
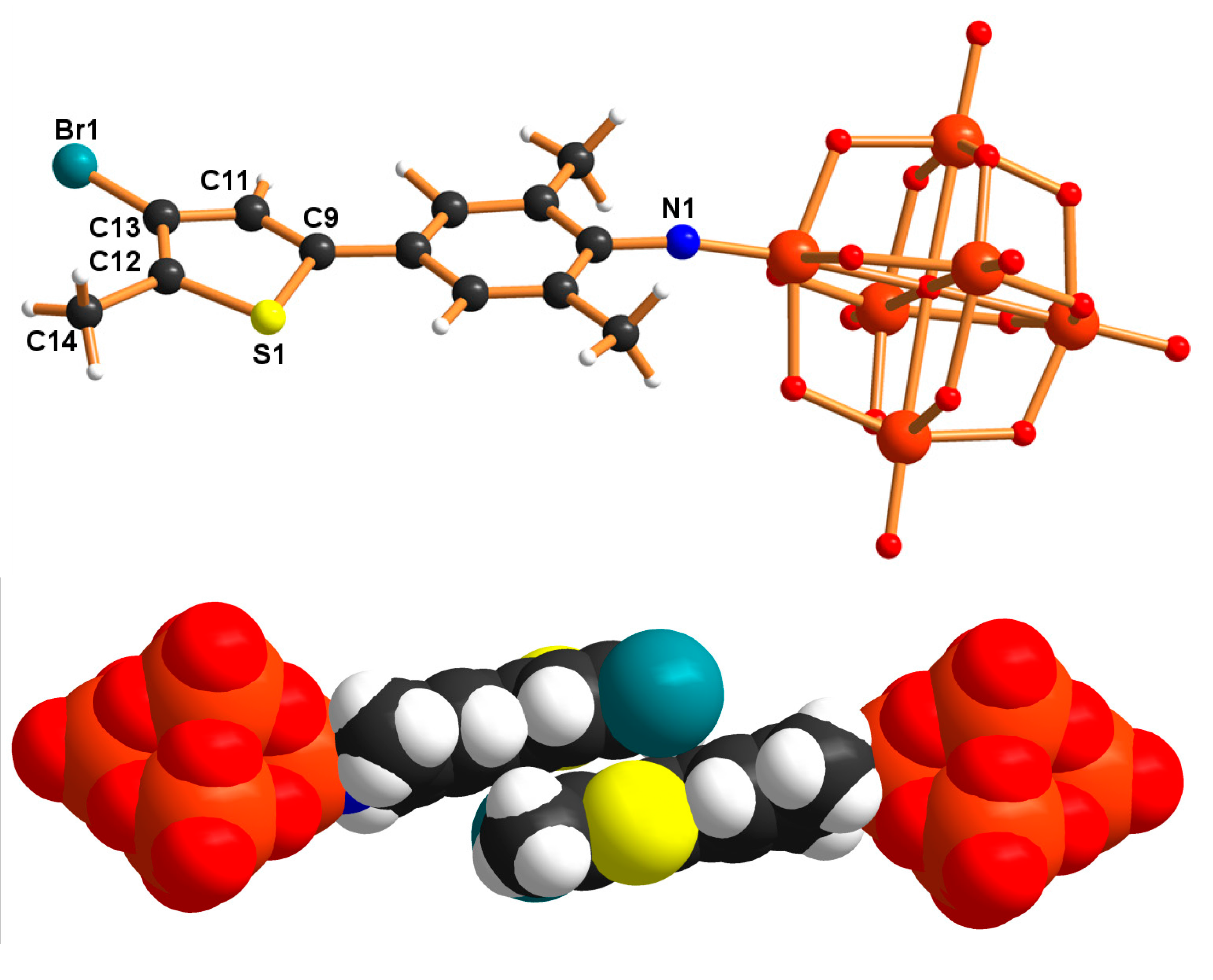
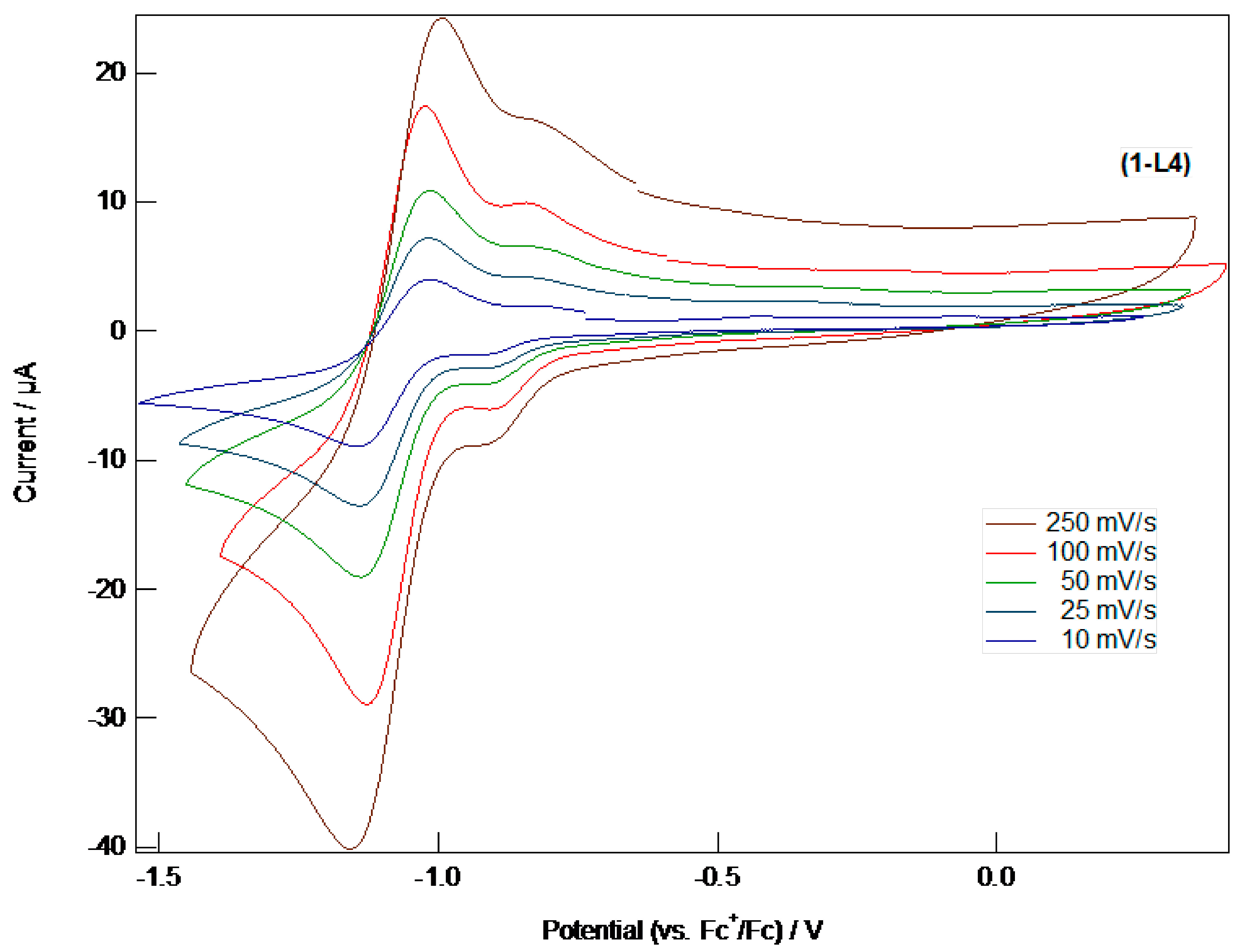
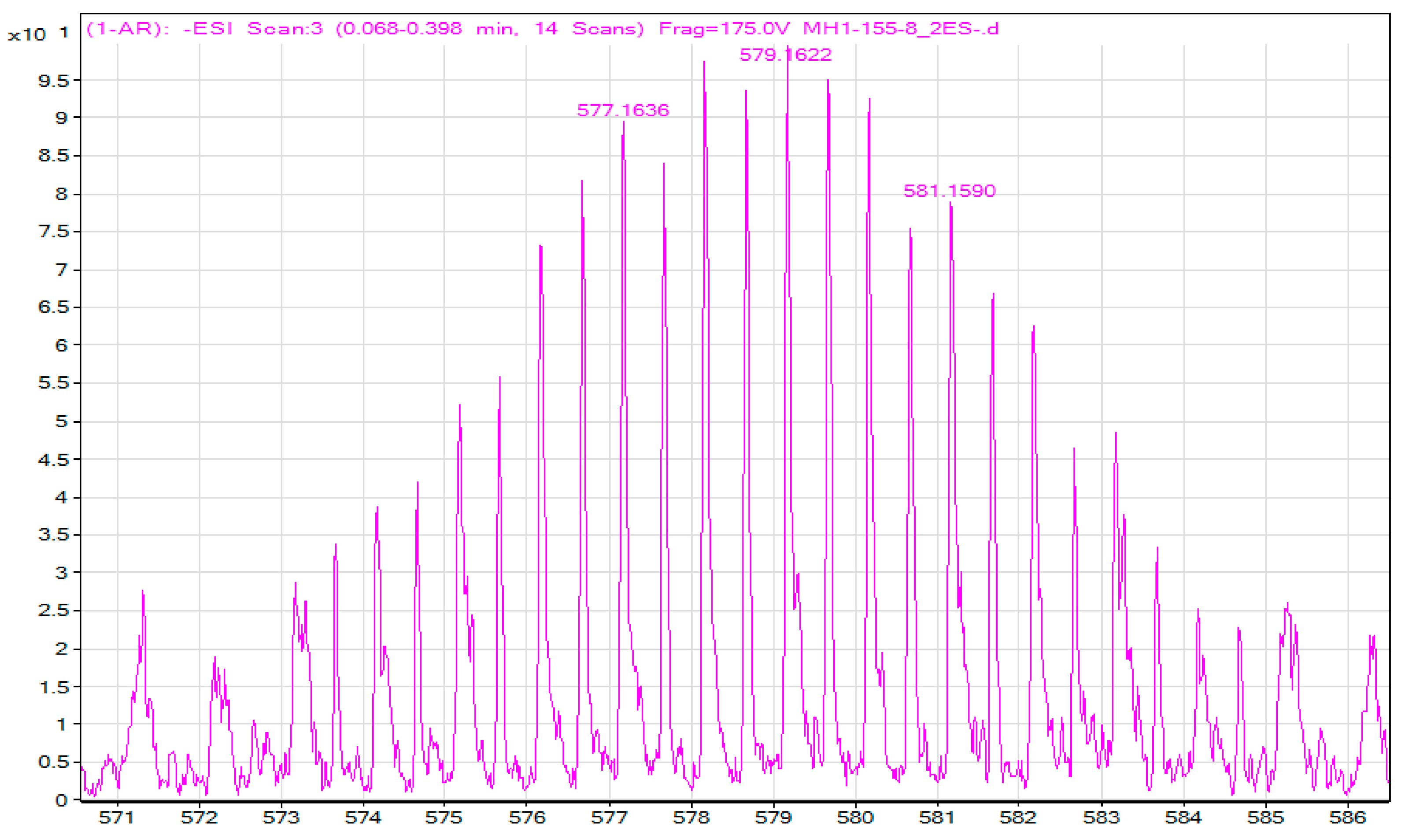
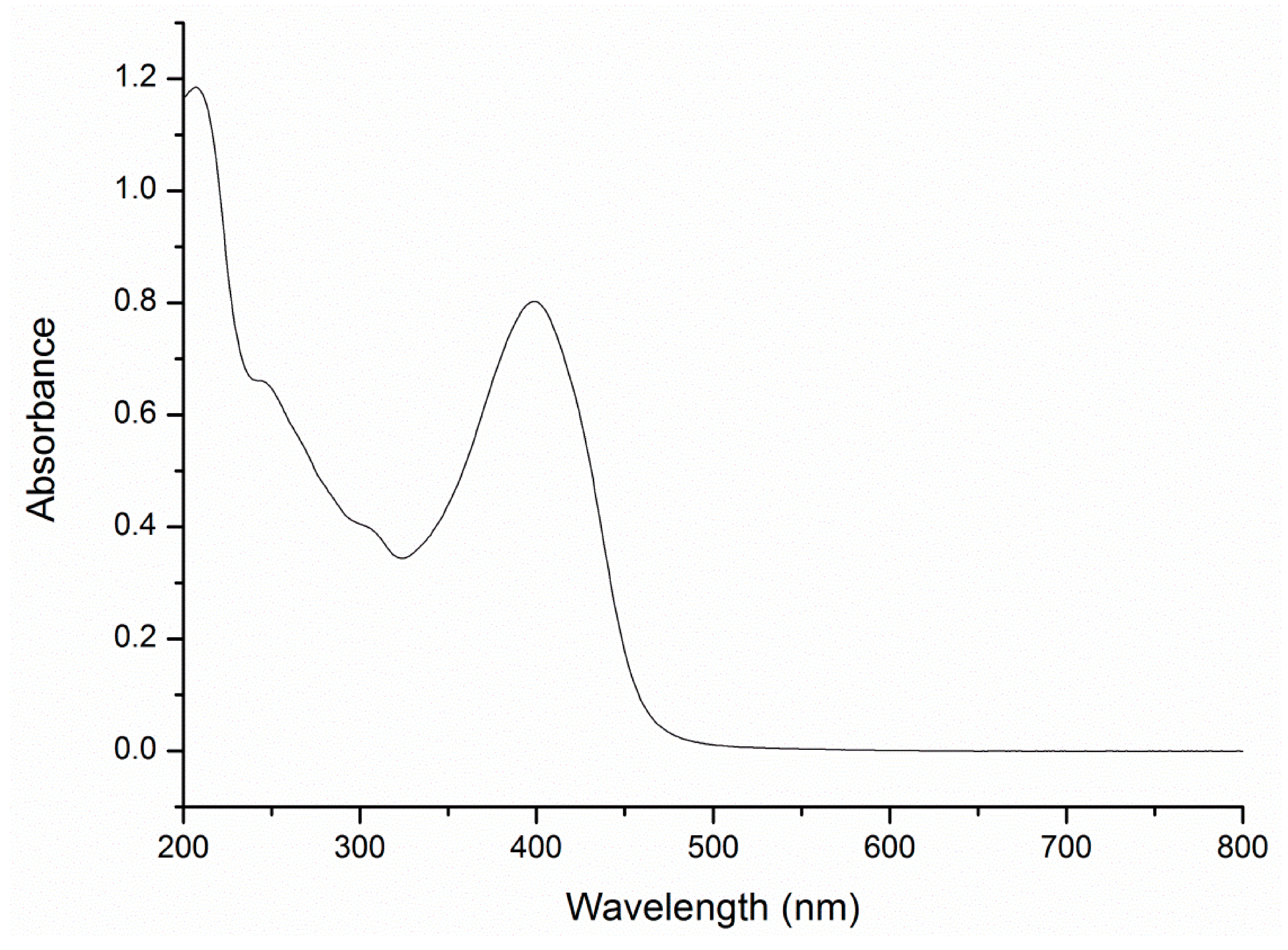
A Heteroaromatically Functionalized Hexamolybdate

{kind=link}
{kind=link}
{kind=link}
{kind=link}
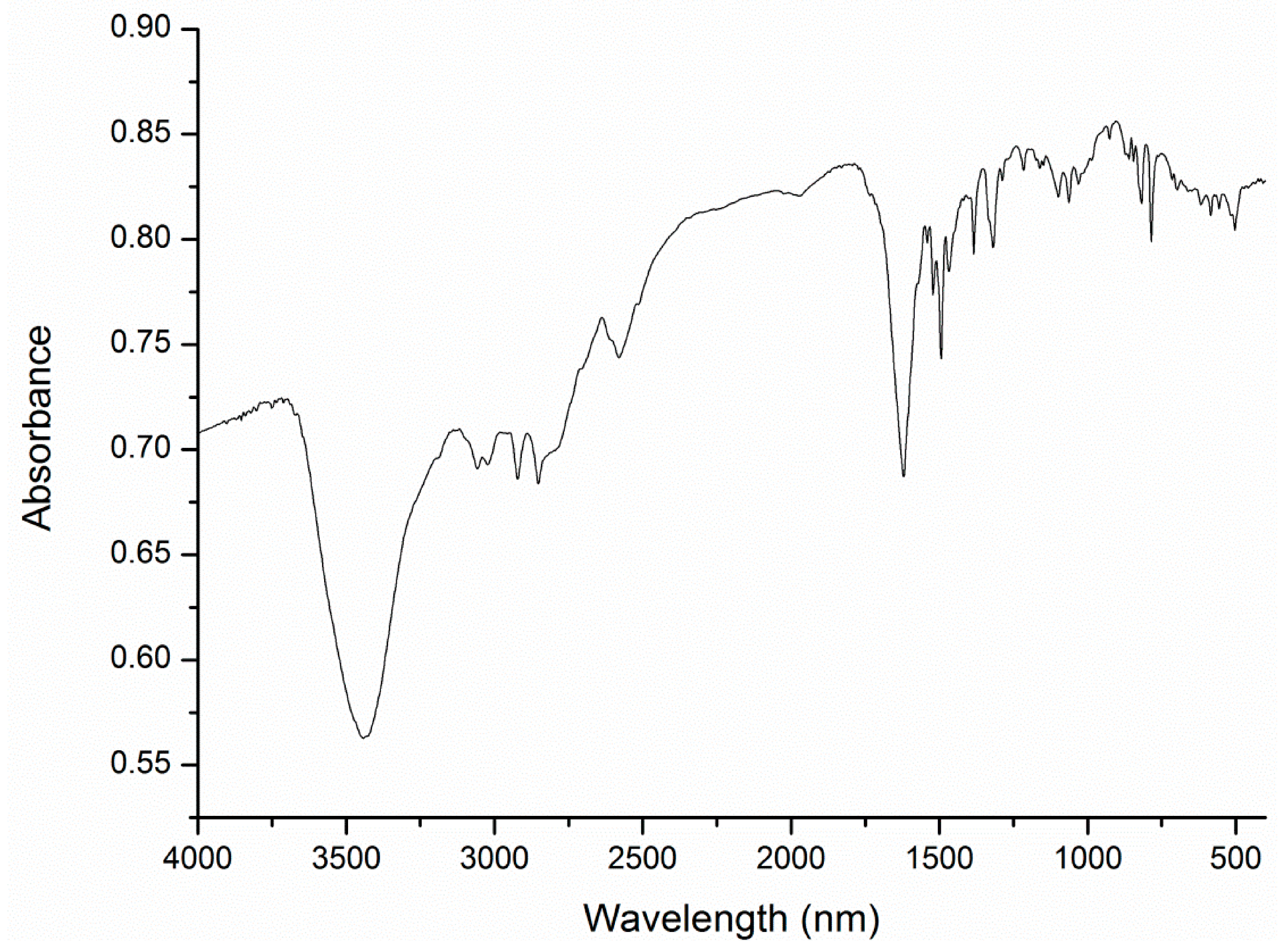{kind=link}
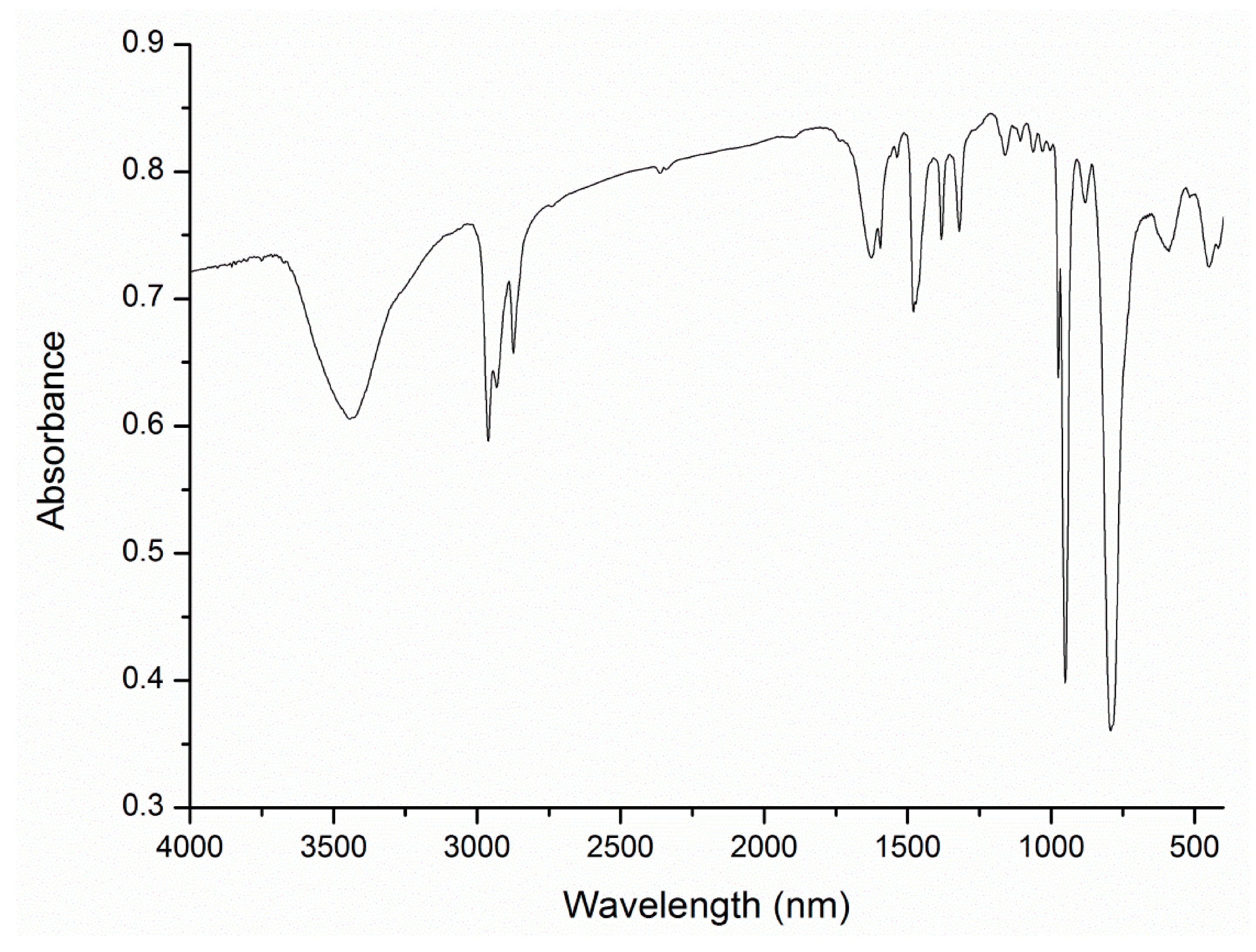{kind=link}
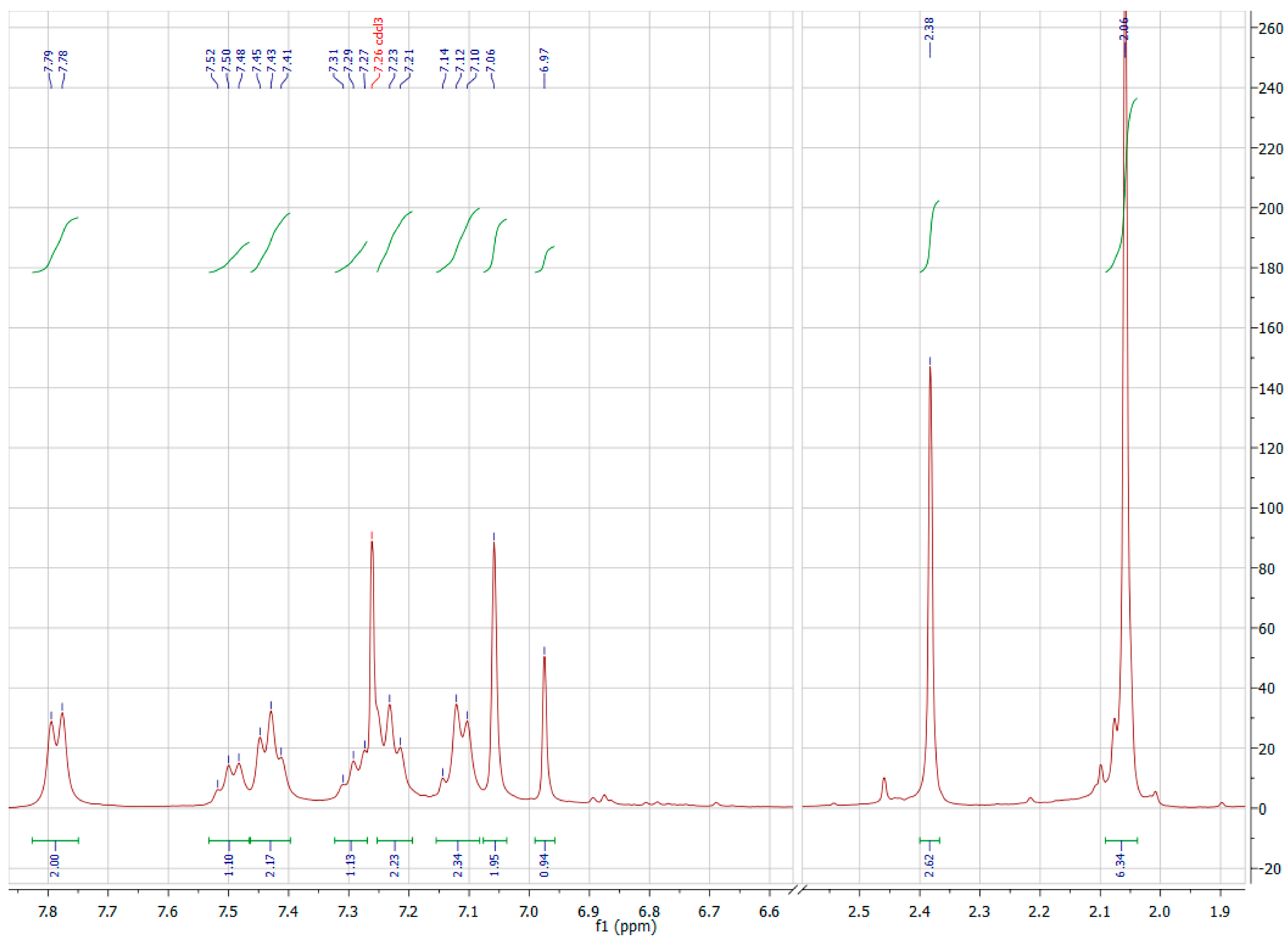{kind=link}
{kind=link}
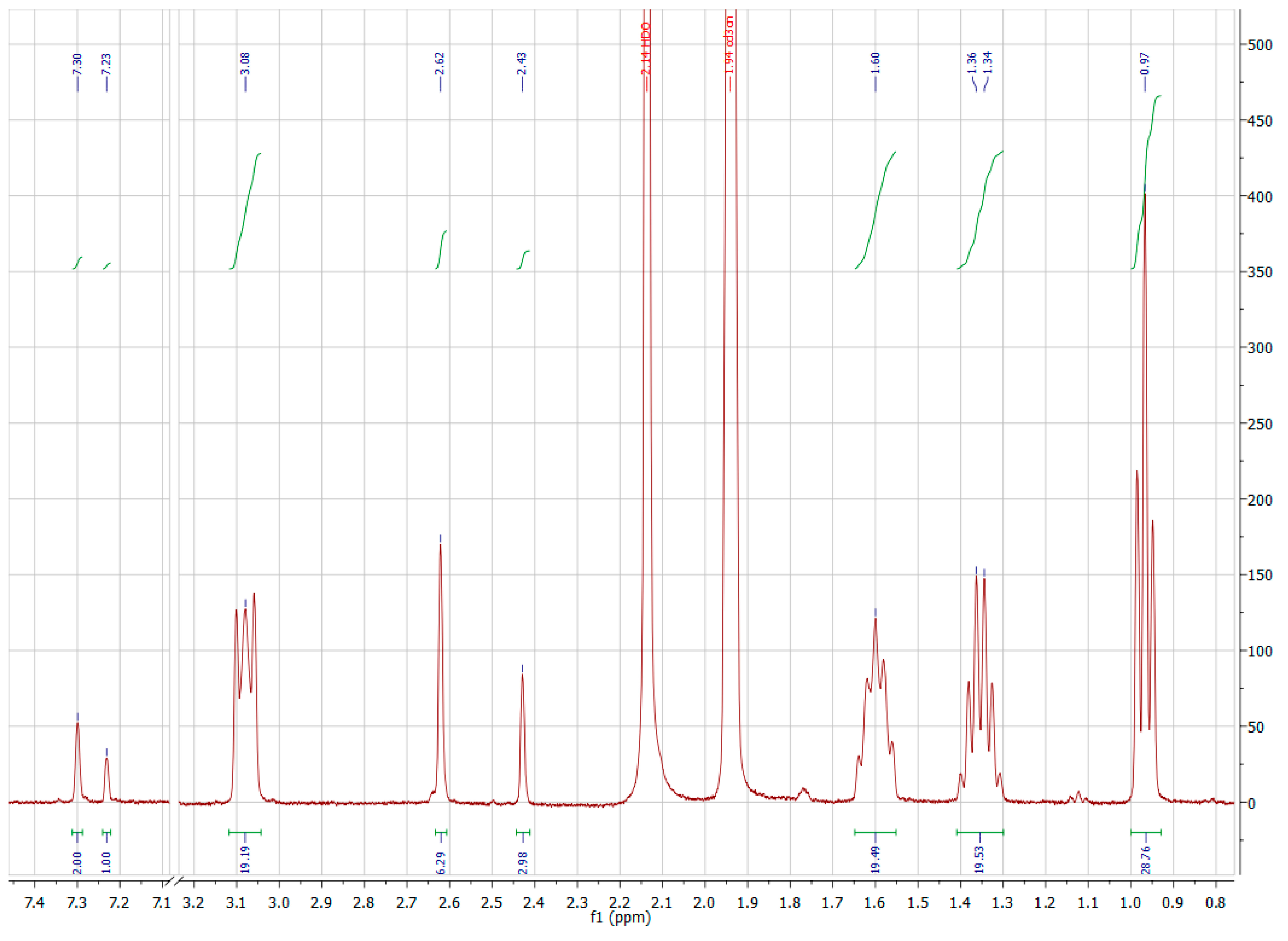{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
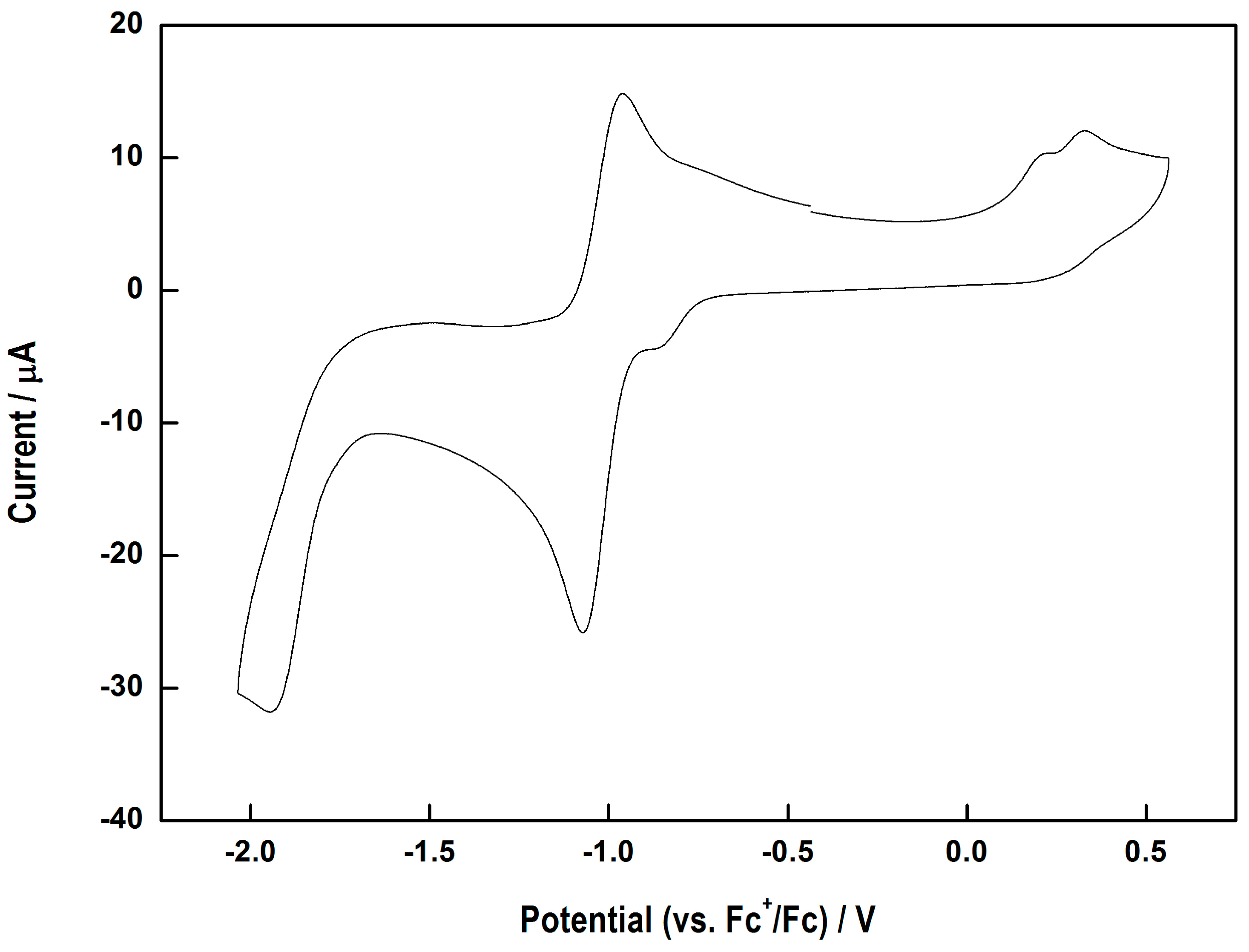
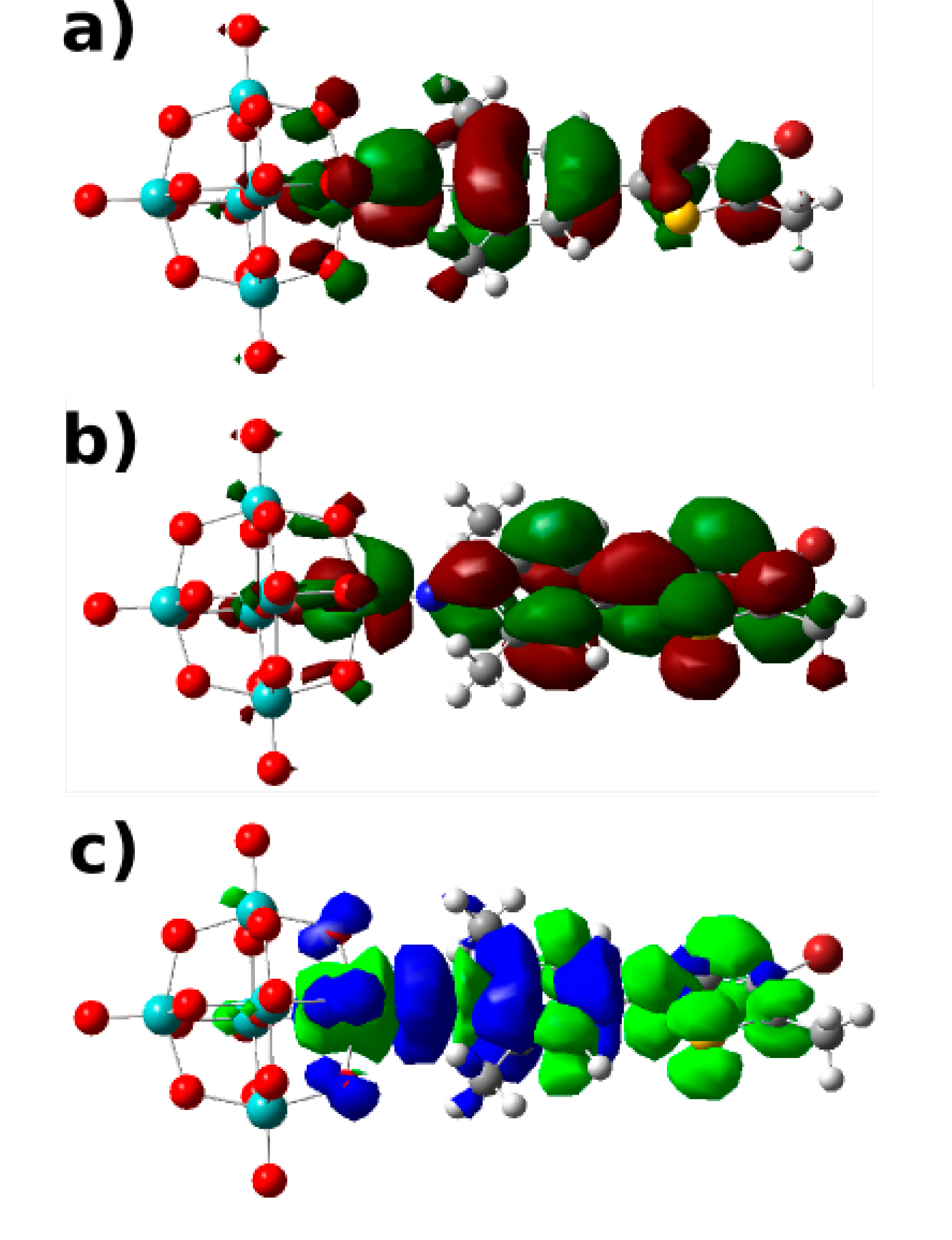
Abstract
Share and Cite
Healey, M.R.; Best, S.P.; Goerigk, L.; Ritchie, C. A Heteroaromatically Functionalized Hexamolybdate. Inorganics 2015, 3, 82-100. https://doi.org/10.3390/inorganics3020082
Healey MR, Best SP, Goerigk L, Ritchie C. A Heteroaromatically Functionalized Hexamolybdate. Inorganics. 2015; 3(2):82-100. https://doi.org/10.3390/inorganics3020082
Chicago/Turabian StyleHealey, Merinda R., Stephen P. Best, Lars Goerigk, and Chris Ritchie. 2015. "A Heteroaromatically Functionalized Hexamolybdate" Inorganics 3, no. 2: 82-100. https://doi.org/10.3390/inorganics3020082
APA StyleHealey, M. R., Best, S. P., Goerigk, L., & Ritchie, C. (2015). A Heteroaromatically Functionalized Hexamolybdate. Inorganics, 3(2), 82-100. https://doi.org/10.3390/inorganics3020082