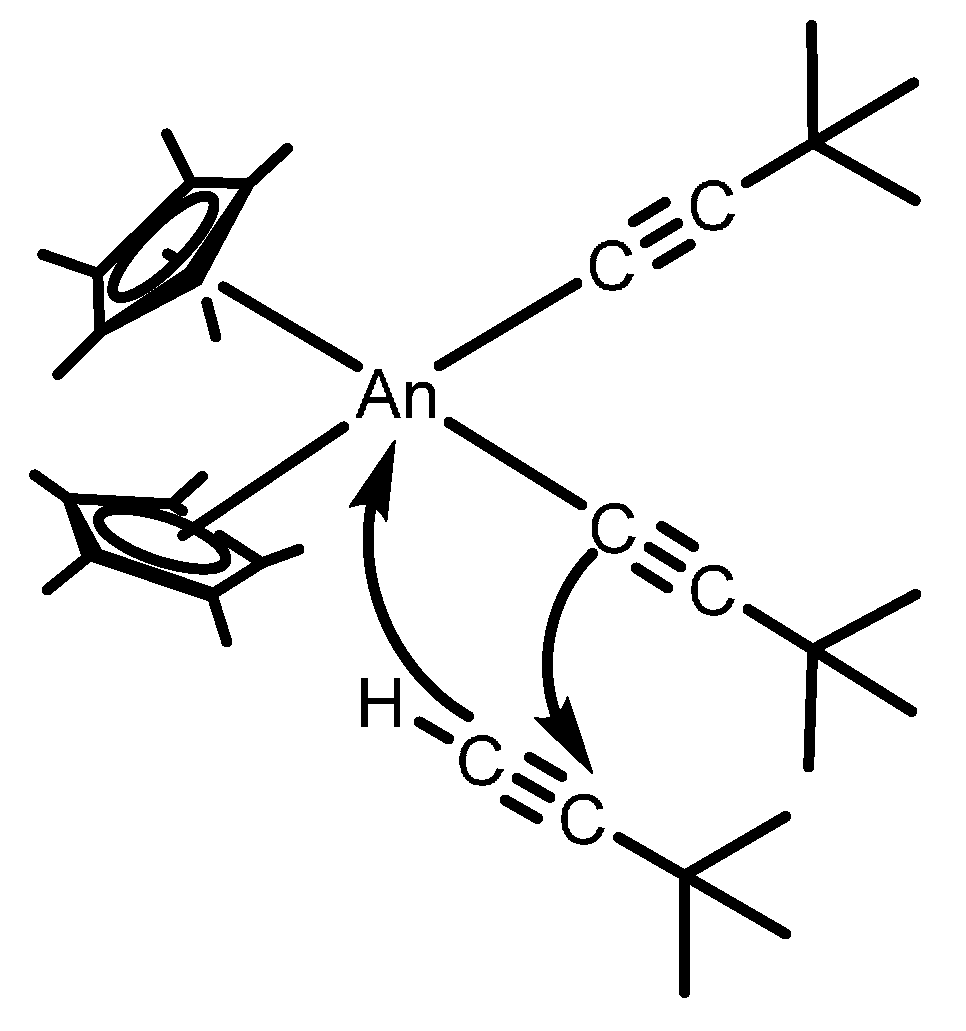
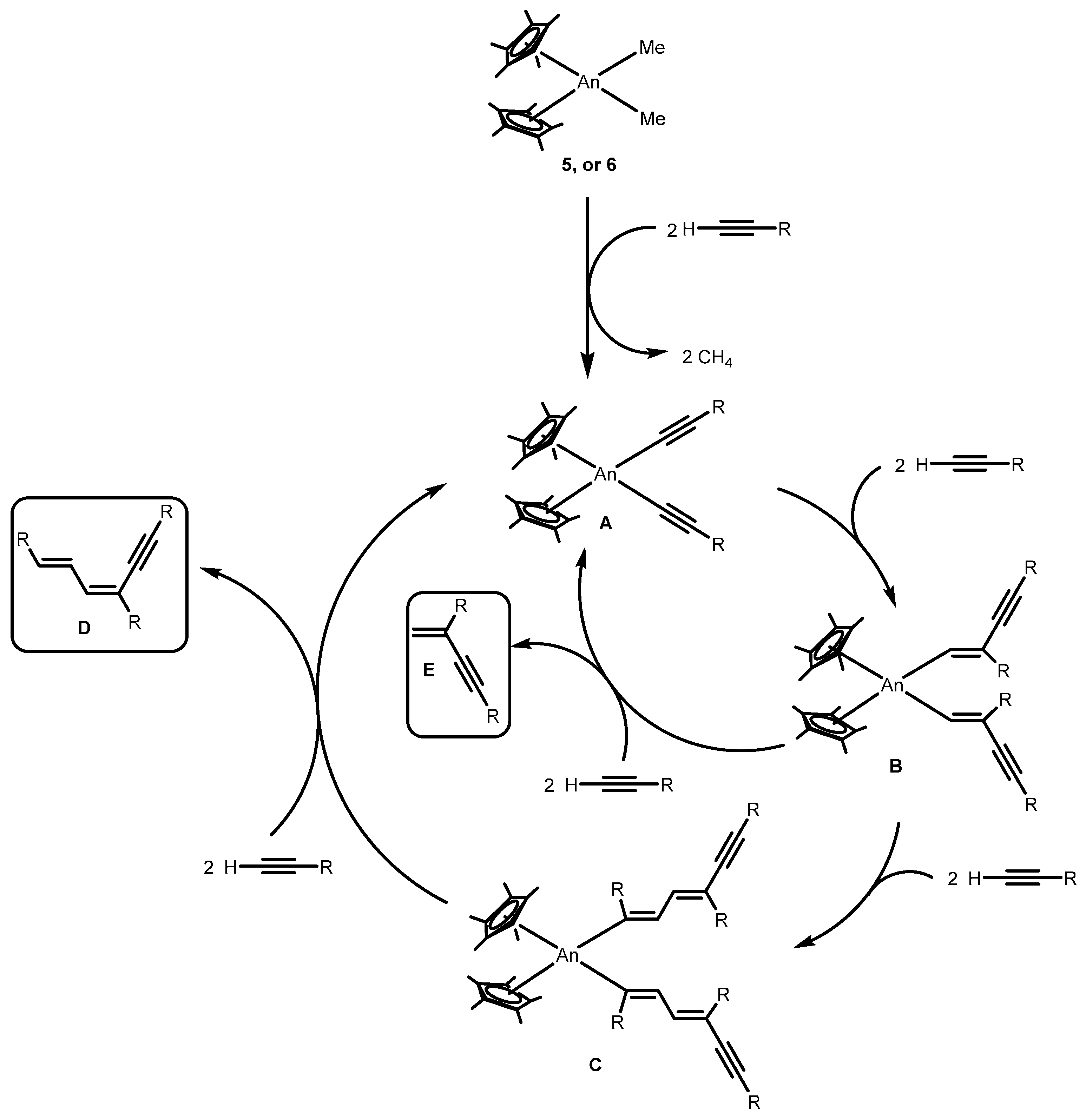
2.2.1. Coupling of Terminal Acetylenes
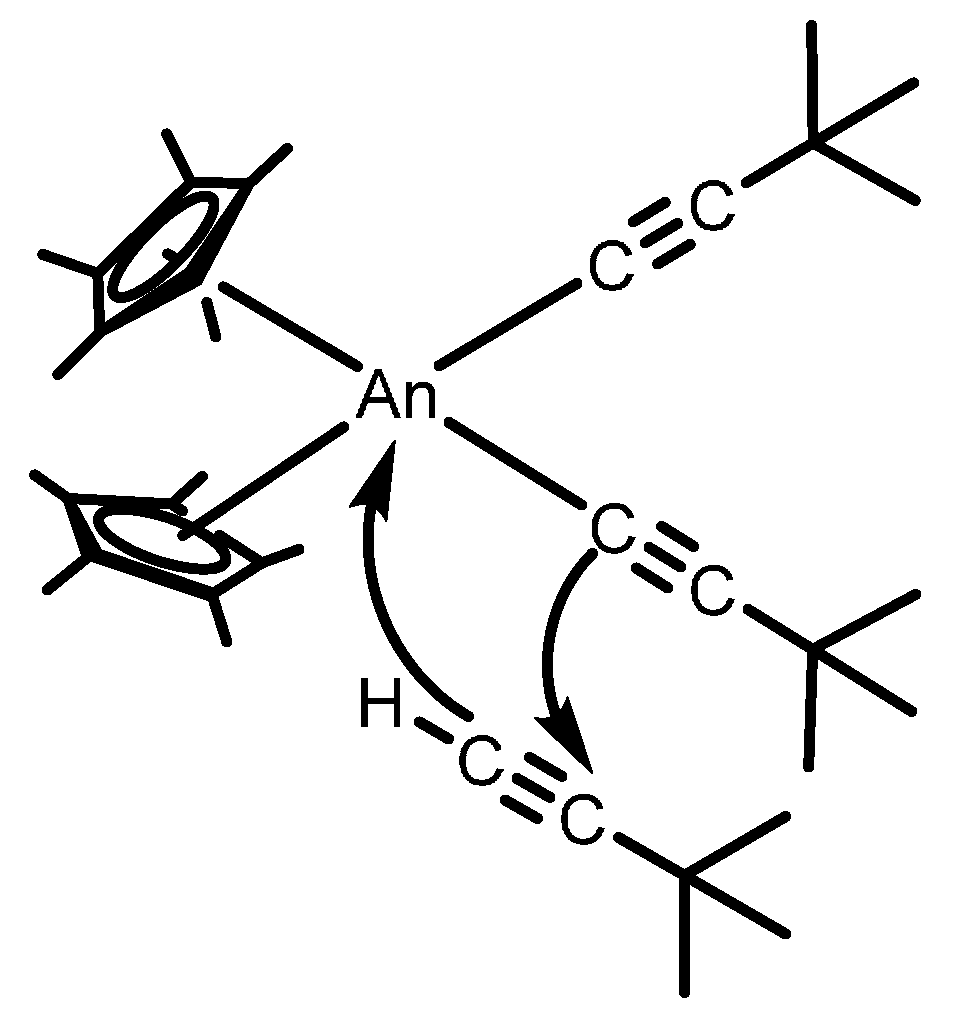
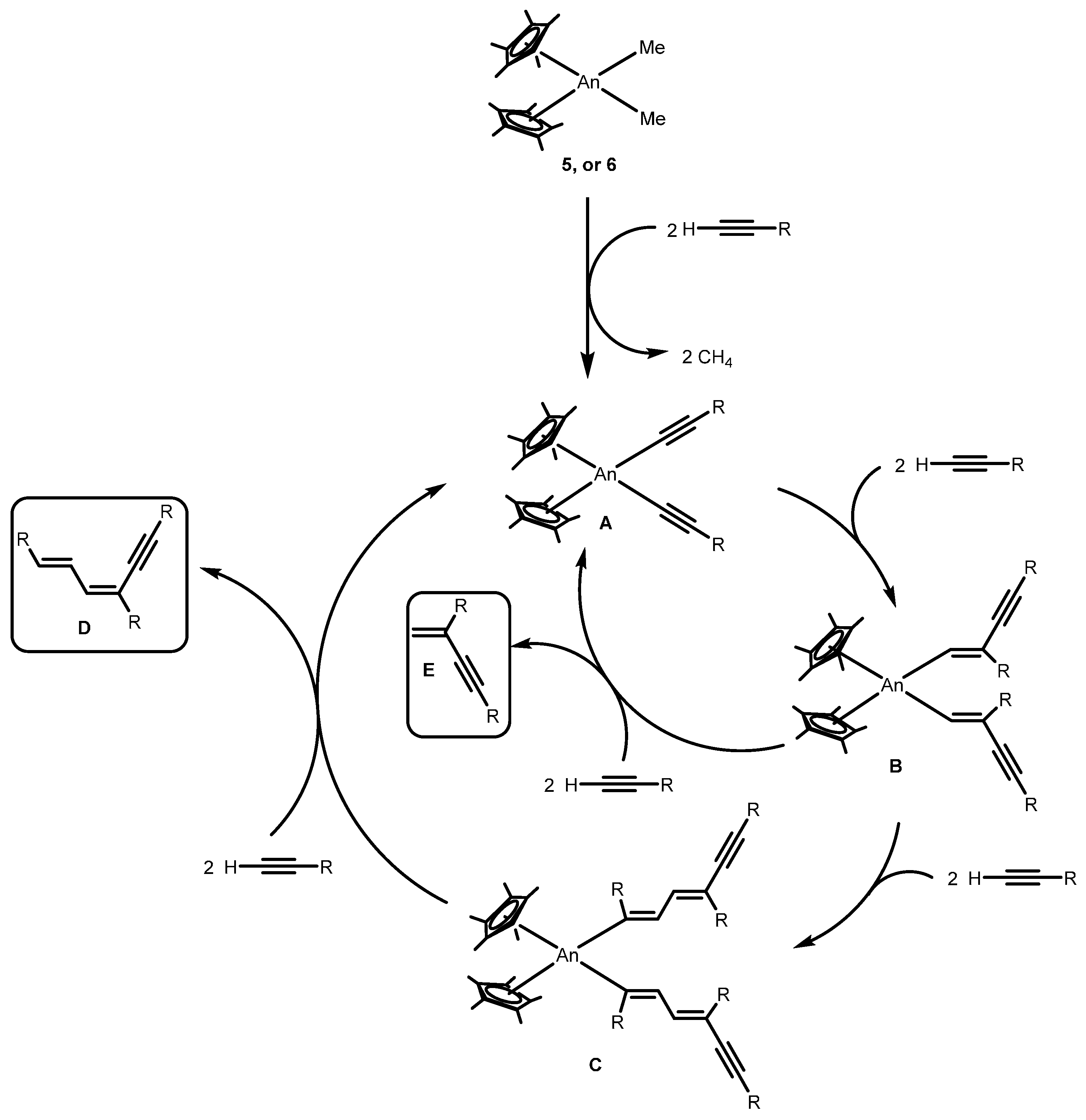
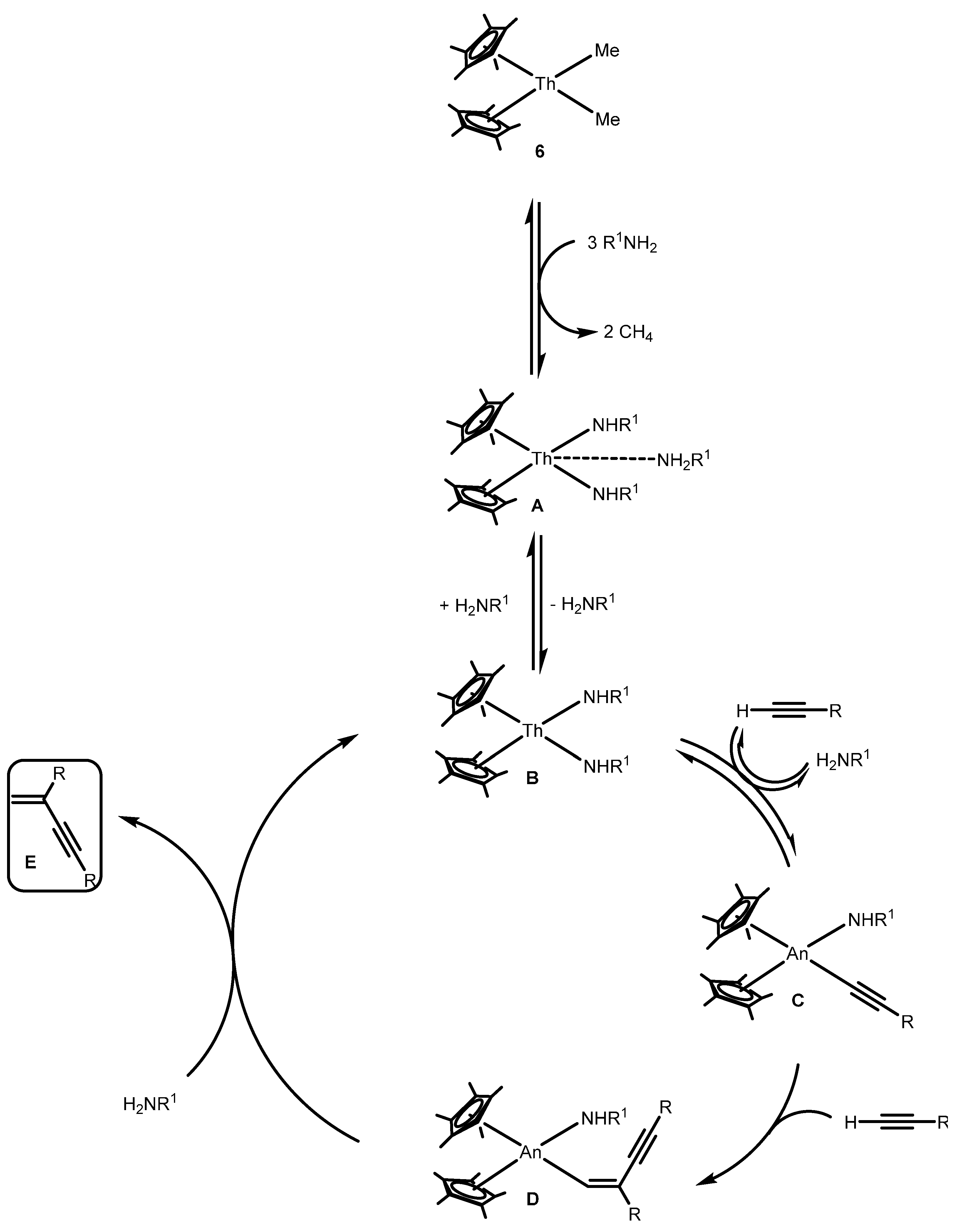
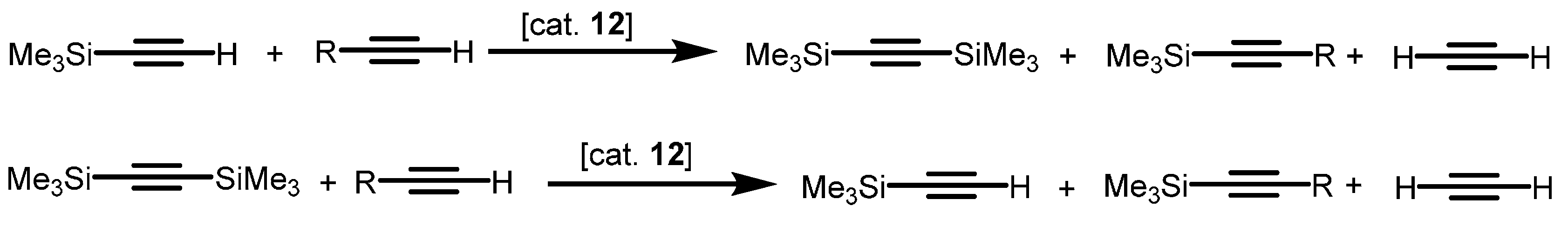
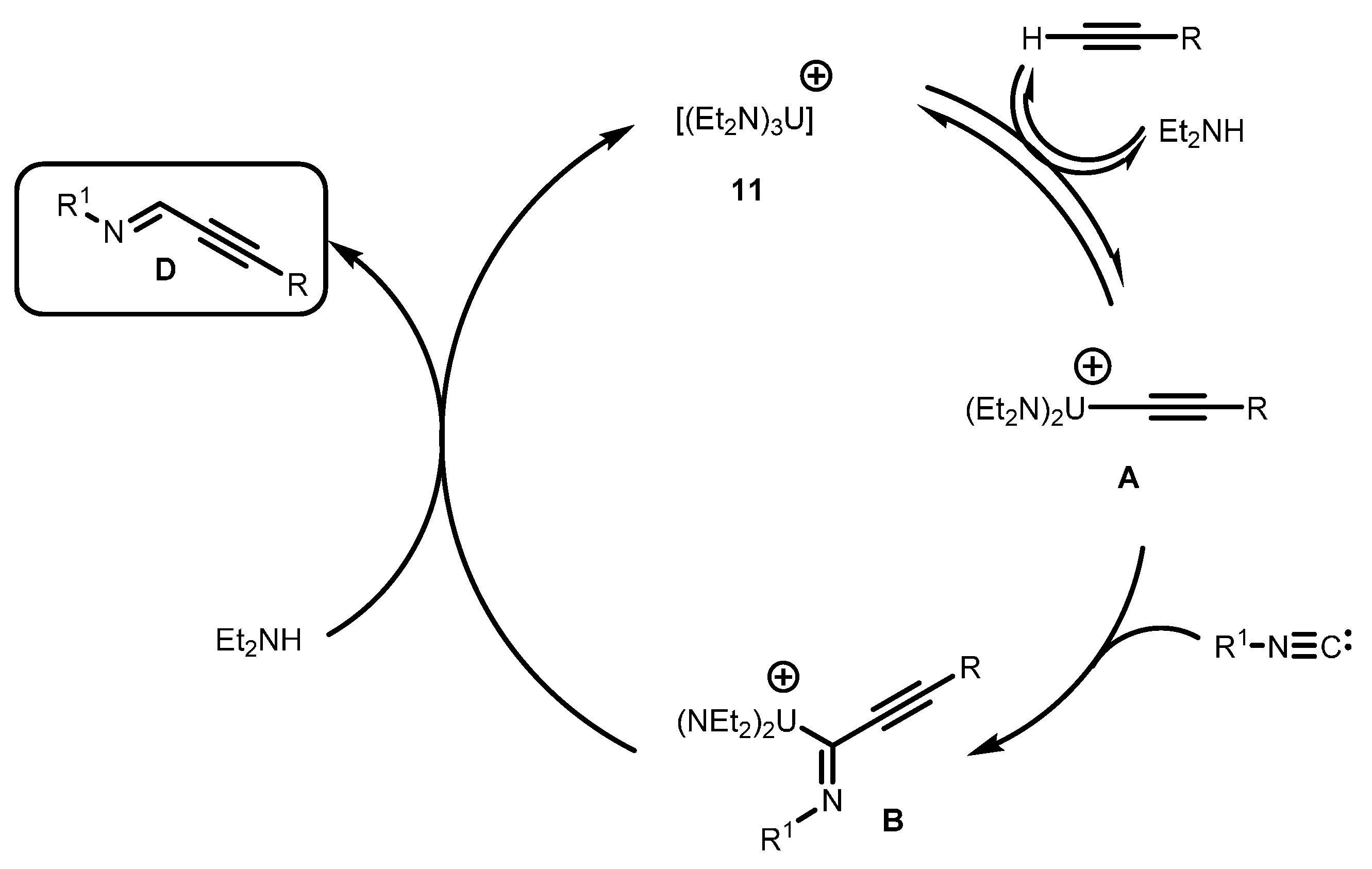
2.2.2. Coupling of Terminal Alkynes and Isonitriles

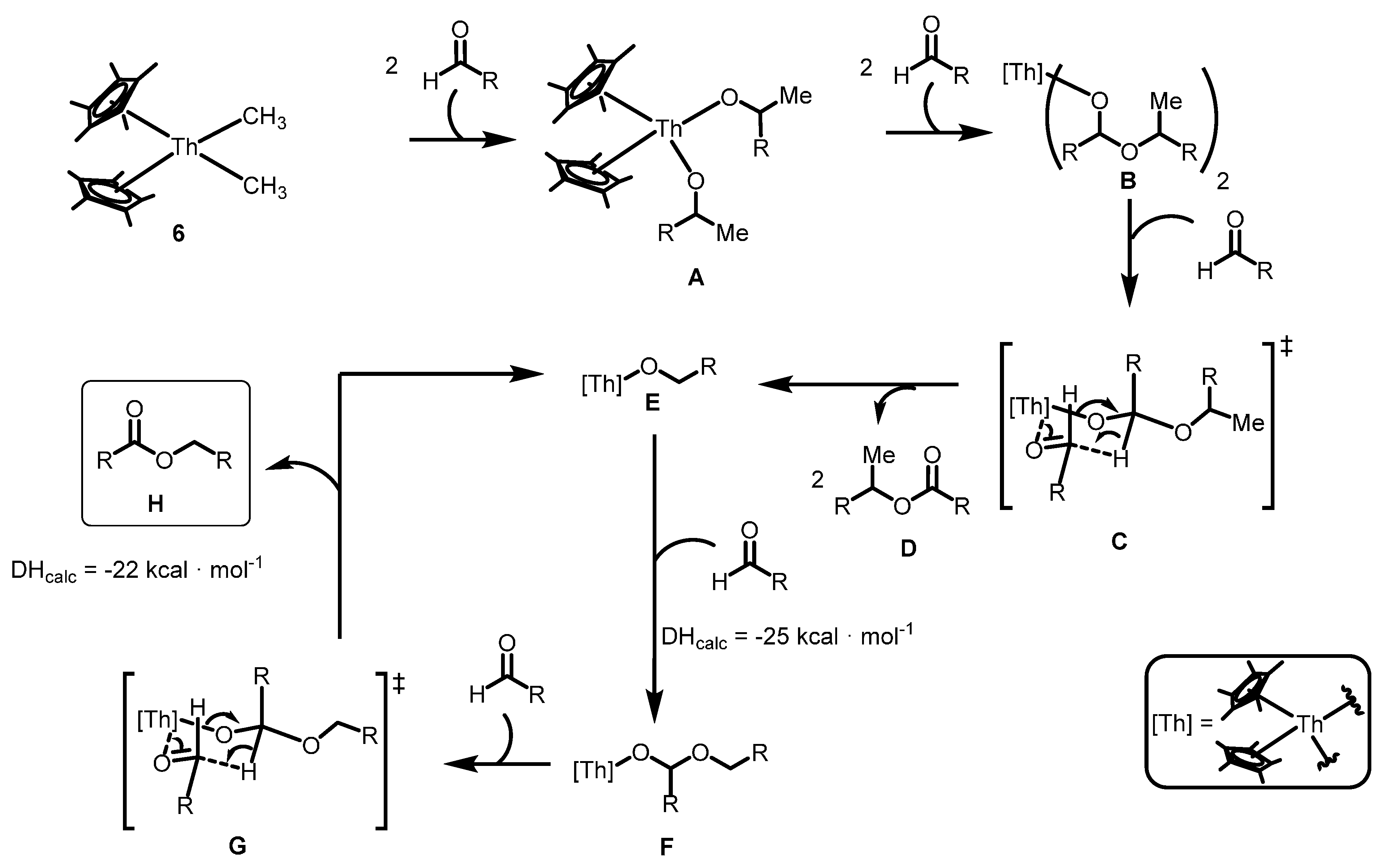
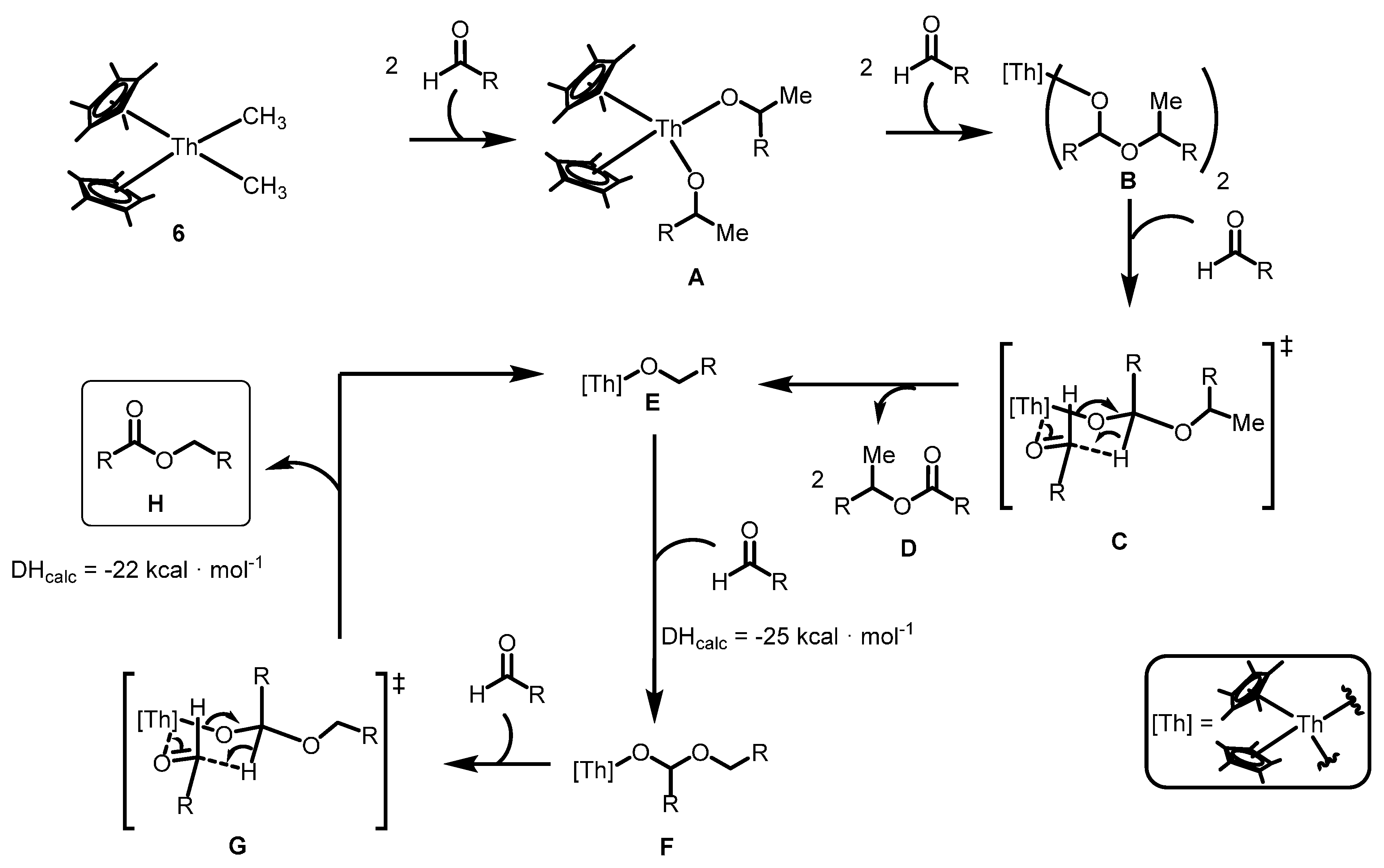
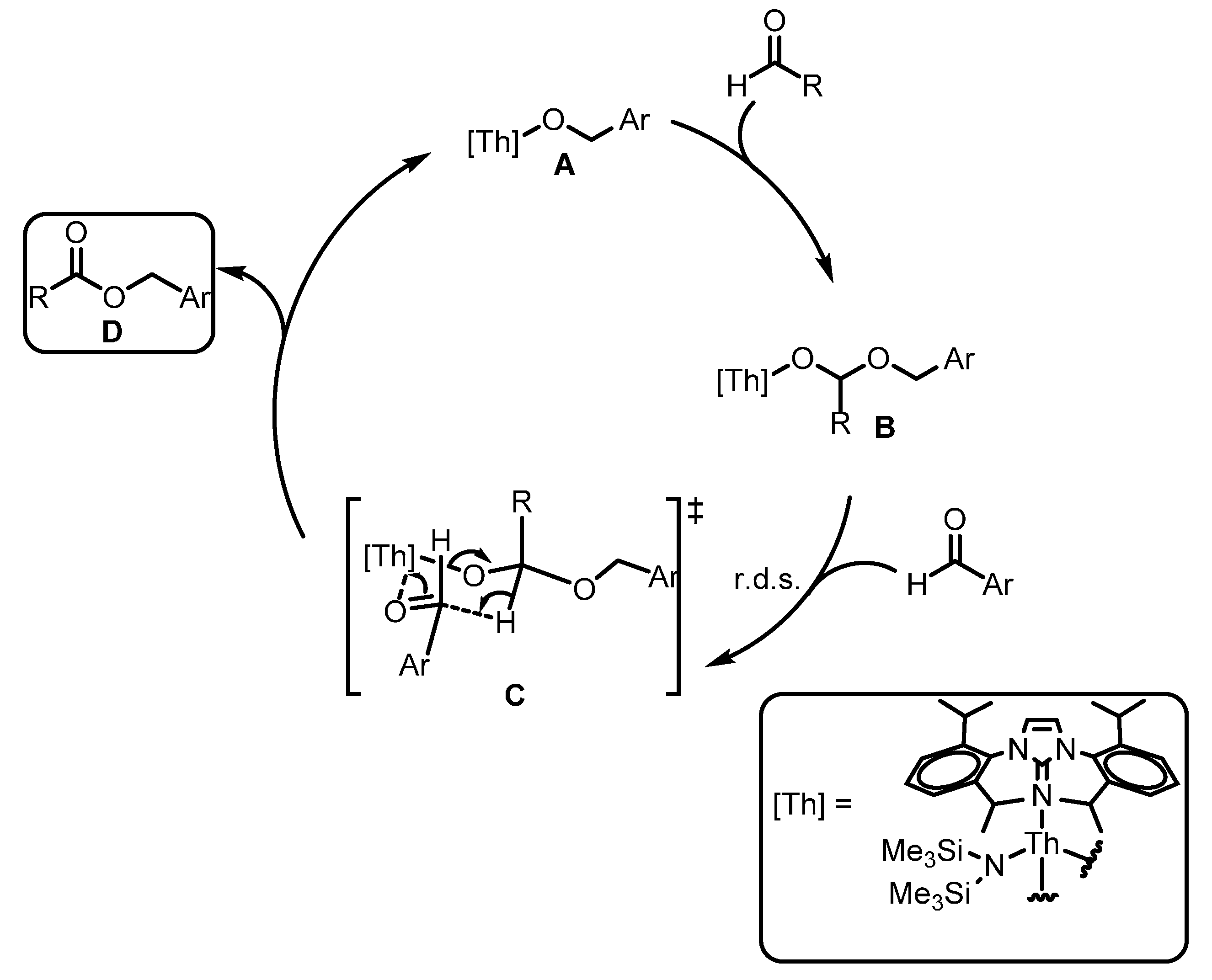
2.2.3. Coupling of Aldehydes: Tishchenko Reaction
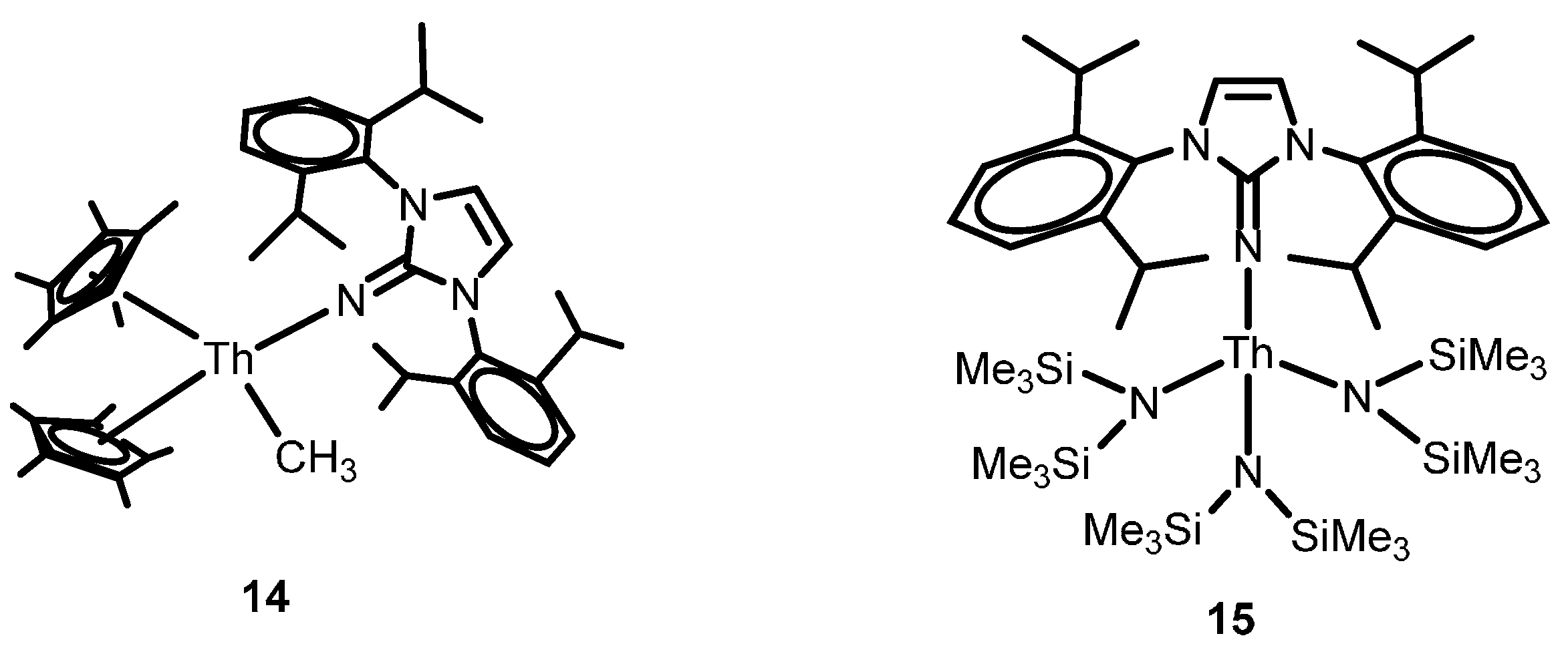
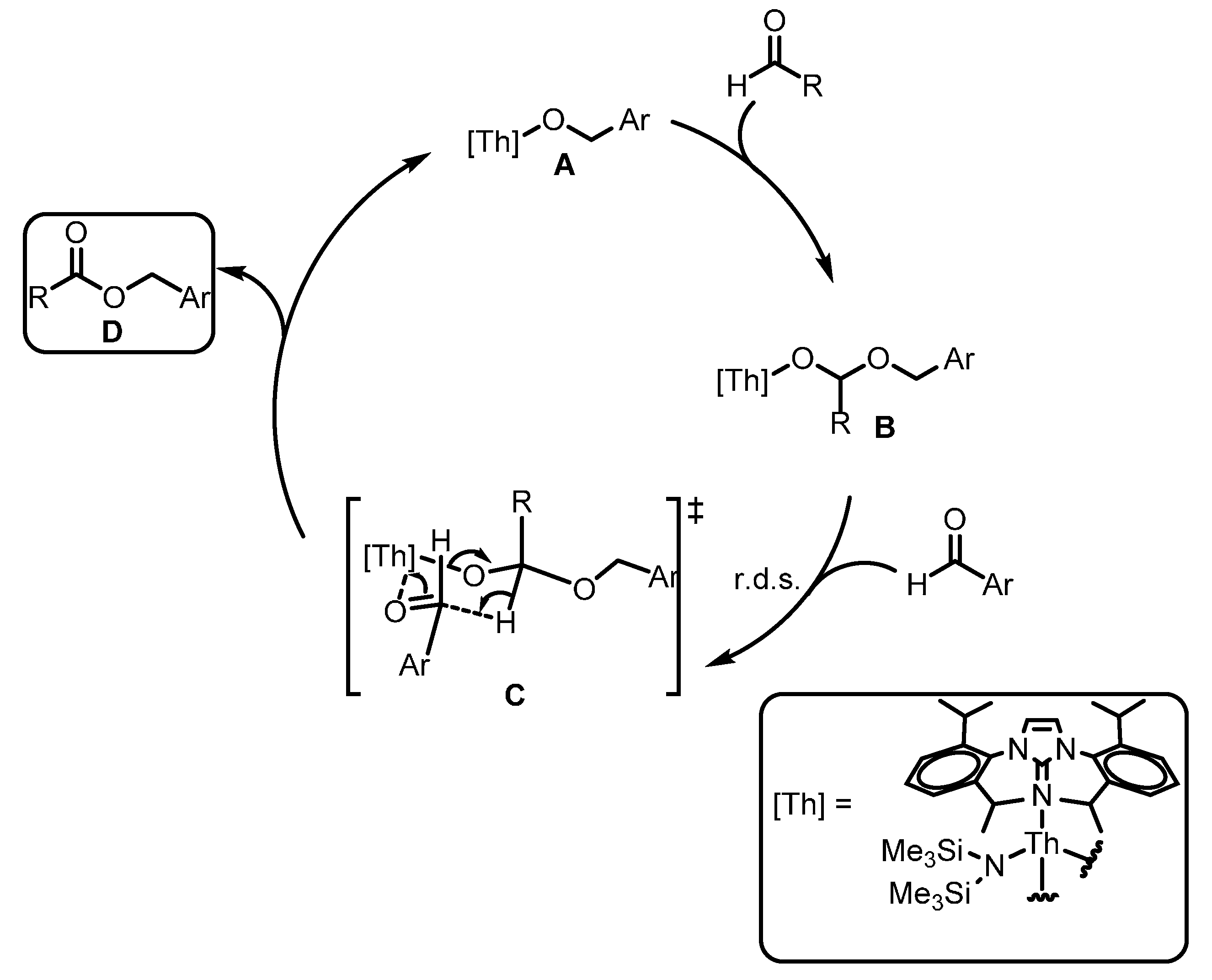
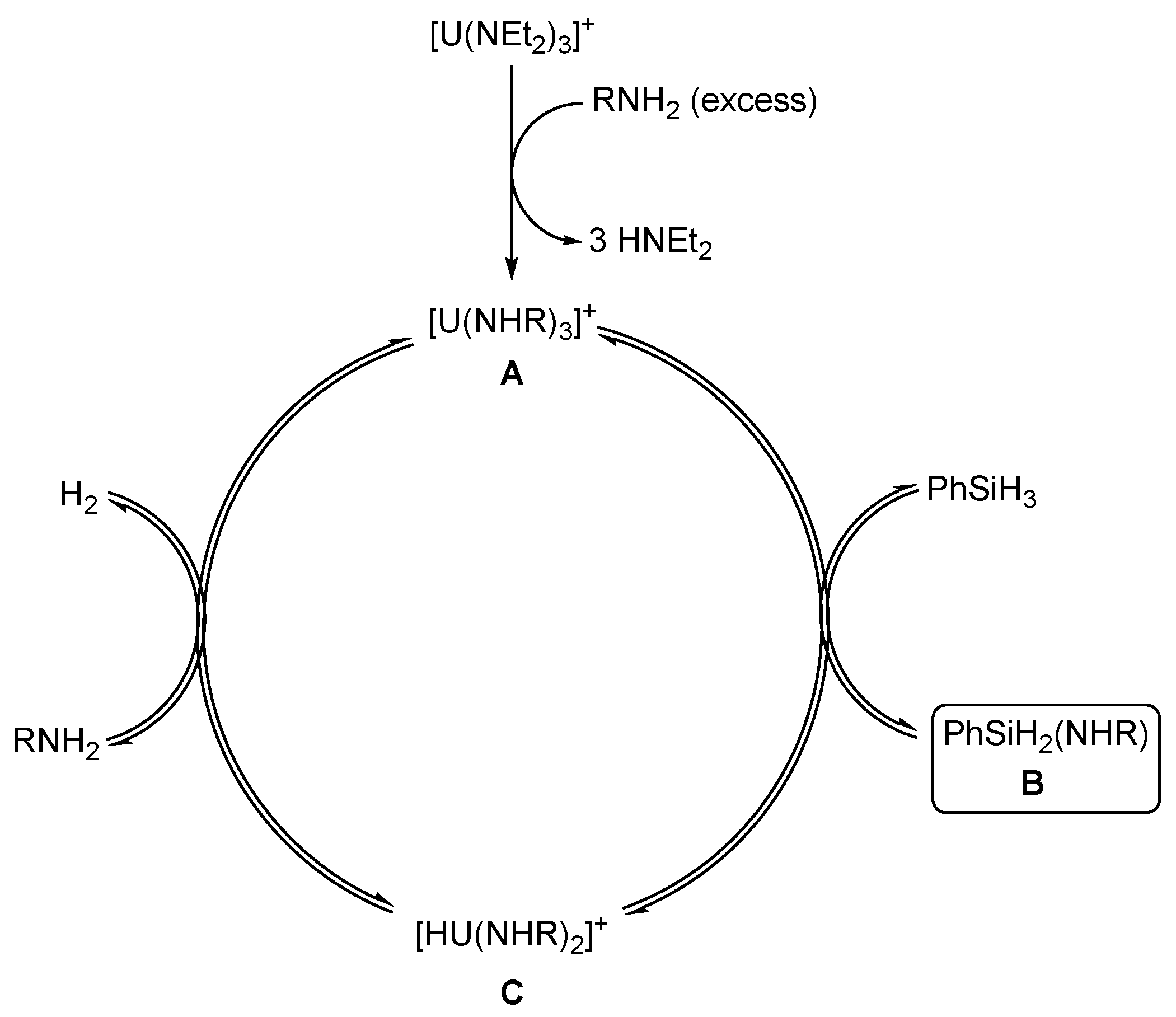
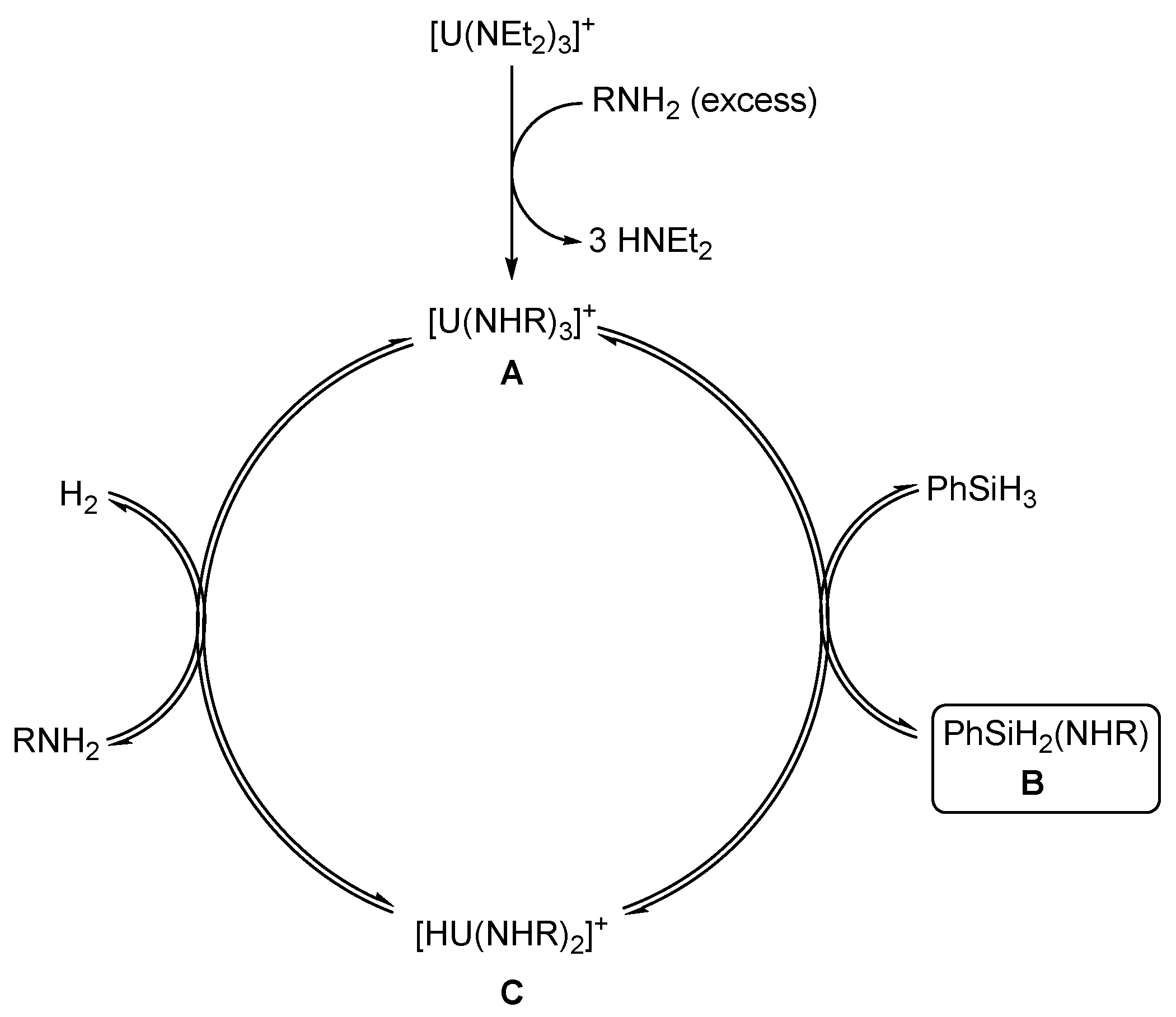
2.2.4. Dehydrocoupling of Amines with Silanes
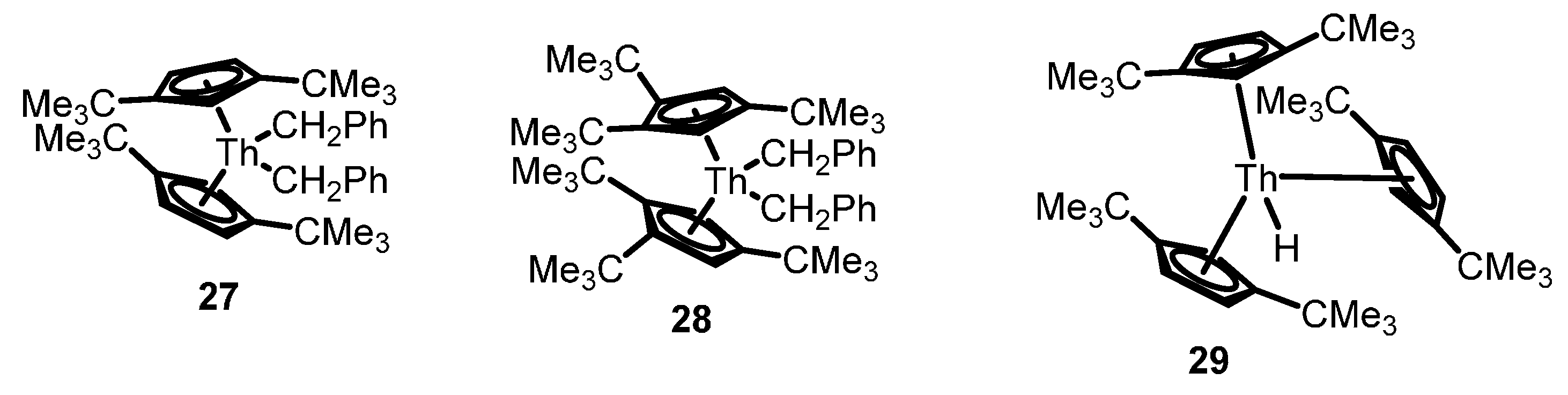
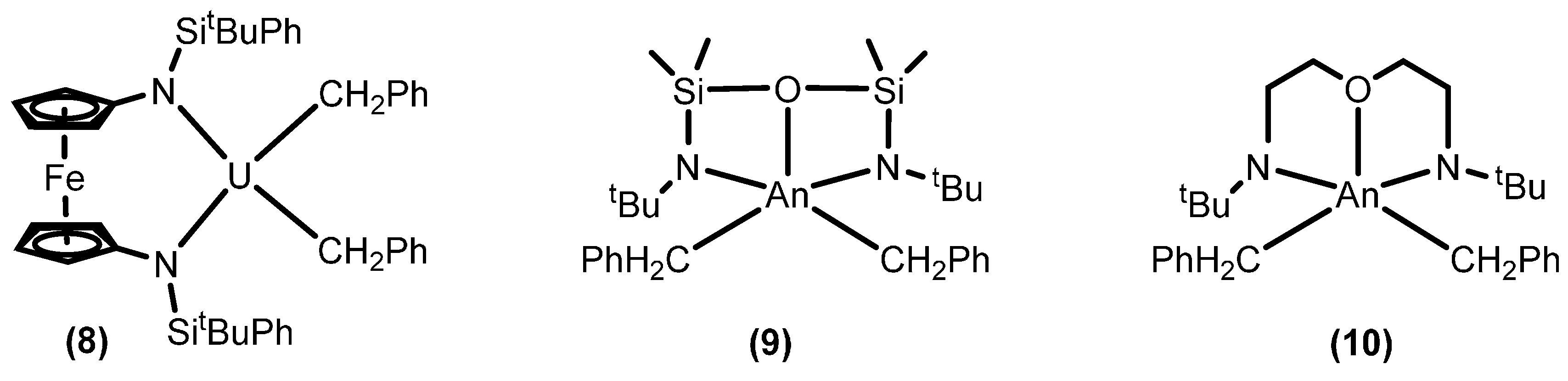
2.3. Polymerization Reactions
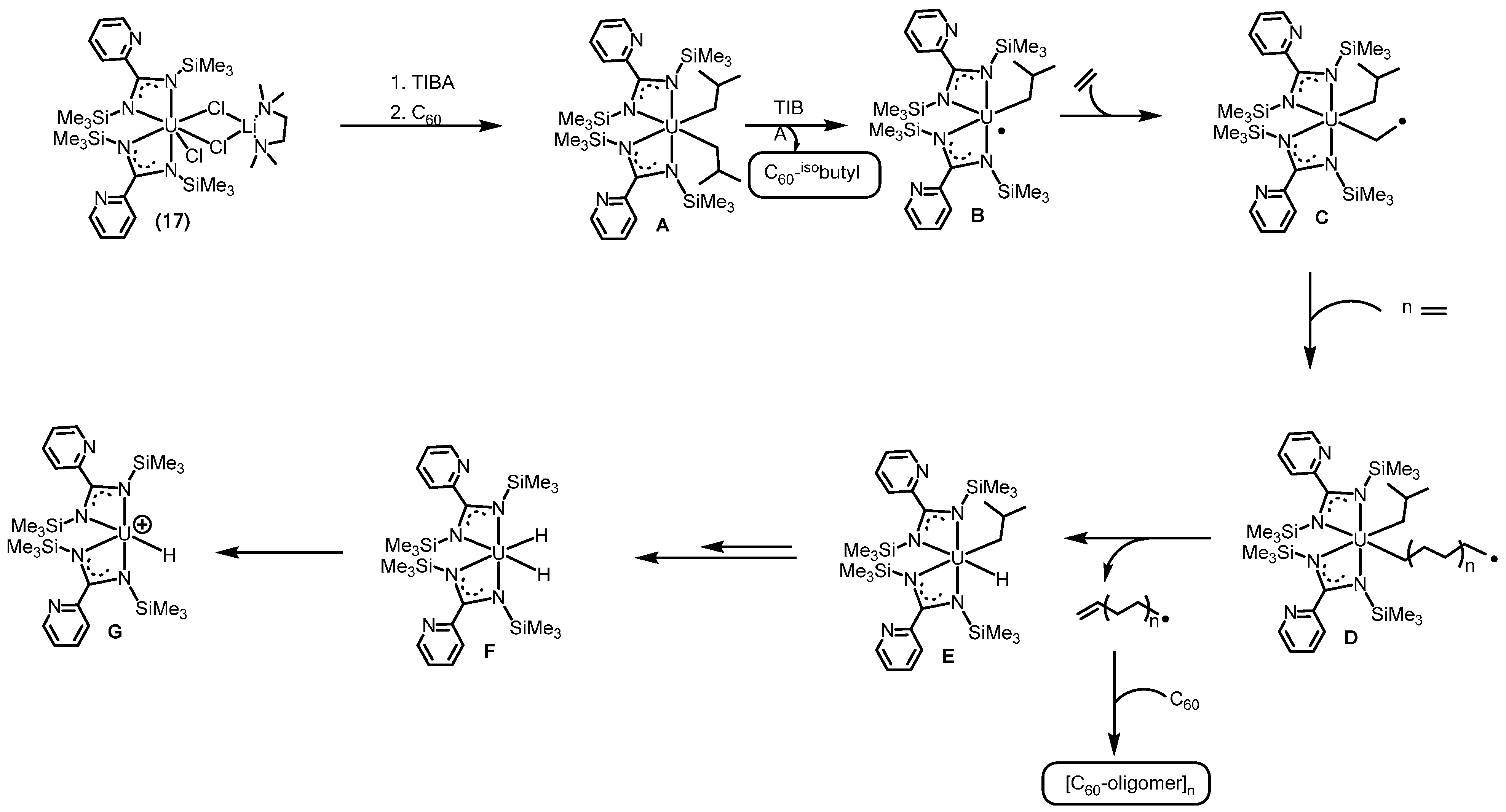
2.3.1. Polymerization of α-Olefins
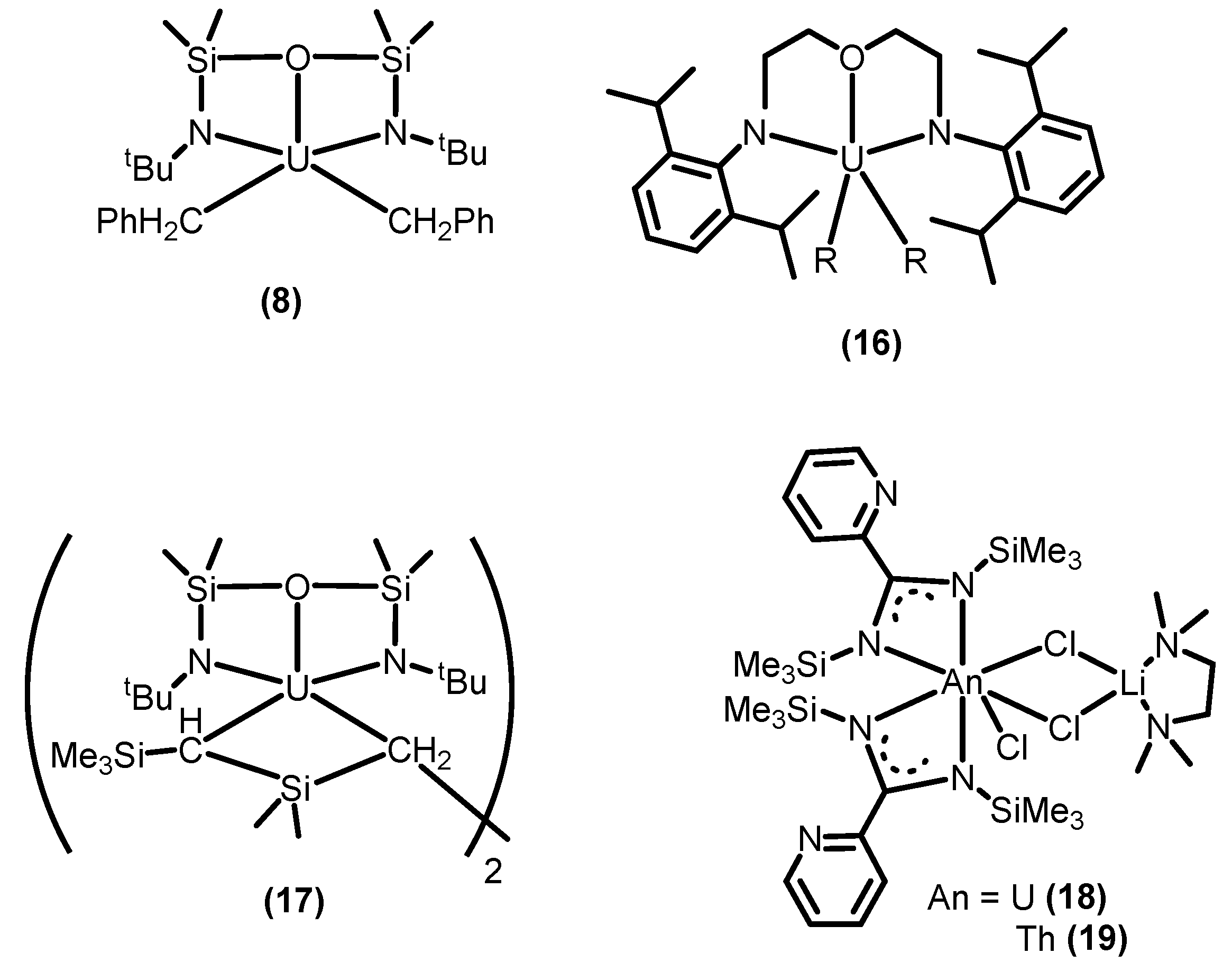
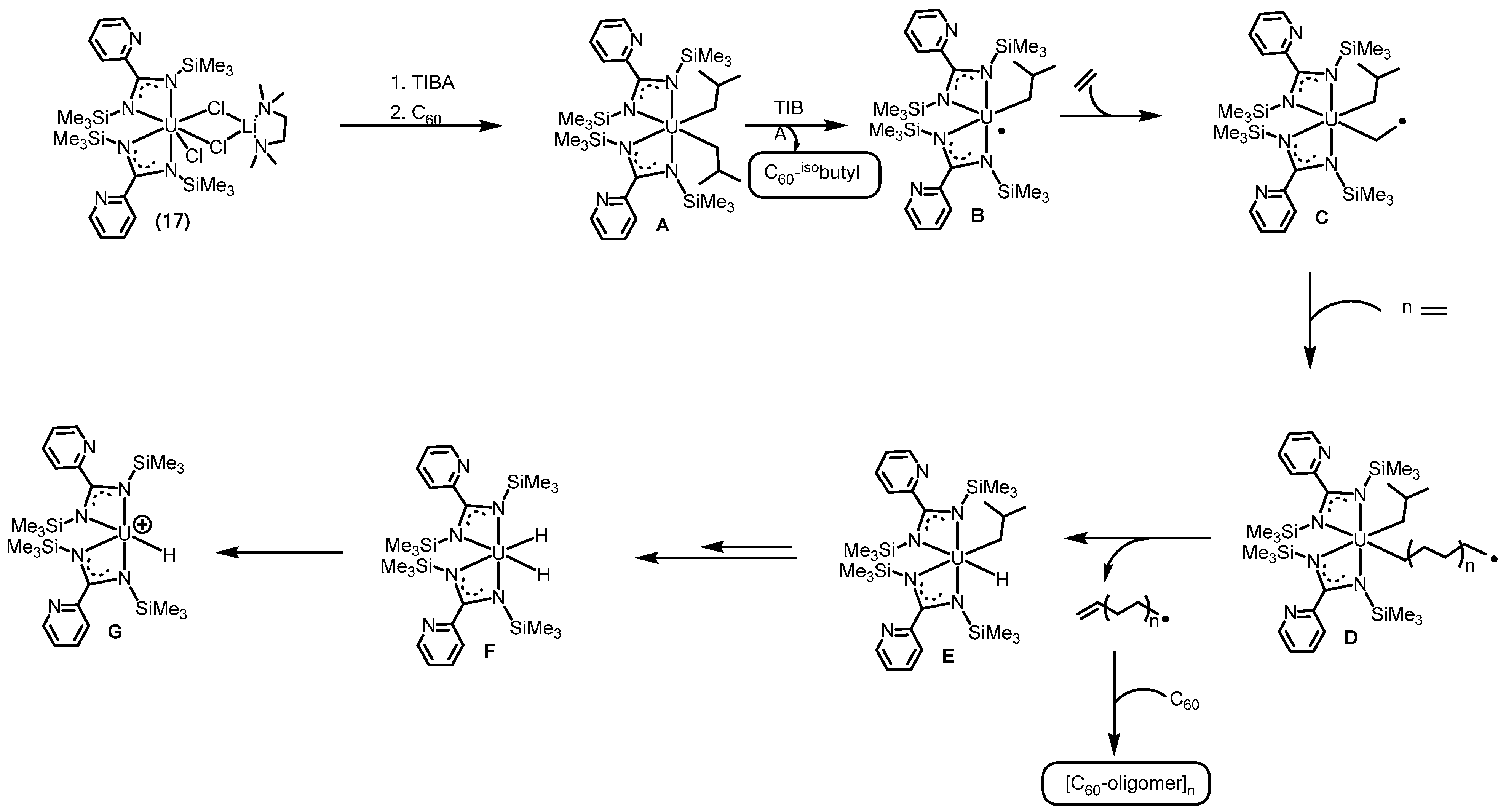
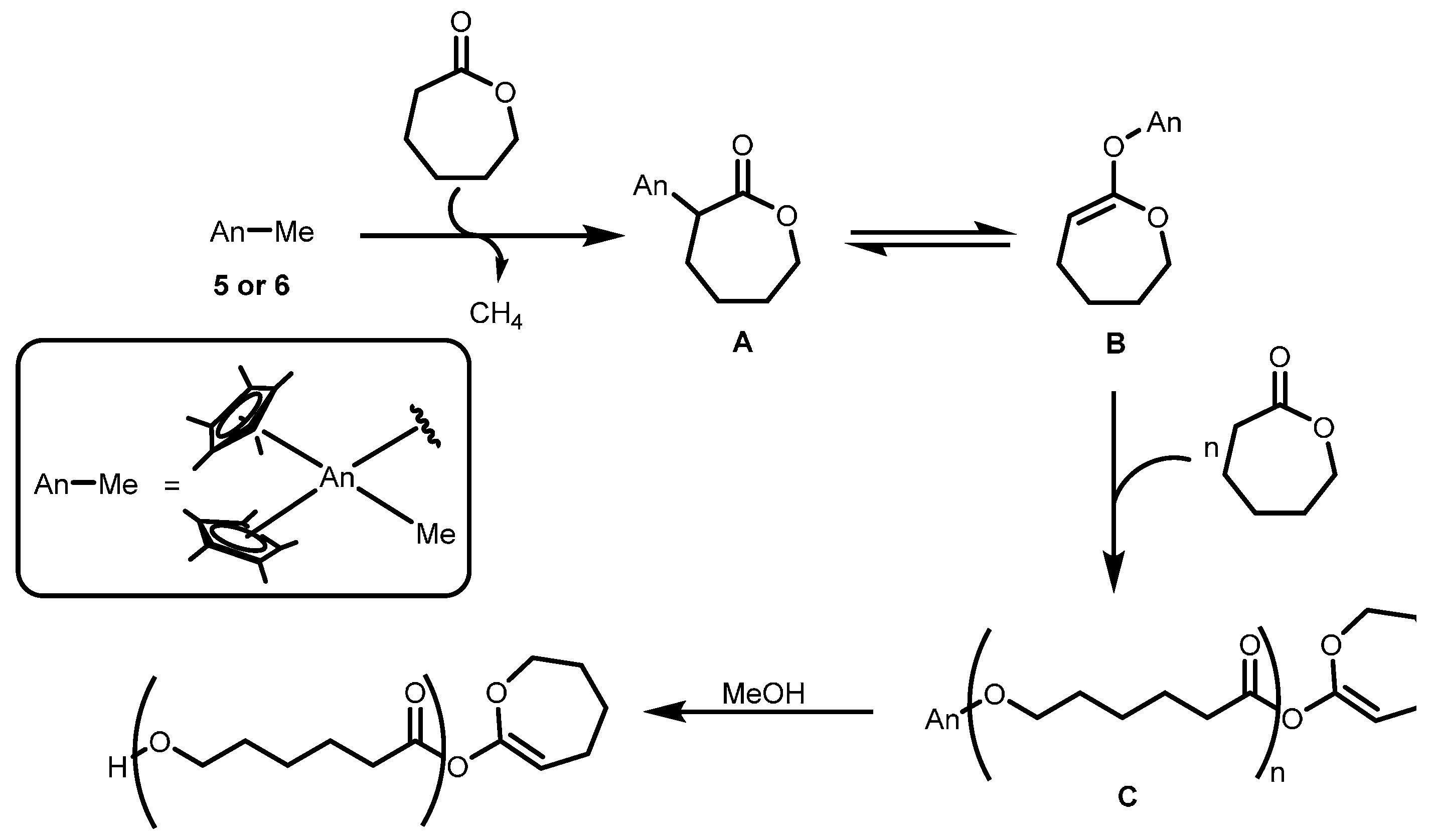
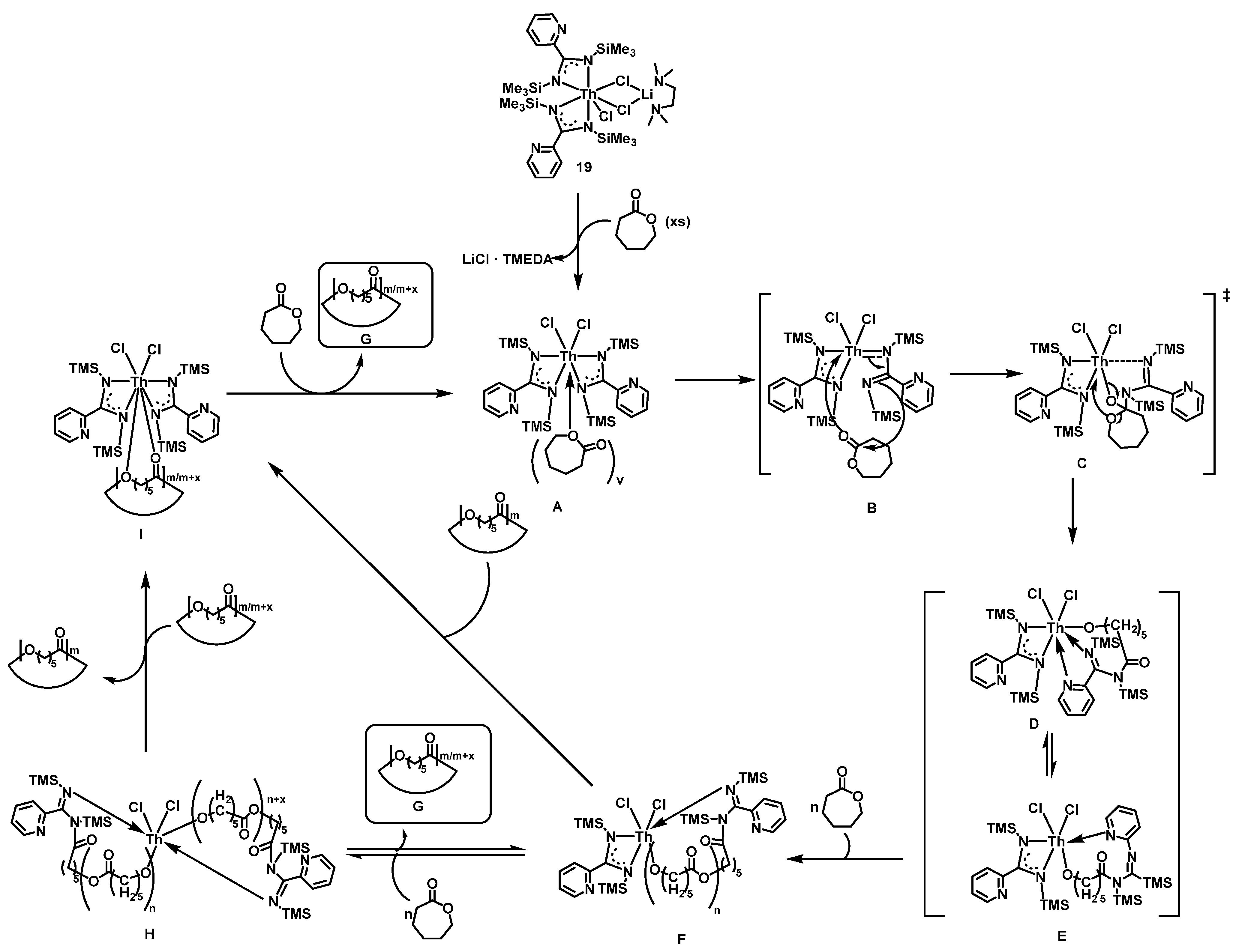
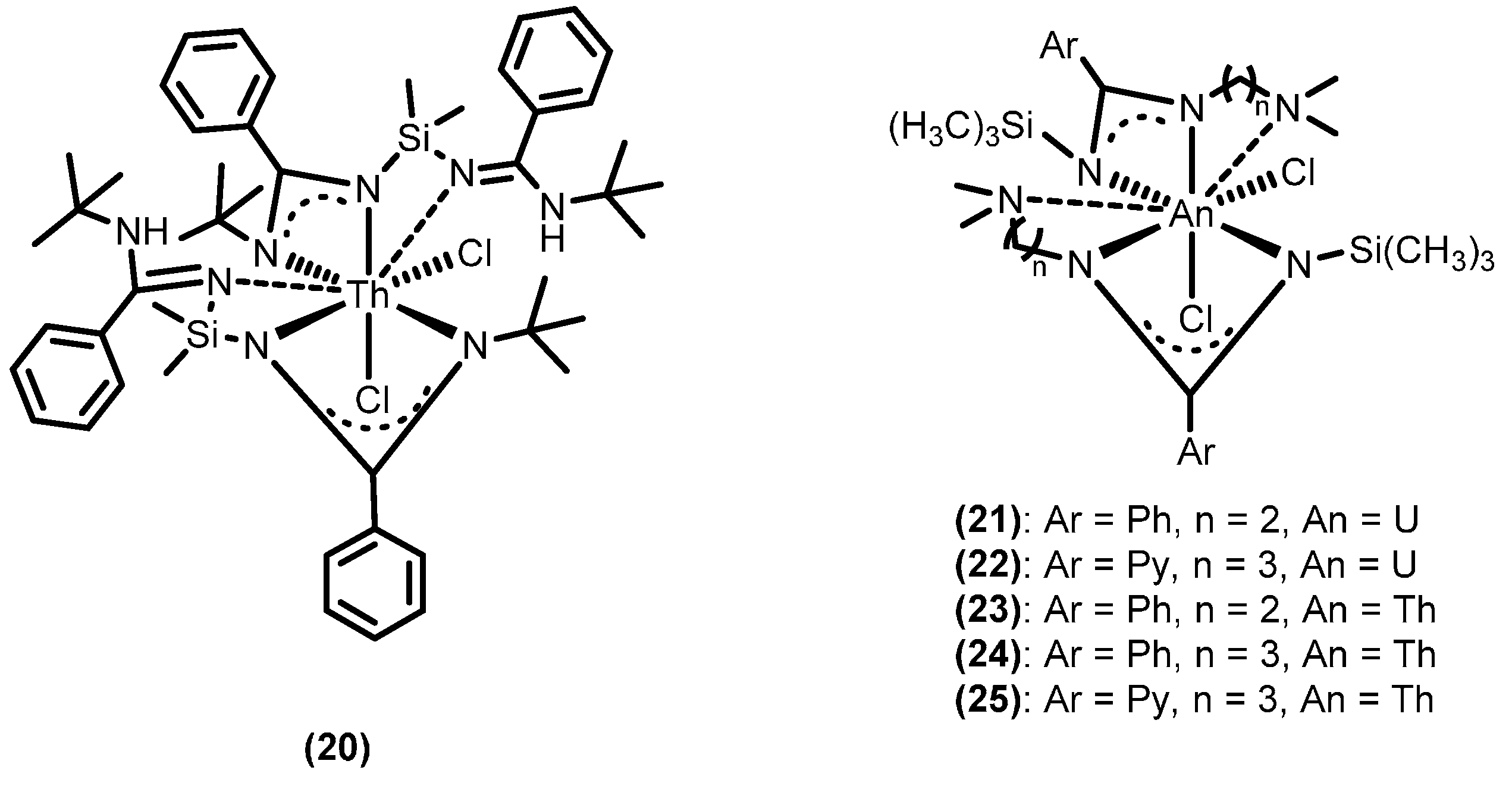
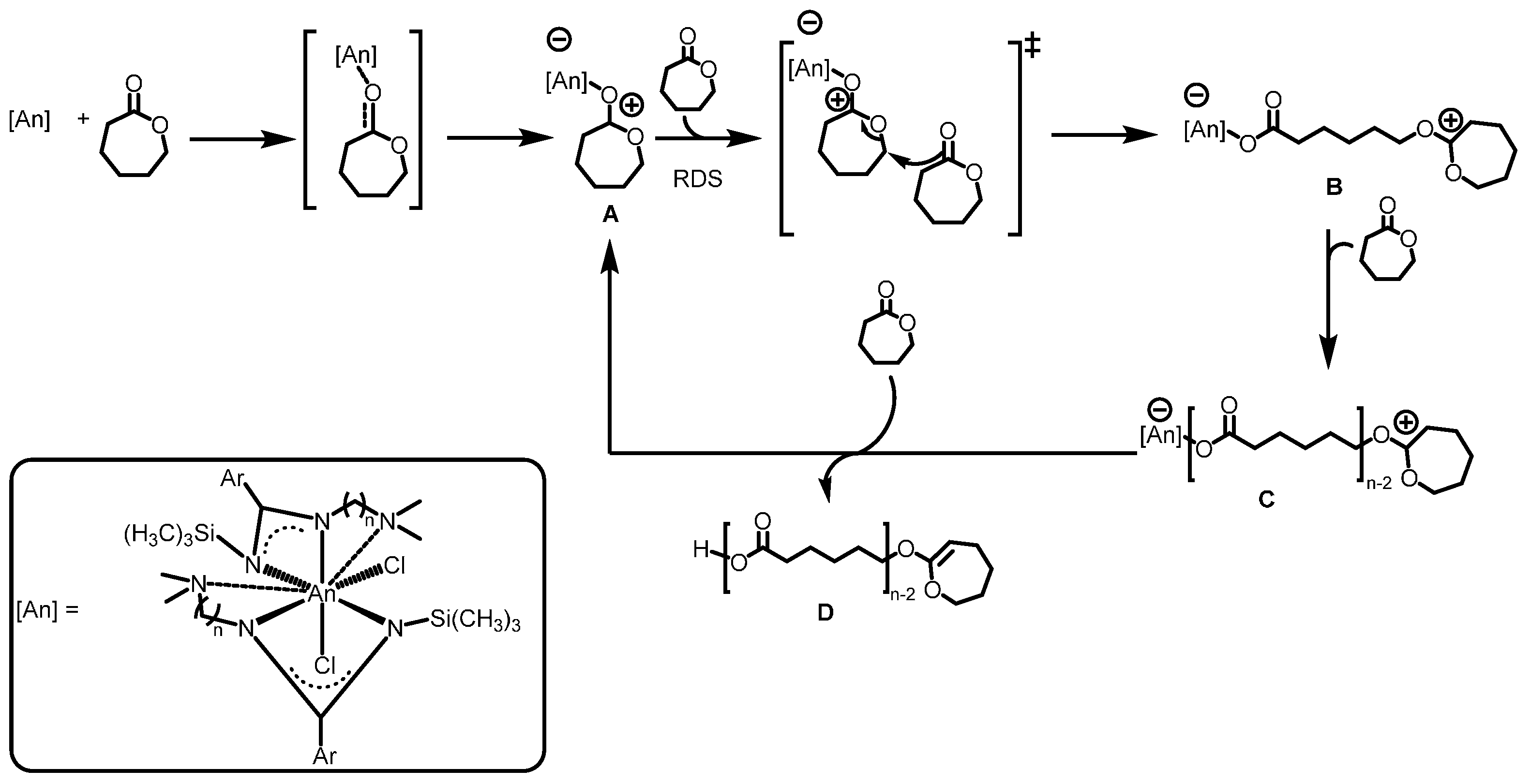
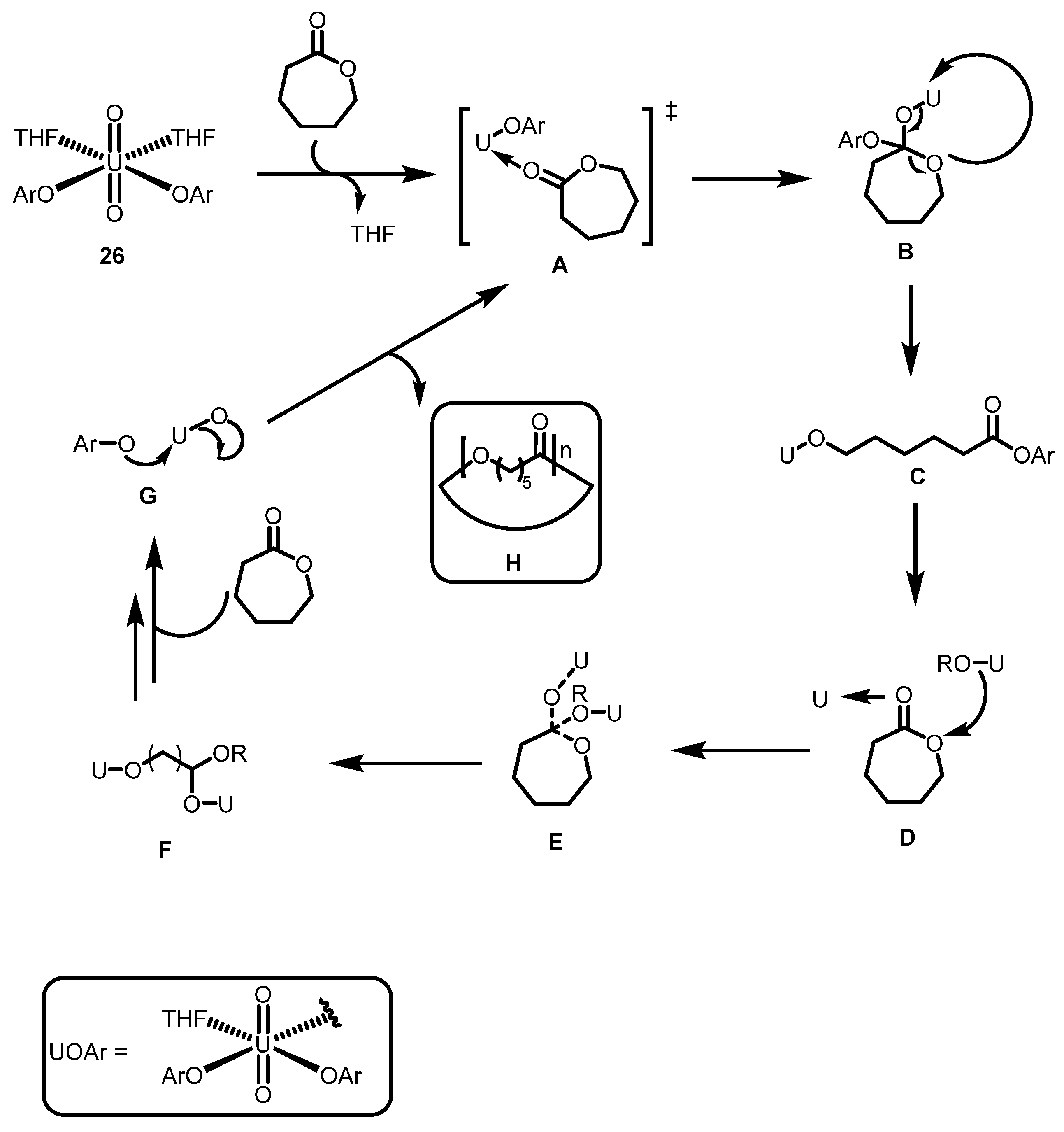
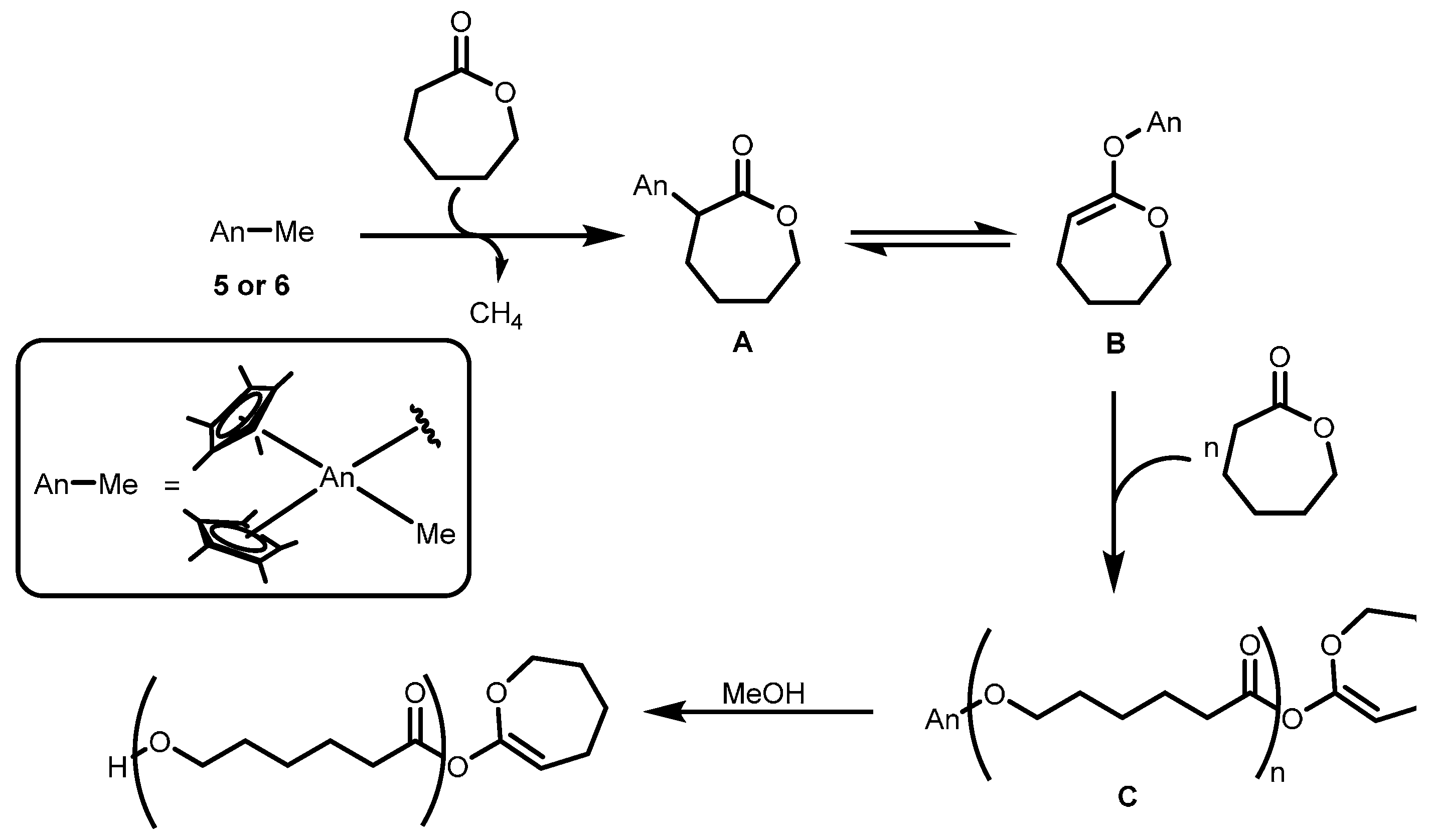
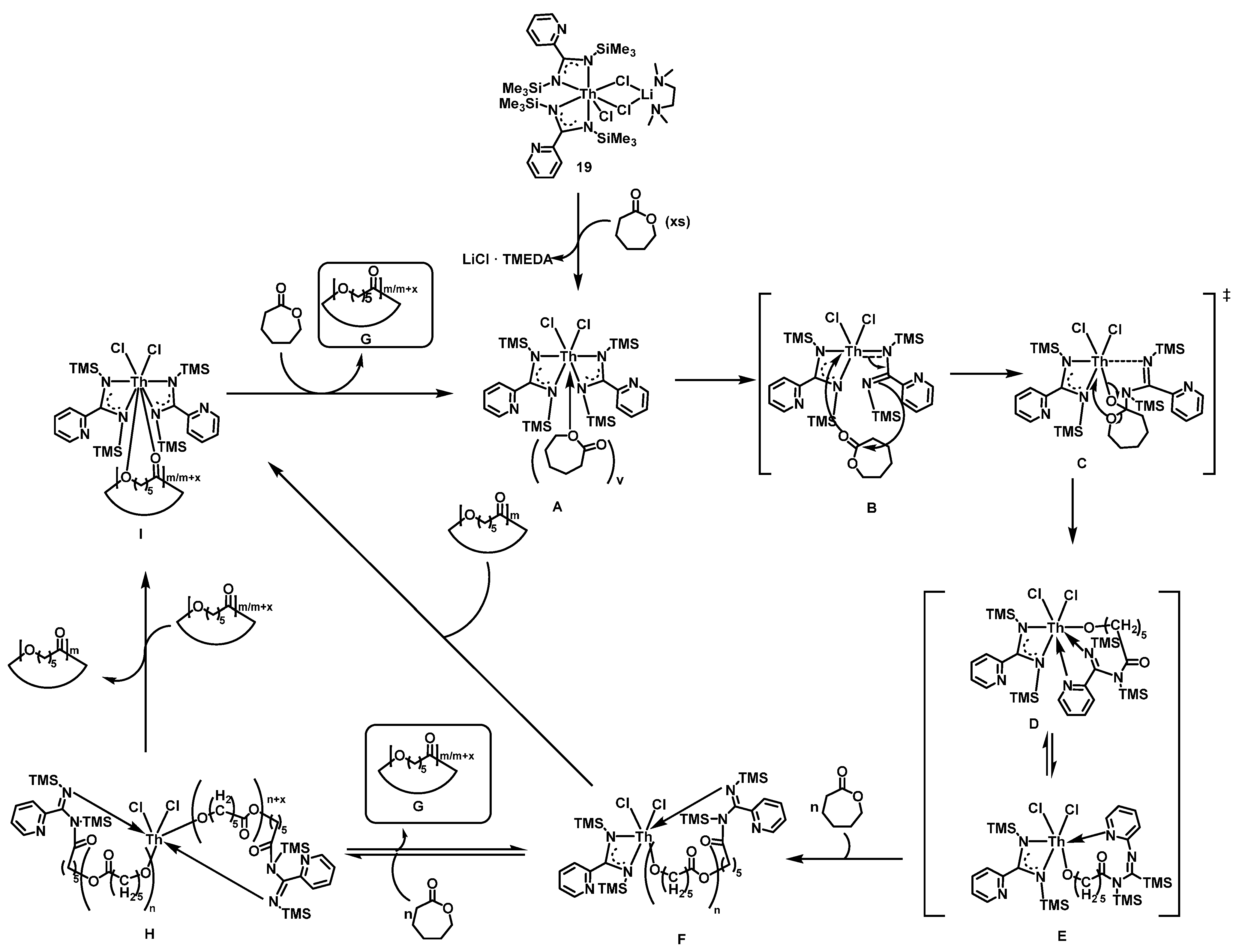
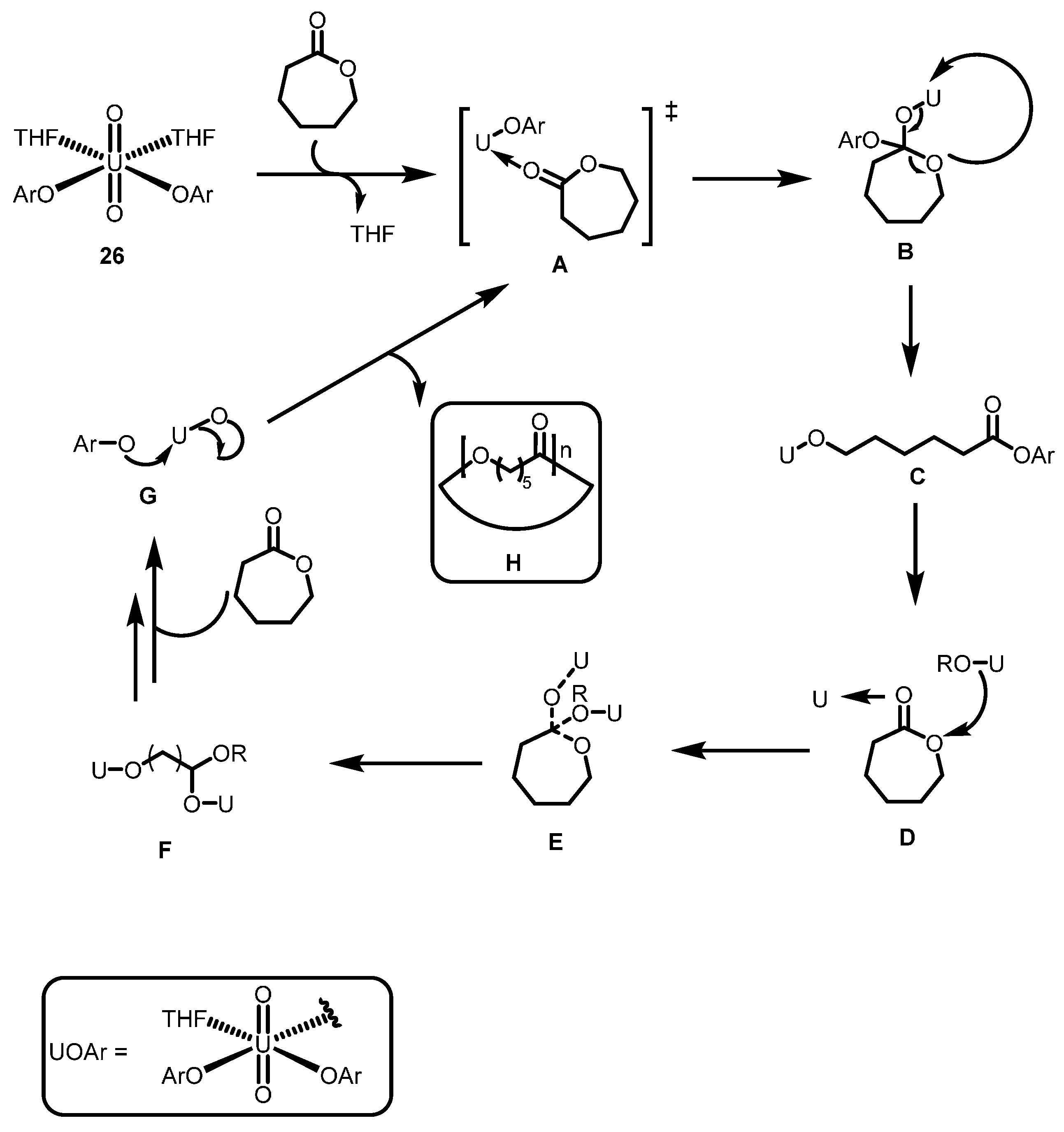
2.3.2. Ring Opening Polymerization of Cyclic Esters
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
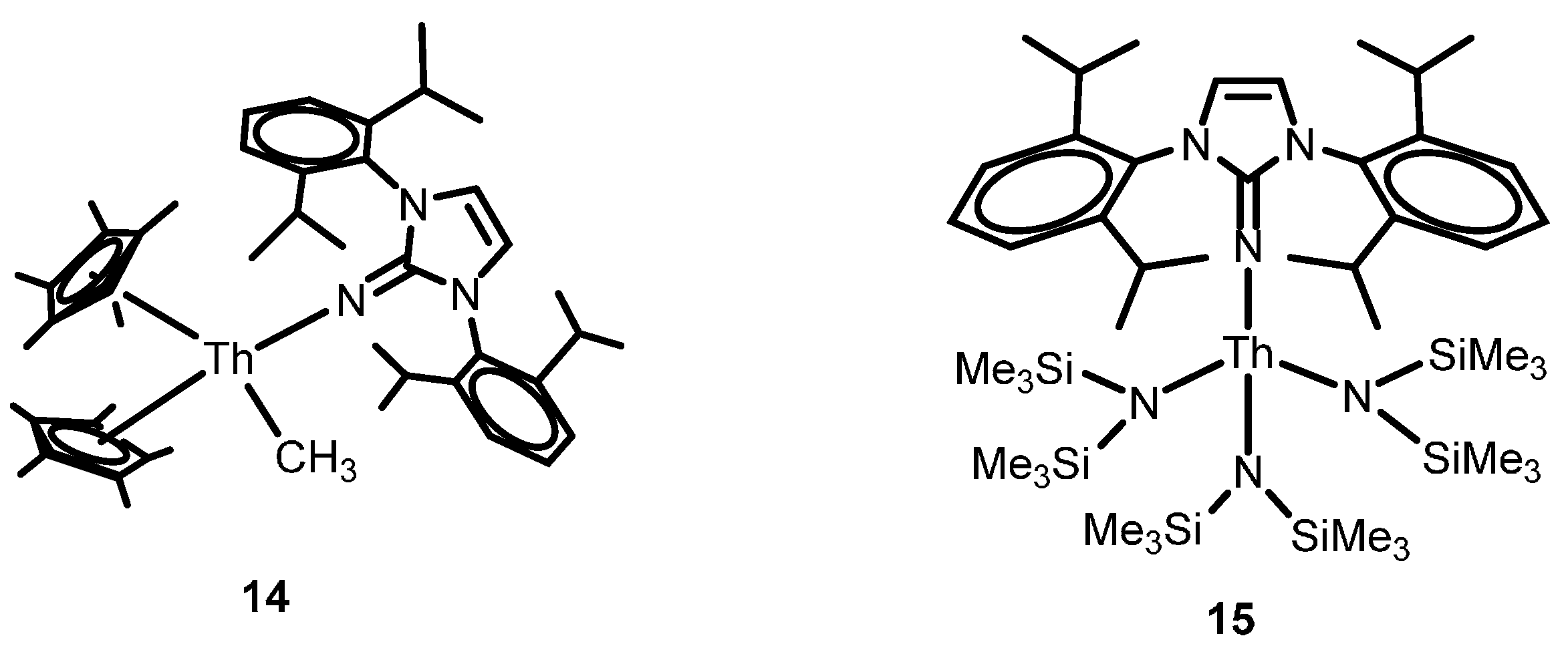{kind=link}
{kind=link}
{kind=link}
{kind=link}
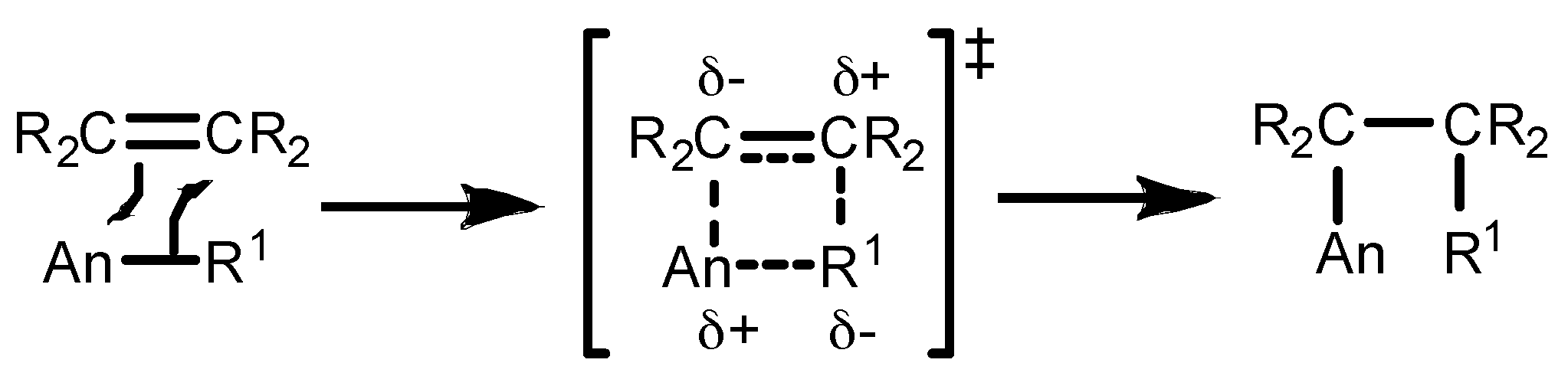{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
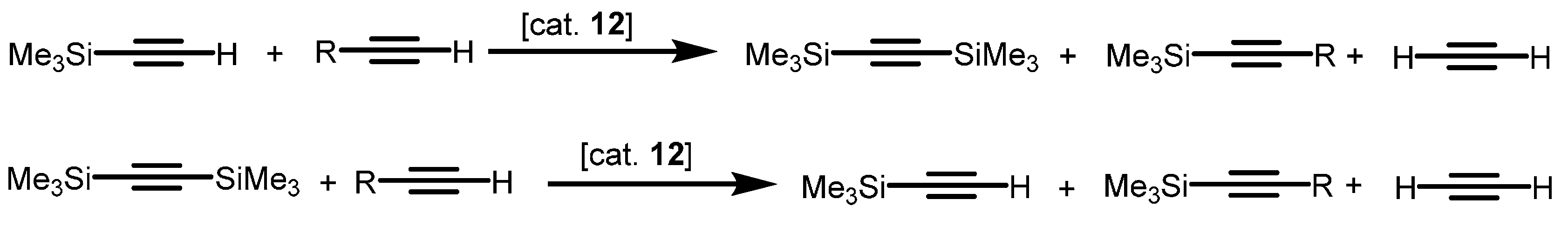{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.4. Further Actinide Mediated Catalytic Transformations
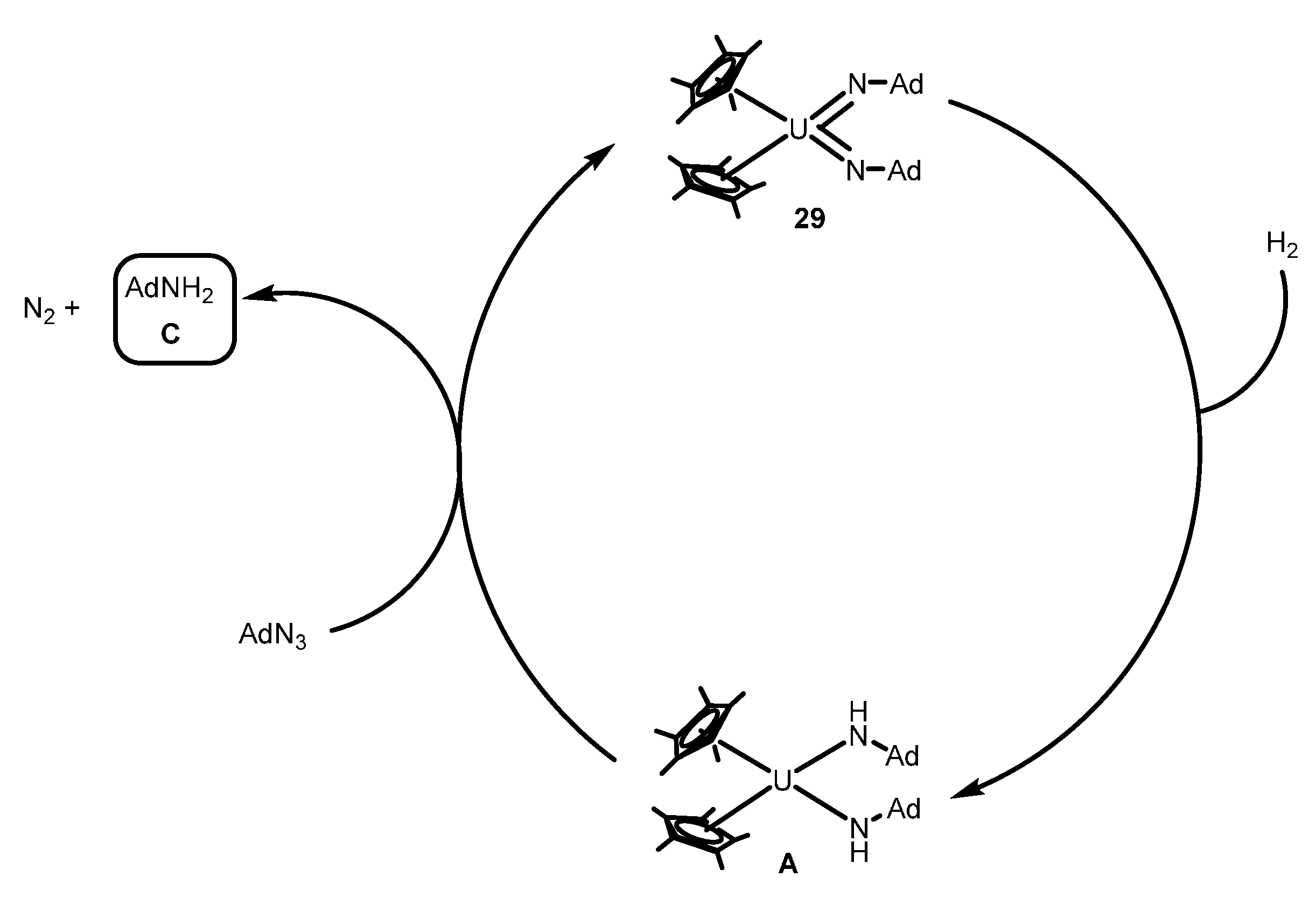
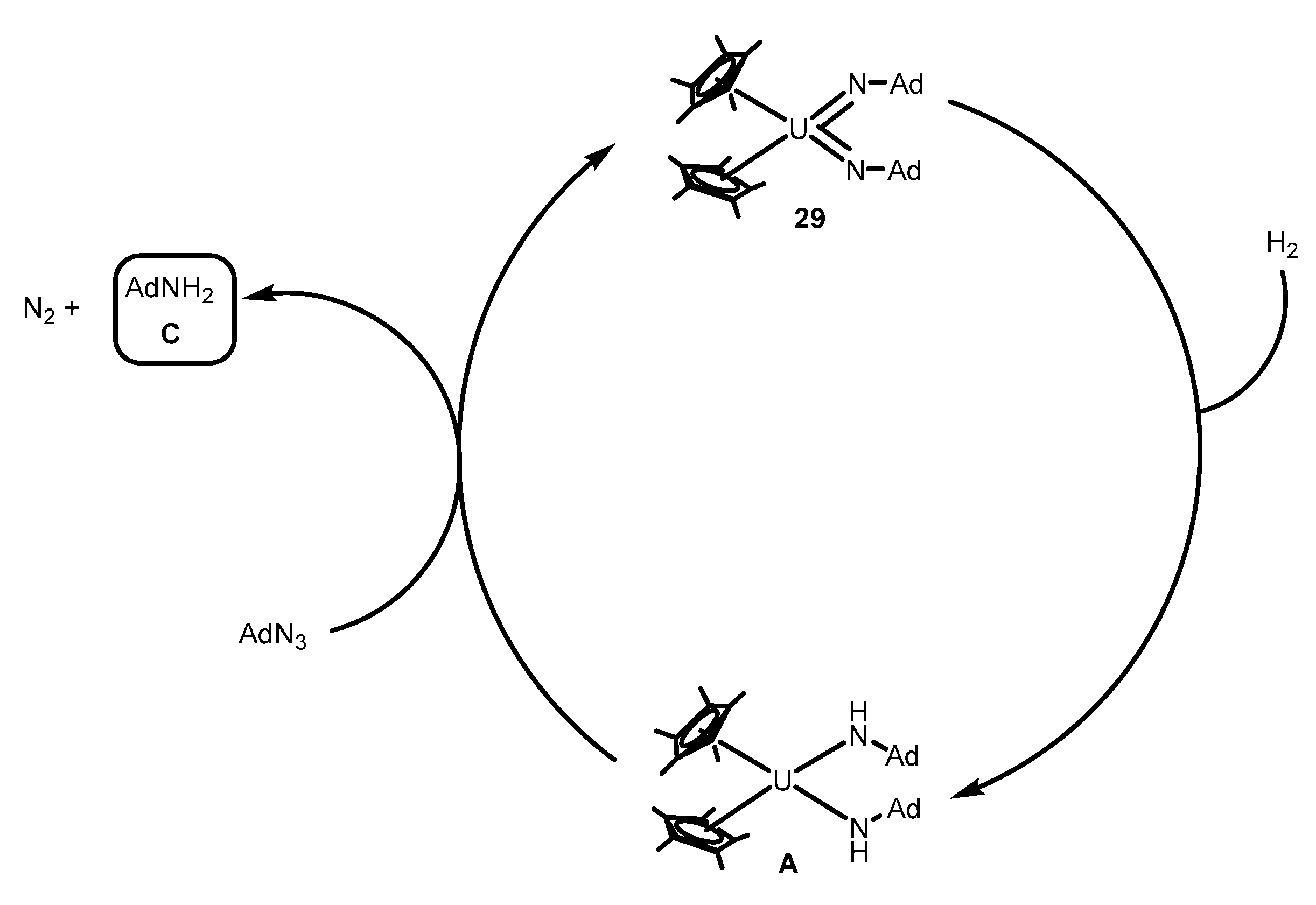
2.4.1. Reduction of Azides and Hydrazines
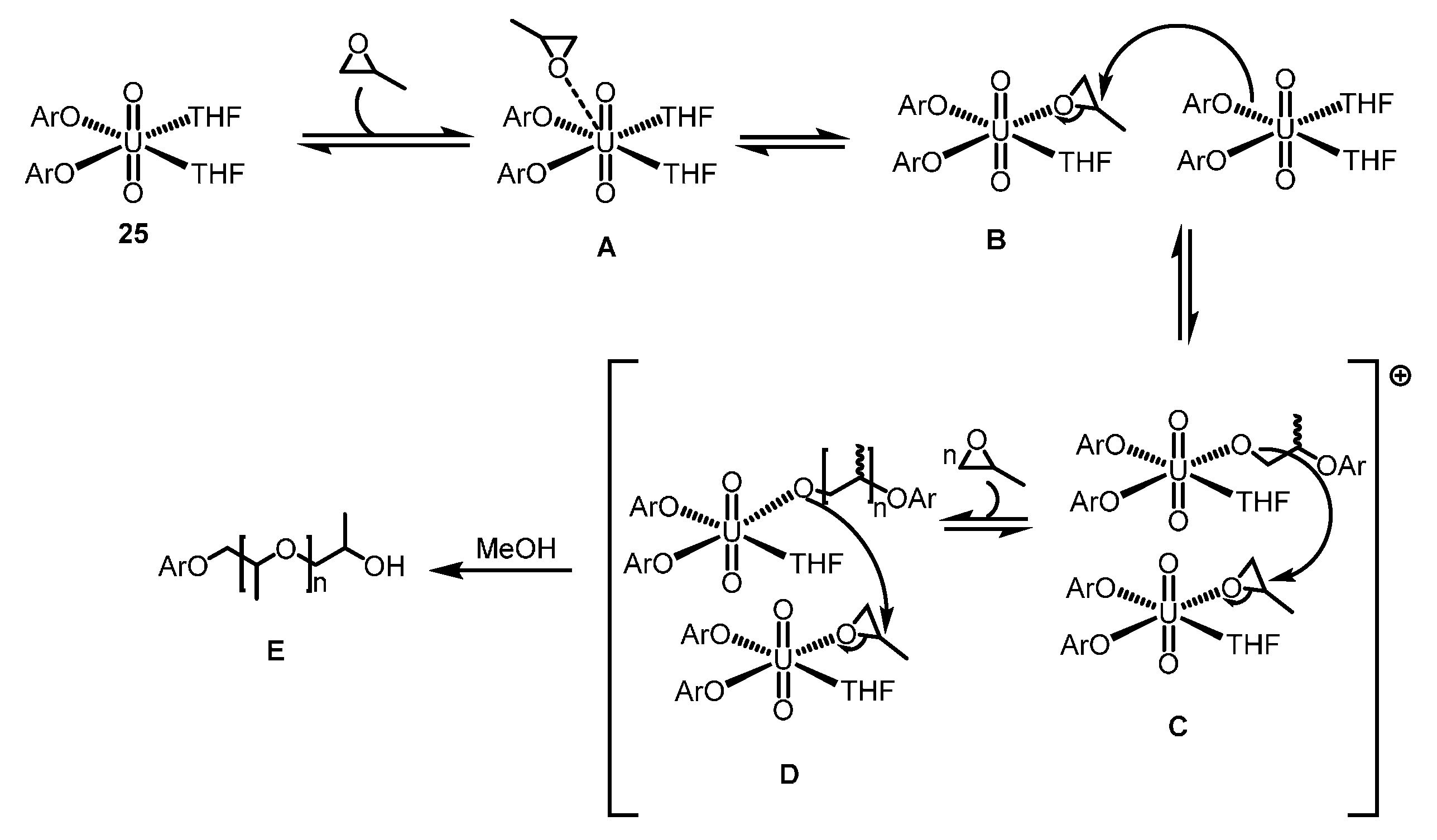
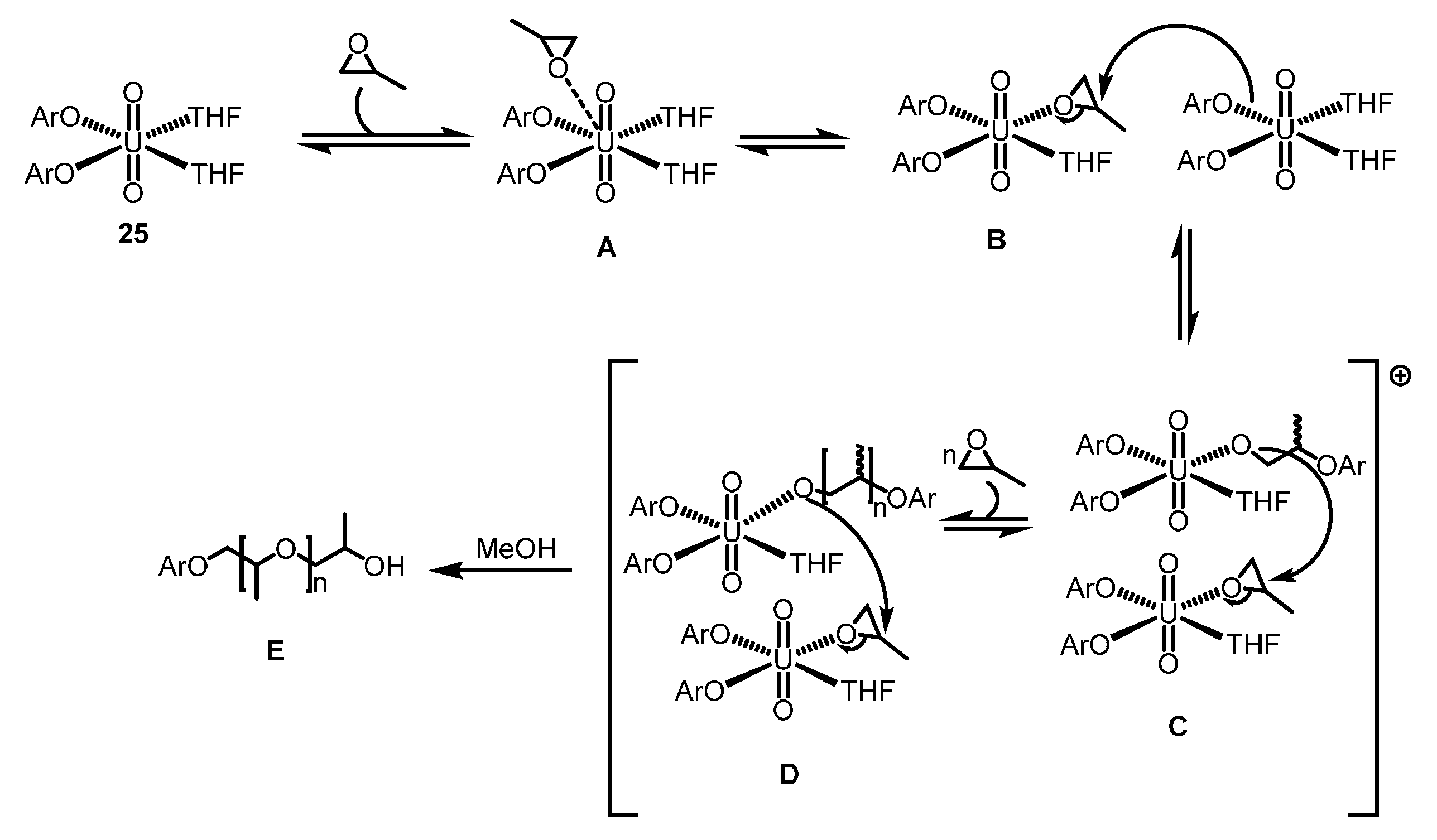
2.4.2. ROP of Epoxides
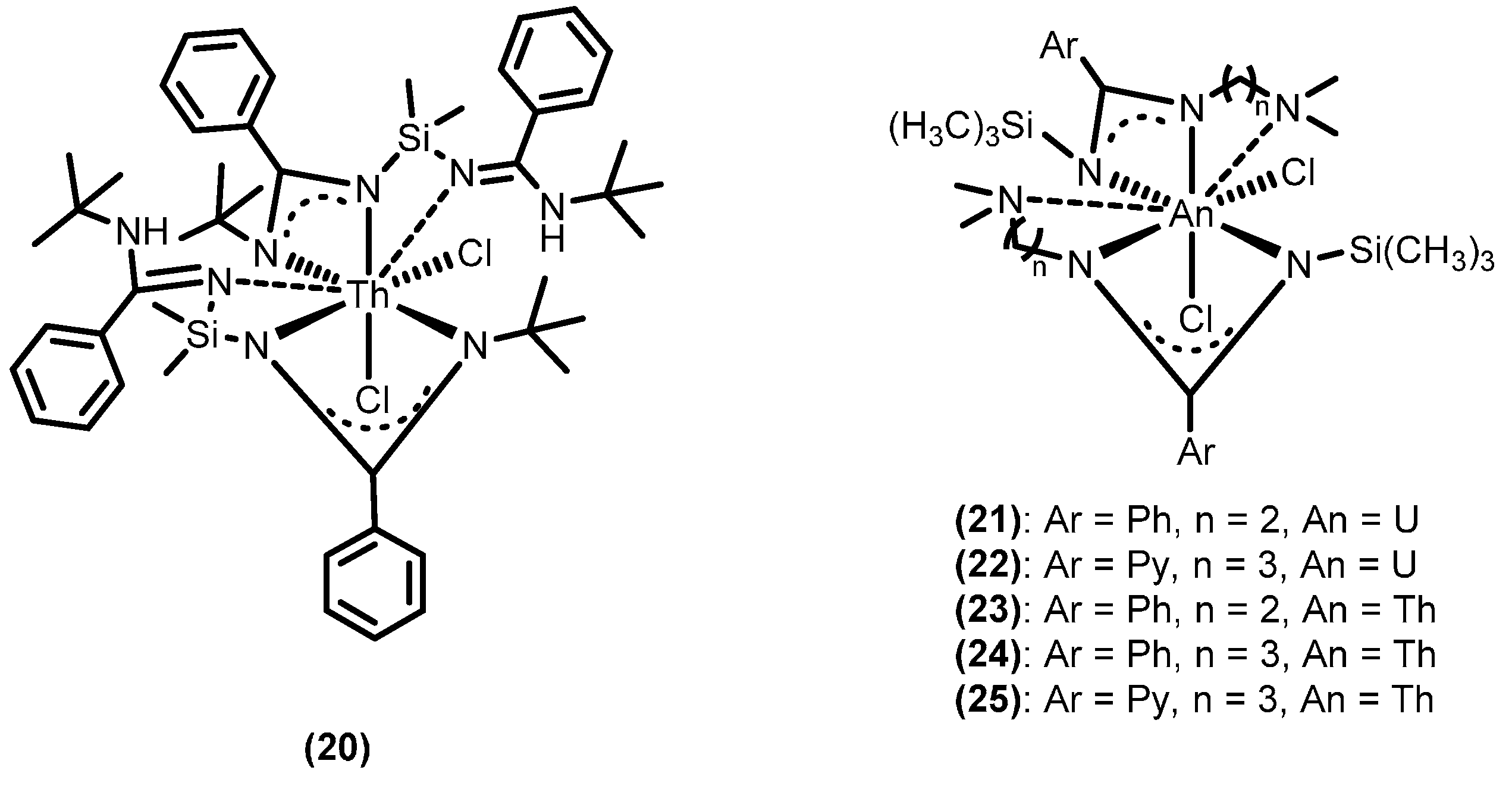
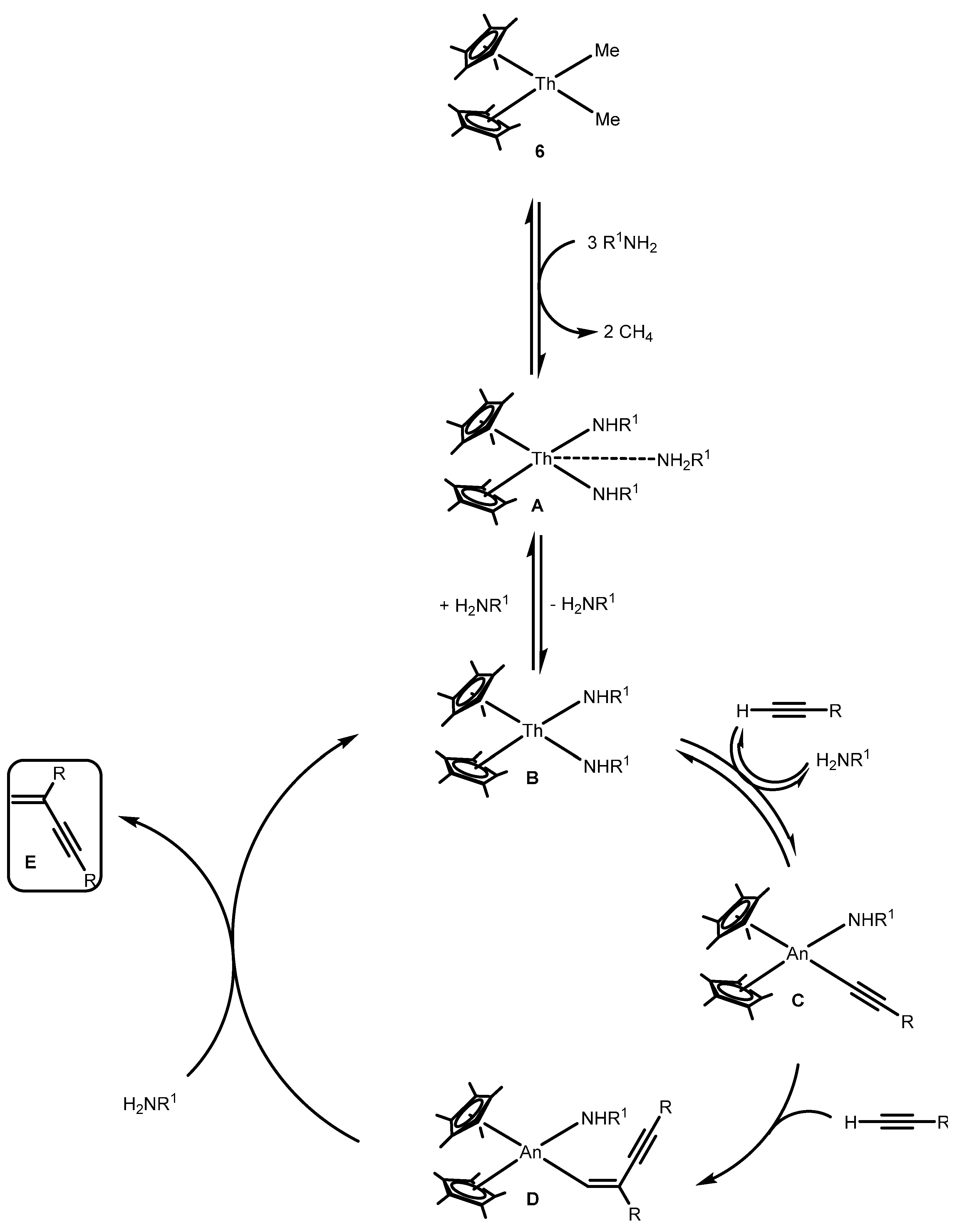
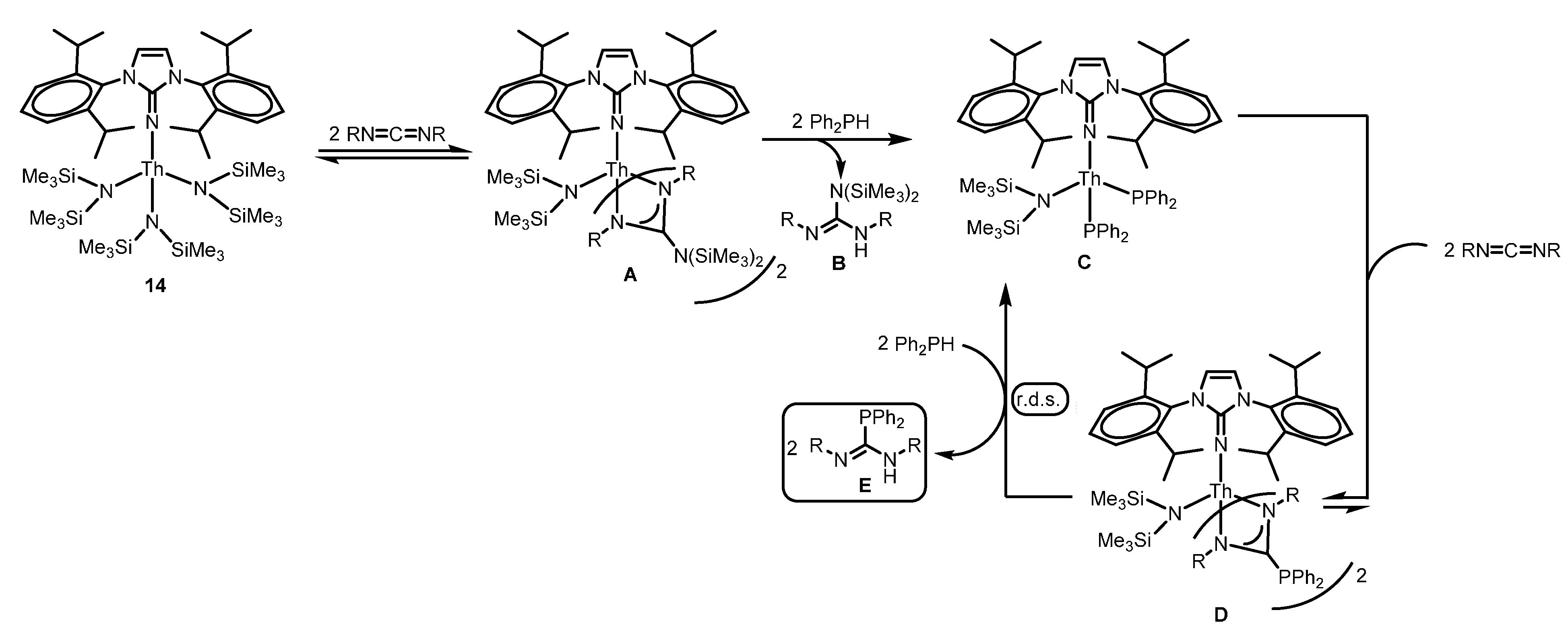
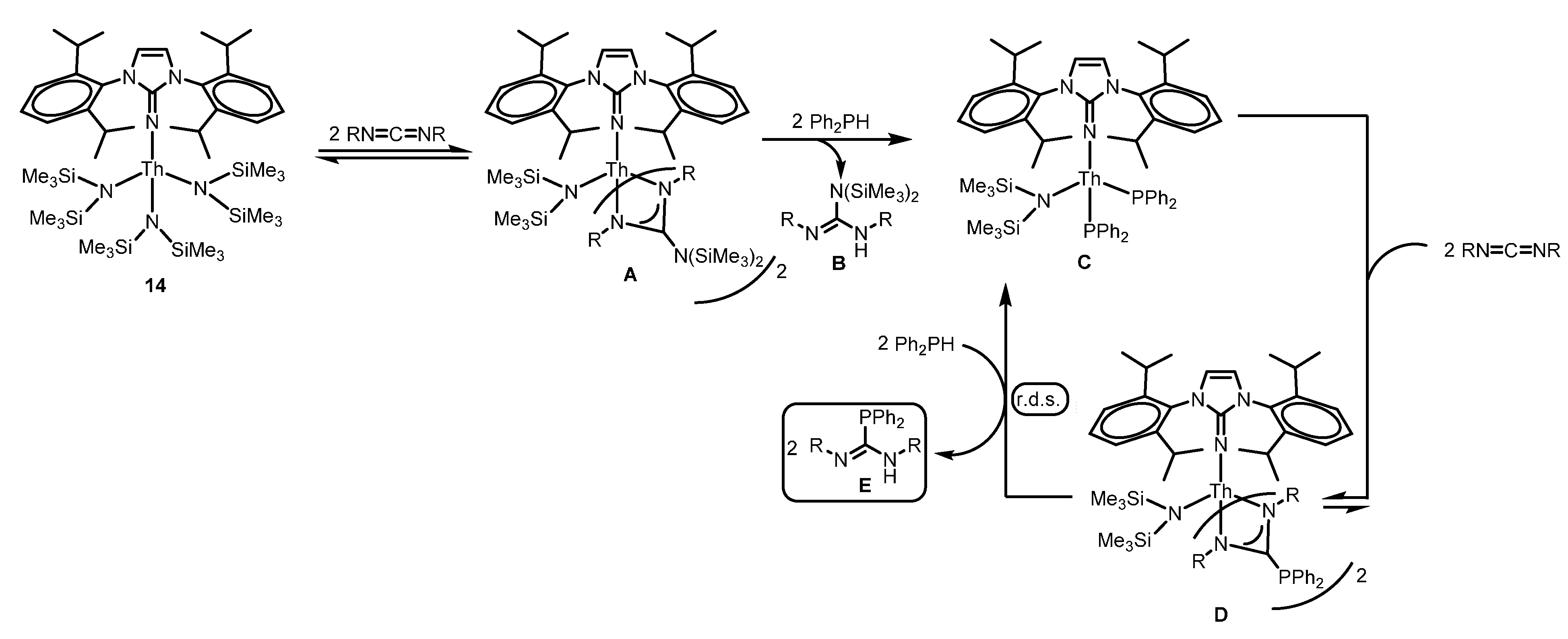
2.4.3. Insertion of Nucleophilic E–H (E = N, P, S) Bonds into Heterocumulenes
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Streitwieser, A.; Mueller-Westerhoff, U. Bis(cyclooctatetraenyl)uranium (uranocene). A new class of sandwich complexes that utilize atomic f orbitals. J. Am. Chem. Soc. 1968, 90, 7364–7364. [Google Scholar] [CrossRef]
- Zalkin, A.; Raymond, K.N. Structure of di-π-cyclooctatetraeneuranium (uranocene). J. Am. Chem. Soc. 1969, 91, 5667–5668. [Google Scholar] [CrossRef]
- Barnea, E.; Eisen, M. Organoactinides in catalysis. Coord. Chem. Rev. 2006, 250, 855–899. [Google Scholar] [CrossRef]
- Andrea, T.; Eisen, M.S. Recent advances in organothorium and organouranium catalysis. Chem. Soc. Rev. 2008, 37, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.R.; Bart, S.C.; Meyer, K.; Cummins, C.C. Towards uranium catalysts. Nature 2008, 455, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Eisen, M.S. Metallocene organoactinide complexes. In Structure and Bonding; Springer Science + Business Media: Berlin, Germany, 2008; pp. 1–85. [Google Scholar]
- Eisen, M.S. Catalytic C–N, C–O, and C–S bond formation promoted by organoactinide complexes. In C–X Bond Formation; Springer Science + Business Media: Berlin, Germany, 2010; pp. 157–184. [Google Scholar]
- Batrice, R.; Karmel, I.; Eisen, M.; Fuerstner, A.; Hall, D.; Marek, I.; Oestreich, M.; Stoltz, B.; Schaumann, E. Product class 13: Organometallic complexes of the actinides. Sci. Synth. Knowl. Updat. 2013, 4, 99–211. [Google Scholar]
- Ephritikhine, M. Recent advances in organoactinide chemistry as exemplified by cyclopentadienyl compounds. Organometallics 2013, 32, 2464–2488. [Google Scholar] [CrossRef]
- Jones, M.B.; Gaunt, A.J. Recent developments in synthesis and structural chemistry of nonaqueous actinide complexes. Chem. Rev. 2013, 113, 1137–1198. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, F.T. Lanthanides and actinides: Annual survey of their organometallic chemistry covering the year 2012. Coord. Chem. Rev. 2014, 261, 73–155. [Google Scholar] [CrossRef]
- Hayes, C.E.; Leznoff, D.B. Actinide coordination and organometallic complexes with multidentate polyamido ligands. Coord. Chem. Rev. 2014, 266–267, 155–170. [Google Scholar] [CrossRef]
- Kaltsoyannis, N.; Kerridge, A. Chemical bonding of lanthanides and actinides. In Fundamental Aspects of Chemical Bonding; Wiley-Blackwell: Oxford, UK, 2014; pp. 337–356. [Google Scholar]
- Meihaus, K.R.; Long, J.R. Actinide-based single-molecule magnets. Dalton Trans. 2015, 44, 2517–2528. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Mazzei, A.; Poggio, S. High 1,4-cis-polybutadiene by uranium catalysts, 1. Tris(π-allyl)uranium halide catalysts. Makromol. Chem. 1974, 175, 2021–2027. [Google Scholar] [CrossRef]
- De Chirico, A.; Lanzani, P.C.; Raggi, E.; Bruzzone, M. High 1,4-cis-polybutadiene by uranium catalysts, 2. Bulk and solution crystallization of polymers. Makromol. Chem. 1974, 175, 2029–2038. [Google Scholar] [CrossRef]
- Guiliani, G.P.; Sorta, E.; Bruzzone, M. High 1,4-cis-polybutadiene by uranium catalyst. III. Strain induced crystallization and processability. Angew. Makromol. Chem. 1976, 50, 87–99. [Google Scholar] [CrossRef]
- Gargani, L.; Giuliani, G.P.; Mistrali, F.; Bruzzone, M. High 1,4-cis-polybutadiene by uranium catalyst. IV. Stretch induced crystallization and ultimate properties. Angew. Makromol. Chem. 1976, 50, 101–113. [Google Scholar] [CrossRef]
- Marks, T.J.; Ernst, R.D. 21—Scandium, yttrium and the lanthanides and actinides. In Comprehensive Organometallic Chemistry; Abel, E.W., Stone, E.G.A., Wilkinson, G., Eds.; Pergamon: Oxford, UK, 1982; pp. 173–270. [Google Scholar]
- Li, X.-F.; Xu, Y.-T.; Feng, X.-Z.; Sun, P.-N. Steric packing and molecular geometry. I. Simulation on tetrahedral structures of weak covalent bonding. Inorg. Chim. Acta 1986, 116, 75–83. [Google Scholar] [CrossRef]
- Li, X.-F.; Feng, X.-Z.; Xu, Y.-T.; Wang, H.-T.; Shi, J.; Liu, L.; Sun, P.-N. Cone packing model—A geometrical approach to coordination and organometallic chemistry of lanthanides and actinides. Inorg. Chim. Acta 1986, 116, 85–93. [Google Scholar] [CrossRef]
- Li, X.-F.; Guo, A.-L. The nature of seat-ligand fitting in coordination space. V. Steric hindrances and reaction mechanisms—A further discussion on the structure and chemistry of compounds containing three π-bonded cyclopentadienyl groups. Inorg. Chim. Acta 1987, 134, 143–153. [Google Scholar] [CrossRef]
- Marçalo, J.; de Matos, A.P. A new definition of coordination number and its use in lanthanide and actinide coordination and organometallic chemistry. Polyhedron 1989, 8, 2431–2437. [Google Scholar] [CrossRef]
- Marks, T.J.; Gagne, M.R.; Nolan, S.P.; Schock, L.E.; Seyam, A.M.; Stern, D. What can metal-ligand bonding energetics teach us about stoichiometric and catalytic organometallic chemistry? Pure Appl. Chem. 1989, 61, 1665–1672. [Google Scholar] [CrossRef]
- Leal, J.P.; Marques, N.; Pires de Matos, A.; Calhorda, M.J.; Galvao, A.M.; Simoes, J.A.M. Uranium-ligand bond dissociation enthalpies in uranium(IV) polypyrazolylborate complexes. Organometallics 1992, 11, 1632–1637. [Google Scholar] [CrossRef]
- King, W.A.; Marks, T.J.; Anderson, D.M.; Duncalf, D.J.; Cloke, F.G.N. Organo-f-element bonding energetics. Large magnitudes of metal arene bond enthalpies in zero-valent lanthanide sandwich complexes. J. Am. Chem. Soc. 1992, 114, 9221–9223. [Google Scholar] [CrossRef]
- Jemine, X.; Goffart, J.; Berthet, J.-C.; Ephritikhine, M. Absolute uranium? Ligand bond-disruption enthalpies of [U(C5H4R)3X] complexes (X = I or H, R = But or SiMe3). Dalton Trans. 1992, 2439–2440. [Google Scholar] [CrossRef]
- Jemine, X.; Goffart, J.; Ephritikhine, M.; Fuger, J. Organo-f-element thermochemistry. Thorium-ligand bond disruption enthalpies in {(CH3)3SiC9H6}3ThX (X = H or D) and in {(CH3)3SiC5H4}3ThH complexes. J. Organomet. Chem. 1993, 448, 95–98. [Google Scholar] [CrossRef]
- Leal, J.P.; Simoes, J.A.M. Uranium-ligand bond-dissociation enthalpies of uranium(IV) poly(pyrazolyl)borate complexes. Dalton Trans. 1994, 2687–2691. [Google Scholar] [CrossRef]
- King, W.A.; Marks, T.J. Metal-silicon bonding energetics in organo-group 4 and organo-f-element complexes. Implications for bonding and reactivity. Inorg. Chim. Acta 1995, 229, 343–354. [Google Scholar] [CrossRef]
- Marks, T.J.; Day, V.W. Fundamental and Technological Aspects of Organo-F-Element Chemistry; Springer Science + Business Media: Berlin, Germany, 1985. [Google Scholar]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-metal-catalyzed addition of heteroatom–hydrogen bonds to alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Gold-catalyzed organic reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef] [PubMed]
- Wobser, S.D.; Marks, T.J. Organothorium-catalyzed hydroalkoxylation/cyclization of alkynyl alcohols. Scope, mechanism, and ancillary ligand effects. Organometallics 2013, 32, 2517–2528. [Google Scholar] [CrossRef]
- Griesbaum, K. Problems and possibilities of the free-radical addition of thiols to unsaturated compounds. Angew. Chem. Int. Ed. Eng. 1970, 9, 273–287. [Google Scholar] [CrossRef]
- Benati, L.; Montevecchi, P.C.; Spagnolo, P. Free-radical reactions of benzenethiol and diphenyl disulphide with alkynes. Chemical reactivity of intermediate 2-(phenylthio)vinyl radicals. J. Chem. Soc. Perkin Trans. 1991, 1, 2103–2109. [Google Scholar] [CrossRef]
- Kondoh, A.; Takami, K.; Yorimitsu, H.; Oshima, K. Stereoselective hydrothiolation of alkynes catalyzed by cesium base: Facile access to (Z)-1-alkenyl sulfides. J. Org. Chem. 2005, 70, 6468–6473. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Mitsudo, T.-A. Metal-catalyzed carbon–sulfur bond formation. Chem. Rev. 2000, 100, 3205–3220. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Fraser, L.R.; Love, J.A. Rhodium-catalyzed alkyne hydrothiolation with aromatic and aliphatic thiols. J. Am. Chem. Soc. 2005, 127, 17614–17615. [Google Scholar] [CrossRef] [PubMed]
- Malyshev, D.A.; Scott, N.M.; Marion, N.; Stevens, E.D.; Ananikov, V.P.; Beletskaya, I.P.; Nolan, S.P. Homogeneous nickel catalysts for the selective transfer of a single arylthio group in the catalytic hydrothiolation of alkynes. Organometallics 2006, 25, 4462–4470. [Google Scholar] [CrossRef]
- Delp, S.A.; Munro-Leighton, C.; Goj, L.A.; Ramírez, M.A.; Gunnoe, T.B.; Petersen, J.L.; Boyle, P.D. Addition of S–H bonds across electron-deficient olefins catalyzed by well-defined copper(I) thiolate complexes. Inorg. Chem. 2007, 46, 2365–2367. [Google Scholar] [CrossRef] [PubMed]
- Shoai, S.; Bichler, P.; Kang, B.; Buckley, H.; Love, J.A. Catalytic alkyne hydrothiolation with alkanethiols using wilkinson’s catalyst. Organometallics 2007, 26, 5778–5781. [Google Scholar] [CrossRef]
- Kondoh, A.; Yorimitsu, H.; Oshima, K. Palladium-catalyzed anti-hydrothiolation of 1-alkynylphosphines. Org. Lett. 2007, 9, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Fraser, L.R.; Bird, J.; Wu, Q.; Cao, C.; Patrick, B.O.; Love, J.A. Synthesis, structure, and hydrothiolation activity of rhodium pyrazolylborate complexes. Organometallics 2007, 26, 5602–5611. [Google Scholar] [CrossRef]
- Weiss, C.J.; Wobser, S.D.; Marks, T.J. Organoactinide-mediated hydrothiolation of terminal alkynes with aliphatic, aromatic, and benzylic thiols. J. Am. Chem. Soc. 2009, 131, 2062–2063. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.J.; Wobser, S.D.; Marks, T.J. Lanthanide- and actinide-mediated terminal alkyne hydrothiolation for the catalytic synthesis of markovnikov vinyl sulfides. Organometallics 2010, 29, 6308–6320. [Google Scholar] [CrossRef]
- Weiss, C.J.; Marks, T.J. Organo-f-element catalysts for efficient and highly selective hydroalkoxylation and hydrothiolation. Dalton Trans. 2010, 39, 6576–6588. [Google Scholar] [CrossRef] [PubMed]
- Haskel, A.; Straub, T.; Eisen, M.S. Organoactinide-catalyzed intermolecular hydroamination of terminal alkynes. Organometallics 1996, 15, 3773–3775. [Google Scholar] [CrossRef]
- Straub, T.; Haskel, A.; Neyroud, T.G.; Kapon, M.; Botoshansky, M.; Eisen, M.S. Intermolecular hydroamination of terminal alkynes catalyzed by organoactinide complexes. Scope and mechanistic studies. Organometallics 2001, 20, 5017–5035. [Google Scholar] [CrossRef]
- Stubbert, B.D.; Stern, C.L.; Marks, T.J. Synthesis and catalytic characteristics of novel constrained-geometry organoactinide catalysts. The first example of actinide-mediated intramolecular hydroamination. Organometallics 2003, 22, 4836–4838. [Google Scholar] [CrossRef]
- Stubbert, B.D.; Marks, T.J. Mechanistic investigation of intramolecular aminoalkene and aminoalkyne hydroamination/cyclization catalyzed by highly electrophilic, tetravalent constrained geometry 4d and 5f complexes. Evidence for an M–N σ-bonded insertive pathway. J. Am. Chem. Soc. 2007, 129, 6149–6167. [Google Scholar] [CrossRef] [PubMed]
- Broderick, E.M.; Gutzwiller, N.P.; Diaconescu, P.L. Inter- and intramolecular hydroamination with a uranium dialkyl precursor. Organometallics 2010, 29, 3242–3251. [Google Scholar] [CrossRef]
- Haskel, A.; Straub, T.; Dash, A.K.; Eisen, M.S. Oligomerization and cross-oligomerization of terminal alkynes catalyzed by organoactinide complexes. J. Am. Chem. Soc. 1999, 121, 3014–3024. [Google Scholar] [CrossRef]
- Haskel, A.; Wang, J.Q.; Straub, T.; Neyroud, T.G.; Eisen, M.S. Controlling the catalytic oligomerization of terminal alkynes promoted by organoactinides: A strategy to short oligomers. J. Am. Chem. Soc. 1999, 121, 3025–3034. [Google Scholar] [CrossRef]
- Hayes, C.E.; Platel, R.H.; Schafer, L.L.; Leznoff, D.B. Diamido-ether actinide complexes as catalysts for the intramolecular hydroamination of aminoalkenes. Organometallics 2012, 31, 6732–6740. [Google Scholar] [CrossRef]
- Fengyu, B.; Kanno, K.-I.; Takahashi, T. Early transition metal catalyzed hydrosilation reaction. Trends Org. Chem. 2008, 12, 1–17. [Google Scholar]
- Rooke, D.A.; Menard, Z.A.; Ferreira, E.M. An analysis of the influences dictating regioselectivity in platinum-catalyzed hydrosilylations of internal alkynes. Tetrahedron 2014, 70, 4232–4244. [Google Scholar] [CrossRef]
- Iglesias, M.; Fernández-Alvarez, F.J.; Oro, L.A. Outer-sphere ionic hydrosilylation catalysis. ChemCatChem 2014, 6, 2486–2489. [Google Scholar] [CrossRef]
- Greenhalgh, M.D.; Jones, A.S.; Thomas, S.P. Iron-catalysed hydrofunctionalisation of alkenes and alkynes. ChemCatChem 2014, 7, 190–222. [Google Scholar] [CrossRef]
- Dash, A.K.; Wang, J.Q.; Eisen, M.S. Catalytic hydrosilylation of terminal alkynes promoted by organoactinides. Organometallics 1999, 18, 4724–4741. [Google Scholar] [CrossRef]
- Dash, A.K.; Gourevich, I.; Wang, J.Q.; Wang, J.; Kapon, M.; Eisen, M.S. The catalytic effect in opening an organoactinide metal coordination sphere: Regioselective dimerization of terminal alkynes and hydrosilylation of alkynes and alkenes with PhSiH3 promoted by Me2SiCp"2ThnBu2. Organometallics 2001, 20, 5084–5104. [Google Scholar] [CrossRef]
- Dash, A.K.; Gurevizt, Y.; Wang, J.Q.; Wang, J.; Kapon, M.; Eisen, M.S. Organoactinides—Novel catalysts for demanding chemical transformations. J. Alloys Compd. 2002, 344, 65–69. [Google Scholar] [CrossRef]
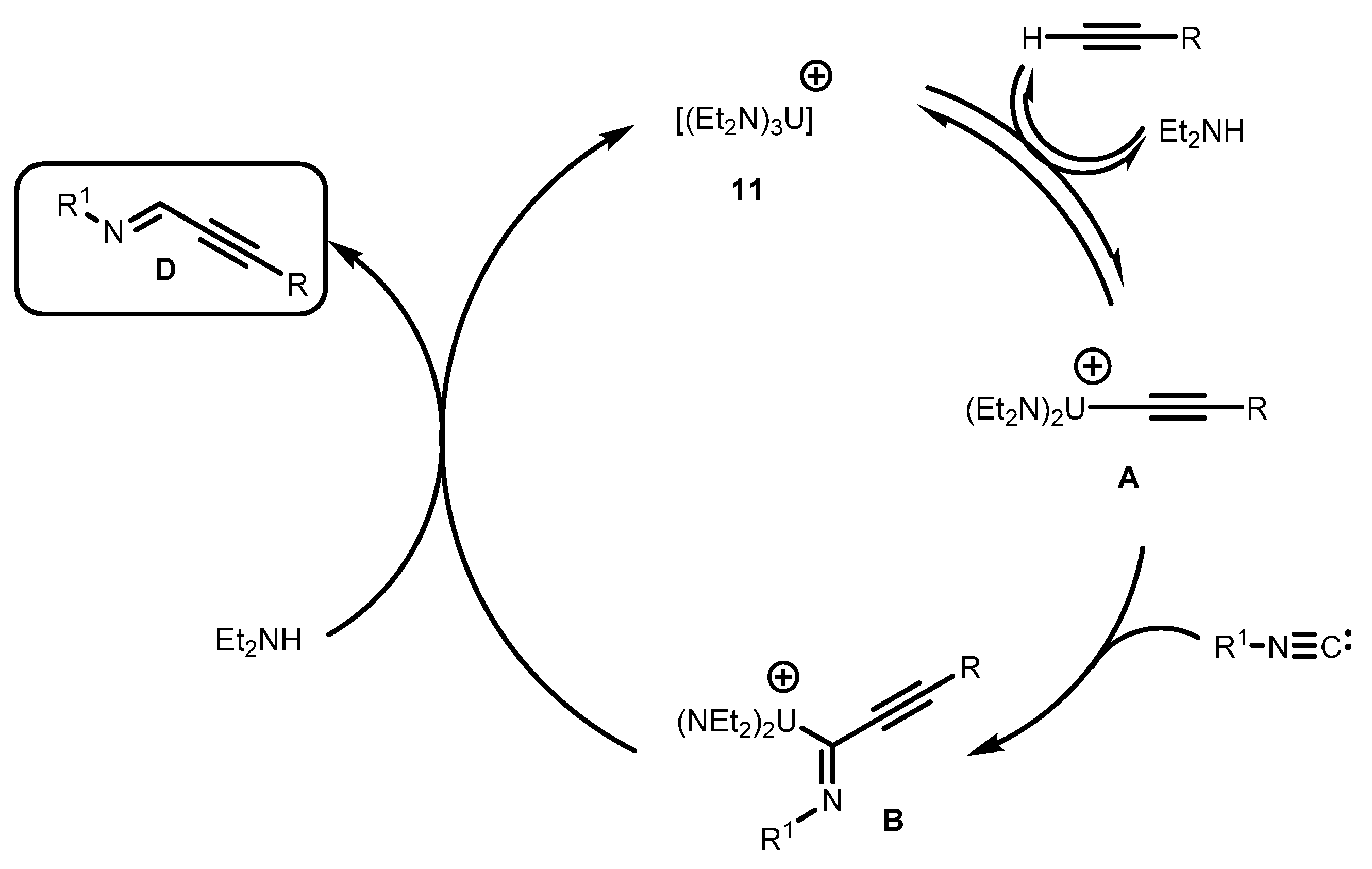
- Dash, A.K.; Wang, J.X.; Berthet, J.C.; Ephritikhine, M.; Eisen, M.S. Diverse catalytic activity of the cationic actinide complex [(Et2N)3U][BPh4] in the dimerization and hydrosilylation of terminal alkynes. Characterization of the first f-element alkyne π-complex [(Et2N)2U(C≡CtBu)(η2-HC≡CtBu)][BPh4]. J. Organomet. Chem. 2000, 604, 83–98. [Google Scholar] [CrossRef]
- Straub, T.; Haskel, A.; Eisen, M.S. Organoactinide-catalyzed oligomerization of terminal acetylenes. J. Am. Chem. Soc. 1995, 117, 6364–6365. [Google Scholar] [CrossRef]
- Wang, J.Q.; Dash, A.K.; Berthet, J.C.; Ephritikhine, M.; Eisen, M.S. Selective dimerization of terminal alkynes promoted by the cationic actinide compound [(Et2N)3U][BPh4]. Formation of the alkyne π-complex [(Et2N)2U(C≡CtBu)(η2-HC≡CtBu)][BPh4]. Organometallics 1999, 18, 2407–2409. [Google Scholar] [CrossRef]
- Wang, J.; Kapon, M.; Berthet, J.C.; Ephritikhine, M.; Eisen, M.S. Cross dimerization of terminal alkynes catalyzed by [(Et2N)3U][BPh4]. Inorg. Chim. Acta 2002, 334, 183–192. [Google Scholar] [CrossRef]
- Wang, J.; Dash, A.K.; Kapon, M.; Berthet, J.-C.; Ephritikhine, M.; Eisen, M.S. Oligomerization and hydroamination of terminal alkynes promoted by the cationic organoactinide compound [(Et2N)3U][BPh4]. Chem. Eur. J. 2002, 8, 5384–5396. [Google Scholar] [CrossRef]
- Kosog, B.; Kefalidis, C.E.; Heinemann, F.W.; Maron, L.; Meyer, K. Uranium(III)-mediated C–C-coupling of terminal alkynes: Formation of dinuclear uranium(IV) vinyl complexes. J. Am. Chem. Soc. 2012, 134, 12792–12797. [Google Scholar] [CrossRef] [PubMed]
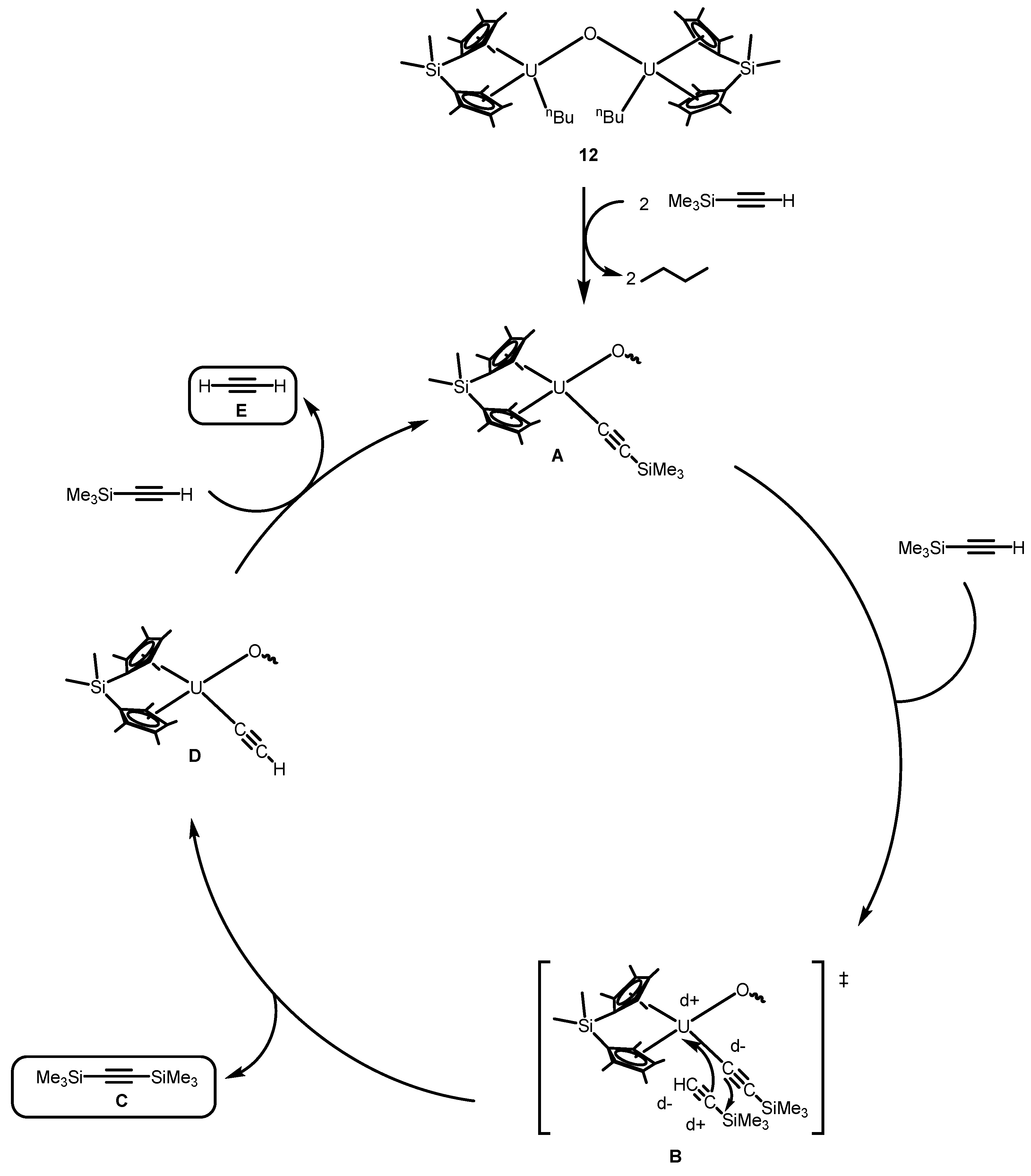
- Wang, J.; Gurevich, Y.; Botoshansky, M.; Eisen, M.S. Unique σ-bond metathesis of silylalkynes promoted by an ansa-dimethylsilyl and oxo-bridged uranium metallocene. J. Am. Chem. Soc. 2006, 128, 9350–9351. [Google Scholar] [CrossRef] [PubMed]
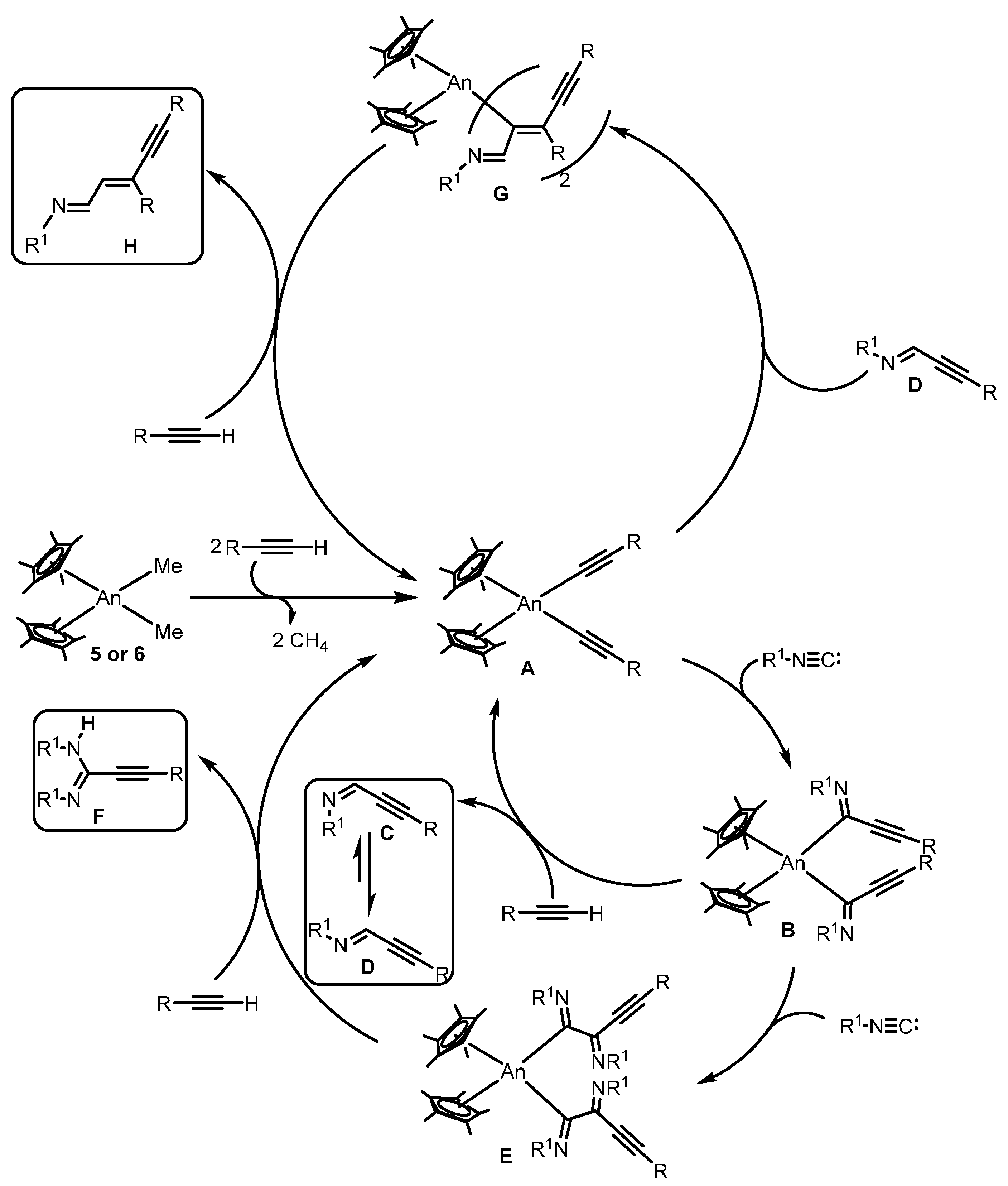
- Barnea, E.; Andrea, T.; Berthet, J.-C.; Ephritikhine, M.; Eisen, M.S. Coupling of terminal alkynes and isonitriles by organoactinide complexes: Scope and mechanistic insights. Organometallics 2008, 27, 3103–3112. [Google Scholar] [CrossRef]
- Lin, Z.; Marks, T.J. Metal, bond energy, and ancillary ligand effects on actinide–carbon σ-bond hydrogenolysis. A kinetic and mechanistic study. J. Am. Chem. Soc. 1987, 109, 7979–7985. [Google Scholar] [CrossRef]
- Andrea, T.; Barnea, E.; Eisen, M.S. Organoactinides promote the tishchenko reaction: The myth of inactive actinide-alkoxo complexes. J. Am. Chem. Soc. 2008, 130, 2454–2455. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Andrea, T.; Brookes, N.J.; Yates, B.F.; Eisen, M.S. Organoactinides promote the dimerization of aldehydes: Scope, kinetics, thermodynamics, and calculation studies. J. Am. Chem. Soc. 2011, 133, 1341–1356. [Google Scholar] [CrossRef] [PubMed]
- Karmel, I.S.R.; Fridman, N.; Tamm, M.; Eisen, M.S. Mixed imidazolin-2-iminato–Cp* thorium(IV) complexes: Synthesis and reactivity toward oxygen-containing substrates. Organometallics 2015, 34, 2933–2942. [Google Scholar] [CrossRef]
- Karmel, I.S.R.; Fridman, N.; Tamm, M.; Eisen, M.S. Mono(imidazolin-2-iminato) actinide complexes: Synthesis and application in the catalytic dimerization of aldehydes. J. Am. Chem. Soc. 2014, 136, 17180–17192. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, B.; Chojnowski, J. Progress in Organosilicon Chemistry; Gordon and Breach Science Publishers: Basel, Switzerland, 1995. [Google Scholar]
- Wang, J.X.; Dash, A.K.; Berthet, J.C.; Ephritikhine, M.; Eisen, M.S. Dehydrocoupling reactions of amines with silanes catalyzed by [(Et2N)3U][BPh4]. J. Organomet. Chem. 2000, 610, 49–57. [Google Scholar] [CrossRef]
- Yang, X.; Stern, C.; Marks, T.J. Models for organometallic molecule-support complexes. Very large counterion modulation of cationic actinide alkyl reactivity. Organometallics 1991, 10, 840–842. [Google Scholar] [CrossRef]
- Jia, L.; Yang, X.; Stern, C.; Marks, T.J. Cationic d0/f0 metallocene catalysts. Properties of binucleaoordinating counteranions derived therefrom. Organometallics 1994, 13, 3755–3757. [Google Scholar] [CrossRef]
- Jia, L.; Yang, X.; Stern, C.L.; Marks, T.J. Cationic metallocene polymerization catalysts based on tetrakis(pentafluorophenyl)borate and its derivatives. Probing the limits of anion “noncoordination” via a synthetic, solution dynamic, structural, and catalytic olefin polymerization study. Organometallics 1997, 16, 842–857. [Google Scholar] [CrossRef]
- Hayes, C.E.; Leznoff, D.B. Diamido-ether uranium(IV) alkyl complexes as single-component ethylene polymerization catalysts. Organometallics 2010, 29, 767–774. [Google Scholar] [CrossRef]
- Domeshek, E.; Batrice, R.J.; Aharonovich, S.; Tumanskii, B.; Botoshansky, M.; Eisen, M.S. Organoactinides in the polymerization of ethylene: Is TIBA a better cocatalyst than MAO? Dalton Trans. 2013, 42, 9069–9078. [Google Scholar] [CrossRef] [PubMed]
- Ikada, Y.; Tsuji, H. Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 2000, 21, 117–132. [Google Scholar] [CrossRef]
- Hedrick, J.L.; Magbitang, T.; Connor, E.F.; Glauser, T.; Volksen, W.; Hawker, C.J.; Lee, V.Y.; Miller, R.D. Application of complex macromolecular architectures for advanced microelectronic materials. Chem. Eur. J. 2002, 8, 3308–3319. [Google Scholar] [CrossRef]
- Joshi, P.; Madras, G. Degradation of polycaprolactone in supercritical fluids. Polym. Degrad. Stab. 2008, 93, 1901–1908. [Google Scholar] [CrossRef]
- Arbaoui, A.; Redshaw, C. Metal catalysts for ε-caprolactone polymerisation. Polym. Chem. 2010, 1, 801–826. [Google Scholar] [CrossRef]
- Villiers, C.; Thuery, P.; Ephritikhine, M. A comparison of analogous 4f- and 5f-element compounds: Syntheses, X-ray crystal structures and catalytic activity of the homoleptic amidinate complexes [M{MeC(NCy)2}3] (M = La, Nd or U). Eur. J. Inorg. Chem. 2004, 2004, 4624–4632. [Google Scholar] [CrossRef]
- Barnea, E.; Moradove, D.; Berthet, J.-C.; Ephritikhine, M.; Eisen, M.S. Surprising activity of organoactinide complexes in the polymerization of cyclic mono- and diesters. Organometallics 2006, 25, 320–322. [Google Scholar] [CrossRef]
- Rabinovich, E.; Aharonovich, S.; Botoshansky, M.; Eisen, M.S. Thorium 2-pyridylamidinates: Synthesis, structure and catalytic activity towards the cyclo-oligomerization of ε-caprolactone. Dalton Trans. 2010, 39, 6667–6676. [Google Scholar] [CrossRef] [PubMed]
- Karmel, I.S.R.; Elkin, T.; Fridman, N.; Eisen, M.S. Dimethylsilyl bis(amidinate)actinide complexes: Synthesis and reactivity towards oxygen containing substrates. Dalton Trans. 2014, 43, 11376–11387. [Google Scholar] [CrossRef] [PubMed]
- Karmel, I.S.R.; Fridman, N.; Eisen, M.S. Actinide amidinate complexes with a dimethylamine side arm: Synthesis, structural characterization, and reactivity. Organometallics 2015, 34, 636–643. [Google Scholar] [CrossRef]
- Das, R.K.; Barnea, E.; Andrea, T.; Kapon, M.; Fridman, N.; Botoshansky, M.; Eisen, M.S. Group 4 lanthanide and actinide organometallic inclusion complexes. Organometallics 2015, 34, 742–752. [Google Scholar] [CrossRef]
- Walshe, A.; Fang, J.; Maron, L.; Baker, R.J. New mechanism for the ring-opening polymerization of lactones? Uranyl aryloxide-induced intermolecular catalysis. Inorg. Chem. 2013, 52, 9077–9086. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Zhao, N.; Chen, L.; Song, H.; Zi, G. Synthesis, structure, and catalytic activity of an organothorium hydride complex. Inorg. Chem. Commun. 2011, 14, 1838–1841. [Google Scholar] [CrossRef]
- Ren, W.; Zhao, N.; Chen, L.; Zi, G. Synthesis, structure, and catalytic activity of benzyl thorium metallocenes. Inorg. Chem. Commun. 2013, 30, 26–28. [Google Scholar] [CrossRef]
- Hayes, C.E.; Sarazin, Y.; Katz, M.J.; Carpentier, J.-F.; Leznoff, D.B. Diamido-ether actinide complexes as initiators for lactide ring-opening polymerization. Organometallics 2013, 32, 1183–1192. [Google Scholar] [CrossRef]
- Peters, R.G.; Warner, B.P.; Burns, C.J. The catalytic reduction of azides and hydrazines using high-valent organouranium complexes. J. Am. Chem. Soc. 1999, 121, 5585–5586. [Google Scholar] [CrossRef]
- Baker, R.J.; Walshe, A. New reactivity of the uranyl ion: Ring opening polymerisation of epoxides. Chem. Commun. 2012, 48, 985–987. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Walshe, A.; Maron, L.; Baker, R.J. Ring-opening polymerization of epoxides catalyzed by uranyl complexes: An experimental and theoretical study of the reaction mechanism. Inorg. Chem. 2012, 51, 9132–9140. [Google Scholar] [CrossRef] [PubMed]
- Behrle, A.C.; Schmidt, J.A.R. Insertion reactions and catalytic hydrophosphination of heterocumulenes using α-metalated N,N-dimethylbenzylamine rare-earth-metal complexes. Organometallics 2013, 32, 1141–1149. [Google Scholar] [CrossRef]
- Tu, J.; Li, W.; Xue, M.; Zhang, Y.; Shen, Q. Bridged bis(amidinate) lanthanide aryloxides: Syntheses, structures, and catalytic activity for addition of amines to carbodiimides. Dalton Trans. 2013, 42, 5890–5901. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xue, M.; Yao, H.; Sun, H.; Zhang, Y.; Shen, Q. Enol-functionalized N-heterocyclic carbene lanthanide amide complexes: Synthesis, molecular structures and catalytic activity for addition of amines to carbodiimides. J. Organomet. Chem. 2012, 713, 27–34. [Google Scholar] [CrossRef]
- Cao, Y.; Du, Z.; Li, W.; Li, J.; Zhang, Y.; Xu, F.; Shen, Q. Activation of carbodiimide and transformation with amine to guanidinate group by Ln(OAr)3(THF)2 (Ln: Lanthanide and yttrium) and Ln(OAr)3(THF)2 as a novel precatalyst for addition of amines to carbodiimides: Influence of aryloxide group. Inorg. Chem. 2011, 50, 3729–3737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, C.; Qian, C.; Han, F.; Xu, F.; Shen, Q. Heterobimetallic dianionic guanidinate complexes of lanthanide and lithium: Highly efficient precatalysts for catalytic addition of amines to carbodiimides to synthesize guanidines. Tetrahedron 2011, 67, 8790–8799. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, H.; Yao, H.; Zhang, Y.; Yao, Y.; Shen, Q. The first bridged lanthanide carbene complex formed through reduction of carbodiimide by diamine-bis(phenolate) ytterbium(II) complex and its reactivity to phenylisocyanate. Chem. Commun. 2011, 47, 3595–3597. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Zhang, J.; Hong, L.; Chen, Z.; Zhou, X. Insertion of isocyanate and isothiocyanate into the Ln–P σ-bond of organolanthanide phosphides. Organometallics 2011, 30, 5809–5814. [Google Scholar] [CrossRef]
- Karmel, I.S.R.; Tamm, M.; Eisen, M.S. Actinide-mediated catalytic addition of E–H bonds (E = N, P, S) to carbodiimides, isocyanates, and isothiocyanates. Angew. Chem. Int. Ed. 2015, 54, 12422–12425. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karmel, I.S.R.; Batrice, R.J.; Eisen, M.S. Catalytic Organic Transformations Mediated by Actinide Complexes. Inorganics 2015, 3, 392-428. https://doi.org/10.3390/inorganics3040392
Karmel ISR, Batrice RJ, Eisen MS. Catalytic Organic Transformations Mediated by Actinide Complexes. Inorganics. 2015; 3(4):392-428. https://doi.org/10.3390/inorganics3040392
Chicago/Turabian StyleKarmel, Isabell S. R., Rami J. Batrice, and Moris S. Eisen. 2015. "Catalytic Organic Transformations Mediated by Actinide Complexes" Inorganics 3, no. 4: 392-428. https://doi.org/10.3390/inorganics3040392
APA StyleKarmel, I. S. R., Batrice, R. J., & Eisen, M. S. (2015). Catalytic Organic Transformations Mediated by Actinide Complexes. Inorganics, 3(4), 392-428. https://doi.org/10.3390/inorganics3040392