

The Effect of Central Metal Ions (Dy, Er, Ni, and V) on the Structural and HSA-Binding Properties of 2-Hydroxy-3-methoxybenzaldehyde Semicarbazone Complexes

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Results and Discussion
2.1. Crystallographic Structure of Selected Compounds
2.2. Hirshfeld Surface Analysis of Selected Compounds
2.3. DFT Optimization of Structure
2.4. QTAIMs Analysis
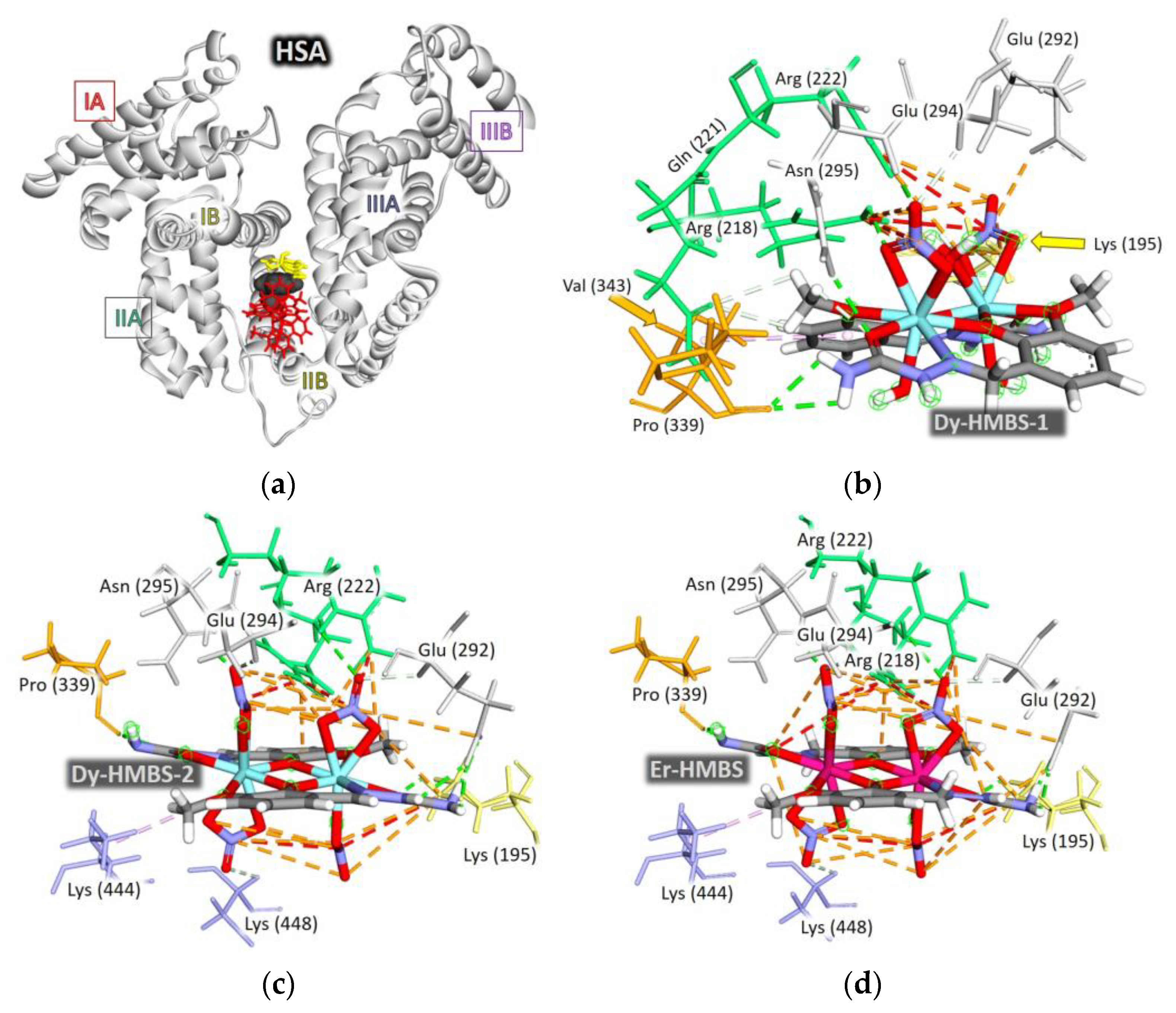
2.5. Molecular Docking Analysis of HSA Binding Properties
2.5.1. Docking Calculations and Binding Site Determination
2.5.2. Binding Energies and Structural Considerations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Ligand | CMI | CN | R(CMI) (pm) | The Structure Used for Docking | ∆Gb (kJ mol−1) |
|---|---|---|---|---|---|---|
| 1 | Dy-HMBS-1 | 2Dy(+3) | 9 | 122.3 CR | [Dy2(HMBS)2(NO3)2(H2O)3]2+ | −37.07 |
| 2 | Dy-HMBS-2 | 2Dy(+3) | 9 | 122.3 CR | [Dy2(HMBSs)2(NO3)4] | −44.35 |
| 3 | Er-HMBS | 2Er(+3) | 9 | 106.2 IR | [Er2(HMBS)2(NO3)4] | −43.60 |
| 4 | Ni-HMBS | 2Ni(+2) | 6 | 83.0 CR | [Ni2(HMBS)2(bpy)2] 2+ | −29.37 |
| 5 | V-HMBS | V(+5) | 5 | 46.0 IR | [VO2(HMBS)] | −29.54 |
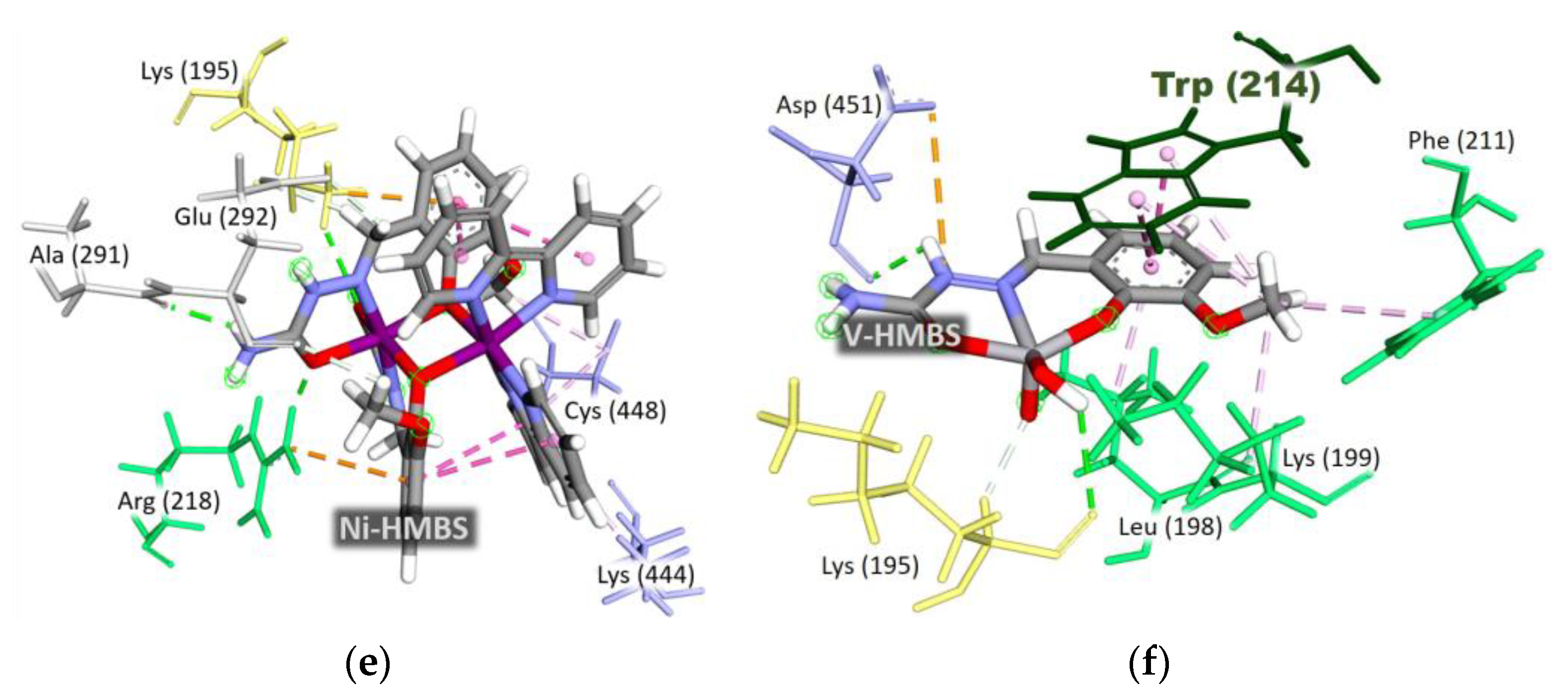
2.5.3. Interaction Patterns and Binding Site Specificity
2.5.4. Hydrophobic Interactions in Non-Nitrate Complexes
2.5.5. Intramolecular Interactions and Structural Stability
3. Materials and Methods
3.1. Cambridge Structural Database (CSD) Search
3.2. Hirshfeld Surface Analysis
3.3. Optimization of Structures and QTAIMs Analysis
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casas, J.S.; García-Tasende, M.S.; Sordo, J. Main group metal complexes of semicarbazones and thiosemicarbazones. A structural review. Coord. Chem. Rev. 2000, 209, 197–261. [Google Scholar] [CrossRef]
- Mustafa, Y.F. Modern Developments in the Application and Function of Metal/Metal Oxide Nanocomposite–Based Antibacterial Agents. Bionanoscience 2023, 13, 840–852. [Google Scholar] [CrossRef]
- Pal, R.; Kumar, V.; Gupta, A.K.; Beniwal, V. Synthesis, characterization and DNA photocleavage study of a novel dehydroacetic acid based hydrazone Schiff’s base and its metal complexes. Med. Chem. Res. 2014, 23, 3327–3335. [Google Scholar] [CrossRef]
- Padhyé, S.; Kauffman, G.B. Transition metal complexes of semicarbazones and thiosemicarbazones. Coord. Chem. Rev. 1985, 63, 127–160. [Google Scholar] [CrossRef]
- Alsoliemy, A.; Alrefaei, A.F.; Almehmadi, S.J.; Almehmadi, S.J.; Hossan, A.; Khalifa, M.E.; El-Metwaly, N.M. Synthesis, characterization and self-assembly of new cholesteryl-substitued sym-tetrazine: Fluorescence, gelation and mesogenic properties. J. Mol. Liq. 2021, 342, 117543. [Google Scholar] [CrossRef]
- Fernández, M.; Becco, L.; Correia, I.; Benítez, J.; Piro, O.E.; Echeverria, G.A.; Medeiros, A.; Comini, M.; Lavaggi, M.L.; González, M.; et al. Oxidovanadium(IV) and dioxidovanadium(V) complexes of tridentate salicylaldehyde semicarbazones: Searching for prospective antitrypanosomal agents. J. Inorg. Biochem. 2013, 127, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, H.; Gambino, D. The Wide Pharmacological Versatility of Semicarbazones, Thiosemicarbazones and Their Metal Complexes. Mini-Reviews Med. Chem. 2004, 4, 31–39. [Google Scholar] [CrossRef]
- Gerasimenko, A.V.; Davidovich, R.L.; Bulimestru, I.G.; Gulea, A.P.; Ng, S.W. Bis(μ-salicylaldehyde semicarbazonato)bis[formatocopper(II)]. Acta Crystallogr. Sect. E Struct. Rep. Online 2005, 61, m1816–m1817. [Google Scholar] [CrossRef]
- Chumakov, Y.M.; Tsapkov, V.I.; Biyushkin, V.N.; Mazus, M.D.; Samus’, N.M. Crystal structure of salicylidenesemicarbazidodimethylformamidoaqua- copper(II) salicylidenesemicarbazidodiaquacopper(II) sulfate trihydrate. Crystallogr. Rep. 1996, 41, 831–836. [Google Scholar] [CrossRef]
- Patole, J.; Dutta, S.; Padhye, S.; Sinn, E. Tuning up superoxide dismutase activity of copper complex of salicylaldehyde semicarbazone by heterocyclic bases pyridine and N-methyl imidazole. Inorganica Chim. Acta 2001, 318, 207–211. [Google Scholar] [CrossRef]
- Lee, W.Y.; Lee, P.P.F.; Yan, Y.K.; Lau, M. Cytotoxic copper(ii) salicylaldehyde semicarbazone complexes: Mode of action and proteomic analysis. Metallomics 2010, 2, 694. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-L.; Liu, B.; Yang, B.-S.; Huang, S.-P. Novel copper(II) complex with unusual π-stacking structure, [Cu(SSC)Cl]2·CH3OH·2H2O (SSC = salicylaldehyde semicarbazone anion). J. Struct. Chem. 2008, 49, 570–574. [Google Scholar] [CrossRef]
- Zhang, Y.-F.; Zhou, K.-L.; Lou, Y.-Y.; Pan, D.; Shi, J.-H. Investigation of the binding interaction between estazolam and bovine serum albumin: Multi-spectroscopic methods and molecular docking technique. J. Biomol. Struct. Dyn. 2017, 35, 3605–3614. [Google Scholar] [CrossRef]
- Noblía, P.; Baran, E.J.; Otero, L.; Draper, P.; Cerecetto, H.; González, M.; Piro, O.E.; Castellano, E.E.; Inohara, T.; Adachi, Y.; et al. New Vanadium(V) Complexes with Salicylaldehyde Semicarbazone Derivatives: Synthesis, Characterization, and in vitro Insulin-Mimetic Activity − Crystal Structure of [V v O 2 (salicylaldehyde semicarbazone)]. Eur. J. Inorg. Chem. 2004, 2004, 322–328. [Google Scholar] [CrossRef]
- Bogdanovic, G.; Leovac, V.; Vojinovic-Jesic, L.; De, B.-S. Crystal structure of tris(pyridine)(salicylaldehyde semicarbazonato(2-))cobalt(III)-trichloropyridinecobaltate(II) at 293 and 120K. J. Serbian Chem. Soc. 2007, 72, 63–71. [Google Scholar] [CrossRef]
- Mir, I.A.; Ain, Q.U.; Qadir, T.; Malik, A.Q.; Jan, S.; Shahverdi, S.; Nabi, S.A. A review of semicarbazone-derived metal complexes for application in biomedicine and related fields. J. Mol. Struct. 2024, 1295, 136216. [Google Scholar] [CrossRef]
- Binil, P.S.; Anoop, M.R.; Suma, S.; Sudarsanakumar, M.R. Growth, spectral, and thermal characterization of 2-hydroxy-3-methoxybenzaldehyde semicarbazone. J. Therm. Anal. Calorim. 2013, 112, 913–919. [Google Scholar] [CrossRef]
- Vomisescu, C.; Bourosh, P.; Kravtsov, V.; Dragancea, D. Nickel(III) Complex Derived from 2-Hydroxy-3-Methoxybenzaldehyde Semicarbazone and 2,2’-Bipyridine. Chem. J. Mold. 2013, 8, 78–82. [Google Scholar] [CrossRef]
- Chellan, P.; Chibale, K.; Smith, G.S. Molecular Structure of an Unexpected Binuclear Salicylaldimine Semicarbazone Palladium(II) Complex. J. Chem. Crystallogr. 2011, 41, 747–750. [Google Scholar] [CrossRef]
- Bag, P.; Rastogi, C.K.; Biswas, S.; Sivakumar, S.; Mereacre, V.; Chandrasekhar, V. Homodinuclear lanthanide {Ln 2 } (Ln = Gd, Tb, Dy, Eu) complexes prepared from an o-vanillin based ligand: Luminescence and single-molecule magnetism behavior. Dalt. Trans. 2015, 44, 4328–4340. [Google Scholar] [CrossRef]
- Biswas, S.; Das, S.; Hossain, S.; Bar, A.K.; Sutter, J.; Chandrasekhar, V. Tetranuclear Lanthanide(III) Complexes Containing a Square-Grid Core: Synthesis, Structure, and Magnetism. Eur. J. Inorg. Chem. 2016, 2016, 4683–4692. [Google Scholar] [CrossRef]
- Biswas, S.; Das, S.; Rogez, G.; Chandrasekhar, V. Hydrazone-Ligand-Based Homodinuclear Lanthanide Complexes: Synthesis, Structure, and Magnetism. Eur. J. Inorg. Chem. 2016, 2016, 3322–3329. [Google Scholar] [CrossRef]
- Hutchings, A.-J.; Habib, F.; Holmberg, R.J.; Korobkov, I.; Murugesu, M. Structural Rearrangement Through Lanthanide Contraction in Dinuclear Complexes. Inorg. Chem. 2014, 53, 2102–2112. [Google Scholar] [CrossRef]
- Li, M.; Wu, H.; Zhang, S.; Sun, L.; Ke, H.; Wei, Q.; Xie, G.; Chen, S.; Gao, S. Fine-Tuning Ligand Fields with Schiff-Base Ligands in Dy 2 Compounds. Eur. J. Inorg. Chem. 2017, 2017, 811–819. [Google Scholar] [CrossRef]
- Taylor, R.; Wood, P.A. A Million Crystal Structures: The Whole Is Greater than the Sum of Its Parts. Chem. Rev. 2019, 119, 9427–9477. [Google Scholar] [CrossRef]
- Milovanović, M.R.; Stanković, I.M.; Živković, J.M.; Ninković, D.B.; Hall, M.B.; Zarić, S.D. Water: New aspect of hydrogen bonding in the solid state. IUCrJ 2022, 9, 639–647. [Google Scholar] [CrossRef]
- Ninković, D.B.; Janjić, G.V.; Zarić, S.D. Crystallographic and ab Initio Study of Pyridine Stacking Interactions. Local Nature of Hydrogen Bond Effect in Stacking Interactions. Cryst. Growth Des. 2012, 12, 1060–1063. [Google Scholar] [CrossRef]
- Milovanović, M.R.; Živković, J.M.; Ninković, D.B.; Stanković, I.M.; Zarić, S.D. How flexible is the water molecule structure? Analysis of crystal structures and the potential energy surface. Phys. Chem. Chem. Phys. 2020, 22, 4138–4143. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Kargar, H.; Ardakani, A.A.; Tahir, M.N.; Ashfaq, M.; Munawar, K.S. Synthesis, spectral characterization, crystal structure and antibacterial activity of nickel(II), copper(II) and zinc(II) complexes containing ONNO donor Schiff base ligands. J. Mol. Struct. 2021, 1233, 130112. [Google Scholar] [CrossRef]
- Sinicropi, M.S.; Ceramella, J.; Iacopetta, D.; Catalano, A.; Mariconda, A.; Rosano, C.; Saturnino, C.; El-Kashef, H.; Longo, P. Metal Complexes with Schiff Bases: Data Collection and Recent Studies on Biological Activities. Int. J. Mol. Sci. 2022, 23, 14840. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Herrera, L.A.; Hernández-Romero, D.; Cruz-Navarro, J.A.; Ramos-Ligonio, Á.; López-Monteon, A.; Rivera-Villanueva, J.M.; Morales-Morales, D.; Colorado-Peralta, R. Transition metal complexes with tetradentate Schiff bases (N2O2) obtained from salicylaldehyde: A review of their possible anticancer properties. Coord. Chem. Rev. 2024, 505, 215698. [Google Scholar] [CrossRef]
- Matsia, S.; Papadopoulos, A.; Hatzidimitriou, A.; Schumacher, L.; Koldemir, A.; Pöttgen, R.; Panagiotopoulou, A.; Chasapis, C.T.; Salifoglou, A. Hybrid Lanthanide Metal–Organic Compounds with Flavonoids: Magneto-Optical Properties and Biological Activity Profiles. Int. J. Mol. Sci. 2025, 26, 1198. [Google Scholar] [CrossRef] [PubMed]
- Jevtovic, V.; Rakić, A.; Alshammari, O.A.O.; Alhar, M.S.; Alenezi, T.; Rakic, V.; Dimić, D. Theoretical Study of the Effects of Different Coordination Atoms (O/S/N) on Crystal Structure, Stability, and Protein/DNA Binding of Ni(II) Complexes with Pyridoxal-Semi, Thiosemi, and Isothiosemicarbazone Ligand Systems. Inorganics 2024, 12, 251. [Google Scholar] [CrossRef]
- Harraf, E.; Bikas, R.; Soltani, B.; Lis, T. Synthesis, spectroscopic properties, crystal structure and Hirshfeld surface analysis of Pr(III), Sm(III), Gd(III), Dy(III), and Ho(III) coordination compounds with Schiff base ligand derived from 4-aminoantipyrine. J. Mol. Struct. 2024, 1316, 139013. [Google Scholar] [CrossRef]
- Macedi, E.; Rossi, P.; Formica, M.; Giorgi, L.; Lippi, M.; Montis, R.; Paderni, D.; Paoli, P.; Fusi, V. Crystal structure, Hirshfeld surface analysis and energy framework calculations of different metal complexes of a biphenol-based ligand: Role of solvent and transition metal ion. J. Mol. Struct. 2024, 1299, 137146. [Google Scholar] [CrossRef]
- Hueso-Ureña, F.; Jiménez-Pulido, S.B.; Fernández-Liencres, M.P.; Fernández-Gómez, M.; Moreno-Carretero, M.N. A new five-coordinated CuIP2NO2 system: XRD structure of 6-acetyl-1,3,7-trimethyl-pteridine-2,4(1H,3H)-dione and its Cu(i) (N5,O61,O4)-tridentate complex with triphenylphosphine. An AIM study of the nature of metal–ligand bonds. Dalt. Trans. 2008, 45, 6461. [Google Scholar] [CrossRef]
- Fabijanić, I.; Matković-Čalogović, D.; Pilepić, V.; Ivanišević, I.; Mohaček-Grošev, V.; Sanković, K. New investigations of the guanine trichloro cuprate(II) complex crystal. J. Mol. Struct. 2017, 1128, 317–324. [Google Scholar] [CrossRef]
- Soliman, S.M.; Albering, J.; Abu-Youssef, M.A.M. Structural analyses of two new highly distorted octahedral copper(II) complexes with quinoline-type ligands; Hirshfeld, AIM and NBO studies. Polyhedron 2017, 127, 36–50. [Google Scholar] [CrossRef]
- Lepetit, C.; Vabre, B.; Canac, Y.; Alikhani, M.E.; Zargarian, D. Pentacoordinated, square pyramidal cationic PCP Ni(II) pincer complexes: ELF and QTAIM topological analyses of nickel–triflate interactions. Theor. Chem. Acc. 2018, 137, 141. [Google Scholar] [CrossRef]
- Kasalović, M.P.; Jelača, S.; Milanović, Ž.; Maksimović-Ivanić, D.; Mijatović, S.; Lađarević, J.; Božić, B.; Marković, Z.; Dunđerović, D.; Rüffer, T.; et al. Novel triphenyltin(iv) compounds with carboxylato N-functionalized 2-quinolones as promising potential anticancer drug candidates: In vitro and in vivo evaluation. Dalt. Trans. 2024, 53, 8298–8314. [Google Scholar] [CrossRef]
- Boulechfar, C.; Ferkous, H.; Boufas, S.; Berredjem, M.; Delimi, A.; Djellali, S.; Djedouani, A.; Bahadi, R.; Laamari, S.; Yadav, K.K.; et al. Synthesis, electrochemical, and quantum chemical studies of some metal complexes: Mn(II), Co(II), and Zn(II) with 2-furaldehyde semicarbazone. J. Mol. Struct. 2023, 1271, 134007. [Google Scholar] [CrossRef]
- Jevtovic, V.; Golubović, L.; Alshammari, O.A.O.; Alhar, M.S.; Alanazi, T.Y.A.; Radulović, A.; Nakarada, Đ.; Dimitrić Marković, J.; Rakić, A.; Dimić, D. Structural, Antioxidant, and Protein/DNA-Binding Properties of Sulfate-Coordinated Ni(II) Complex with Pyridoxal-Semicarbazone (PLSC) Ligand. Inorganics 2024, 12, 280. [Google Scholar] [CrossRef]
- Jevtovic, V.; Golubović, L.; Alshammari, B.; Alshammari, M.R.; Rajeh, S.Y.; Alreshidi, M.A.; Alshammari, O.A.O.; Rakić, A.; Dimić, D. Crystal Structure, Theoretical Analysis, and Protein/DNA Binding Activity of Iron(III) Complex Containing Differently Protonated Pyridoxal–S-Methyl-Isothiosemicarbazone Ligands. Int. J. Mol. Sci. 2024, 25, 7058. [Google Scholar] [CrossRef] [PubMed]
- Jevtovic, V.; Golubović, L.; Alshammari, O.A.O.; Alhar, M.S.; Alanazi, T.Y.A.; Rakic, V.; Ganguly, R.; Dimitrić Marković, J.; Rakić, A.; Dimić, D. The Counterion (SO42− and NO3−) Effect on Crystallographic, Quantum-Chemical, Protein-, and DNA-Binding Properties of Two Novel Copper(II)–Pyridoxal-Aminoguanidine Complexes. Crystals 2024, 14, 814. [Google Scholar] [CrossRef]
- Papadopoulos, Z.; Doulopoulou, E.; Zianna, A.; Hatzidimitriou, A.G.; Psomas, G. Copper(II) Complexes of 5–Fluoro–Salicylaldehyde: Synthesis, Characterization, Antioxidant Properties, Interaction with DNA and Serum Albumins. Molecules 2022, 27, 8929. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Allen, F.H.; Davies, J.E.; Galloy, J.J.; Johnson, O.; Kennard, O.; Macrae, C.F.; Mitchell, E.M.; Mitchell, G.F.; Smith, J.M.; Watson, D.G. The development of versions 3 and 4 of the Cambridge Structural Database System. J. Chem. Inf. Comput. Sci. 1991, 31, 187–204. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Grabowsky, S.; Dean, P.M.; Skelton, B.W.; Sobolev, A.N.; Spackman, M.A.; White, A.H. Crystal packing in the 2-R,4-oxo-[1,3-a/b]-naphthodioxanes—Hirshfeld surface analysis and melting point correlation. CrystEngComm 2012, 14, 1083–1093. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Savin, A.; Preuss, H. Energy-adjusted pseudopotentials for the rare earth elements. Theor. Chim. Acta 1989, 75, 173–194. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Todd, A. AIMAll, version 19.10.12; Keith, T.K., Ed.; Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural Basis of the Drug-binding Specificity of Human Serum Albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A versatile graphical tool for assisting molecular docking with Autodock Vina and Autodock4. Biol. Direct 2020, 15, 12. [Google Scholar] [CrossRef] [PubMed]
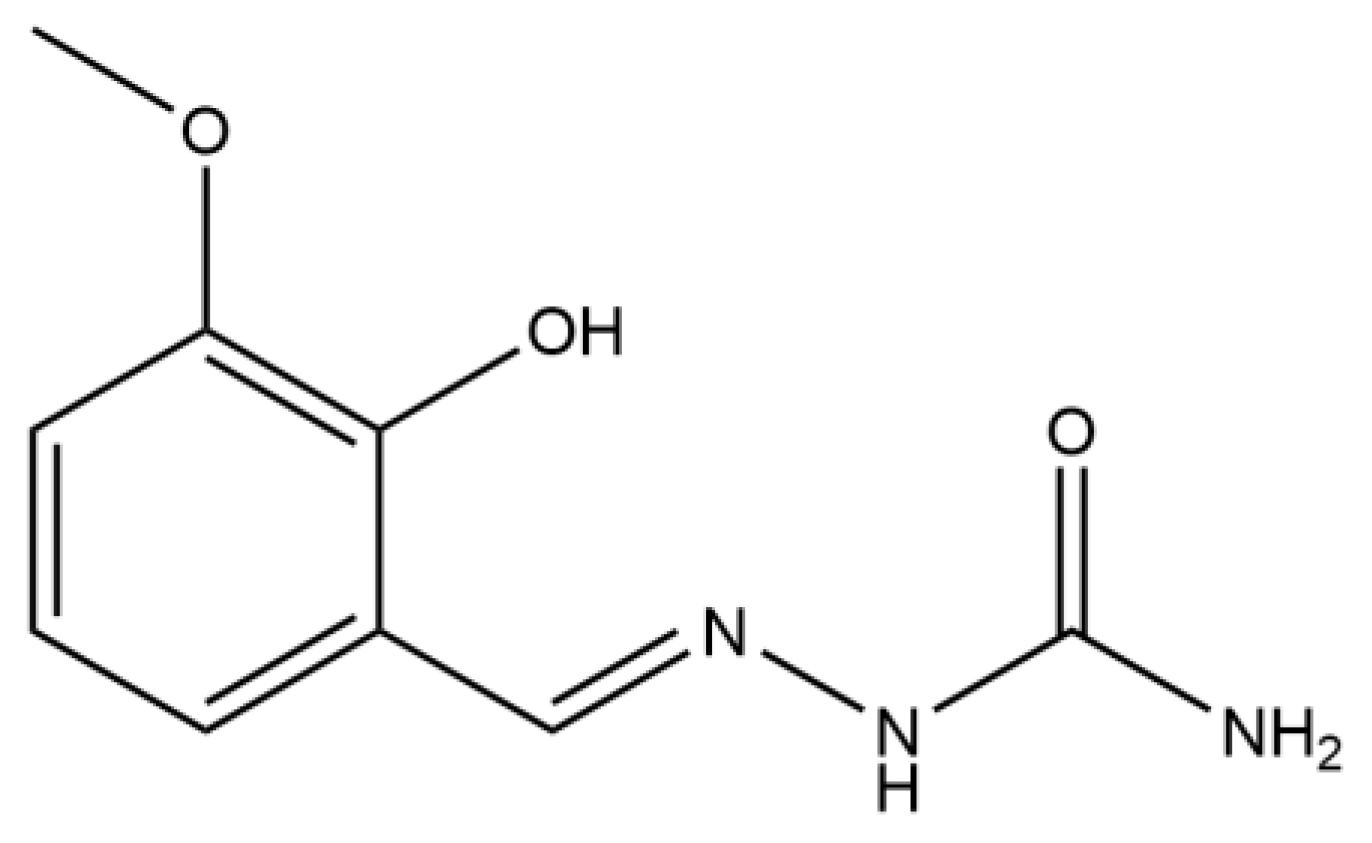
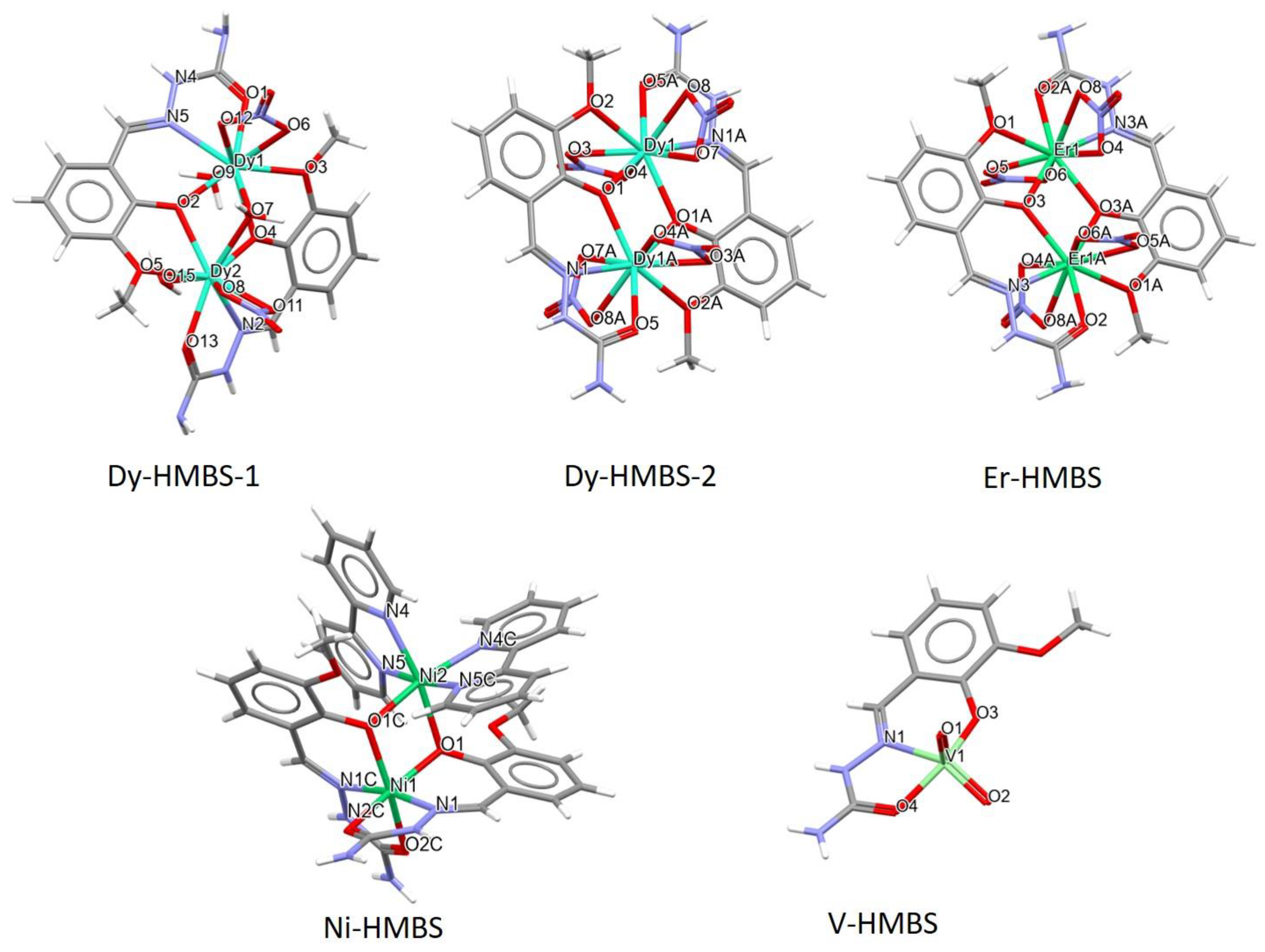
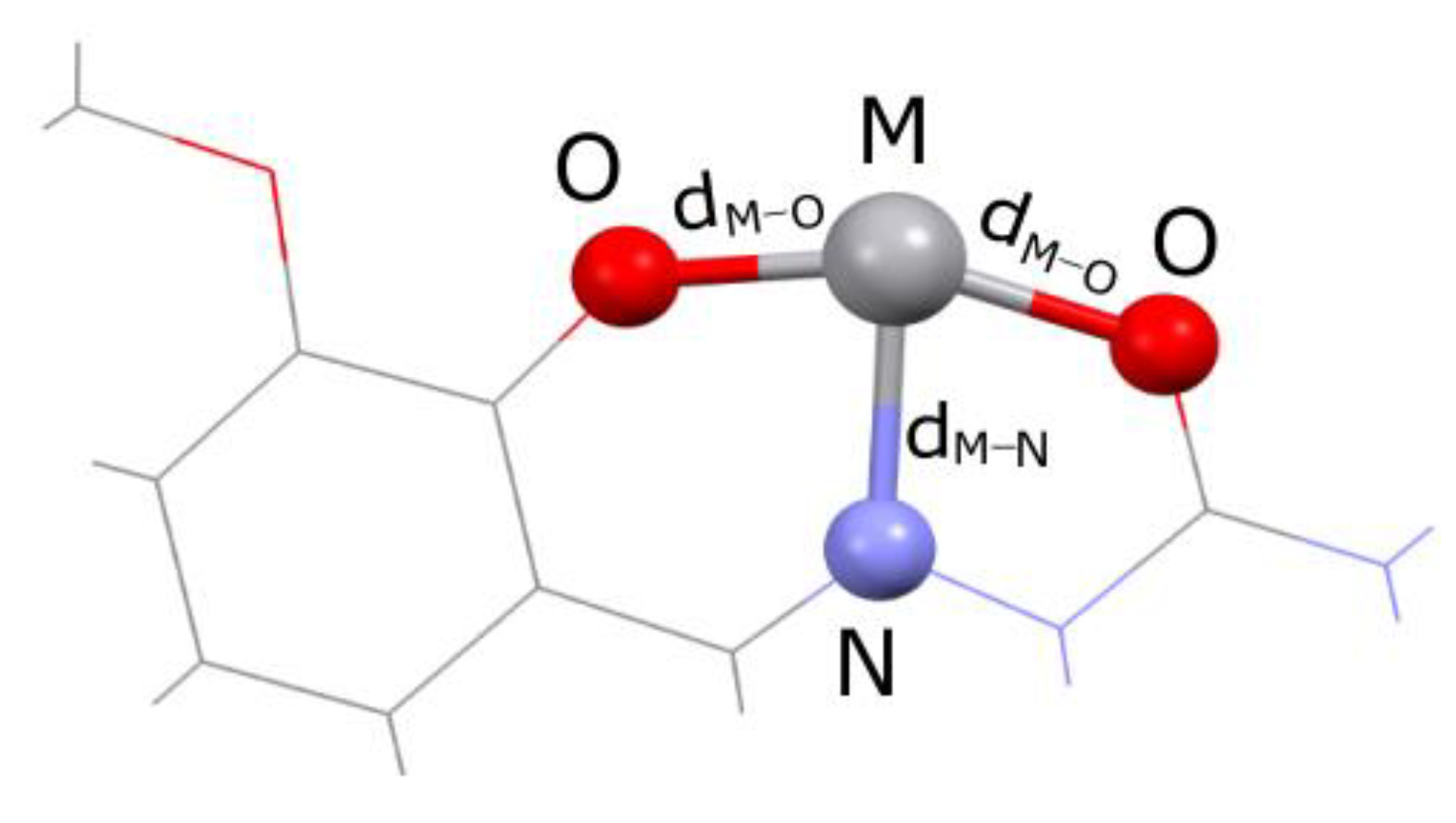
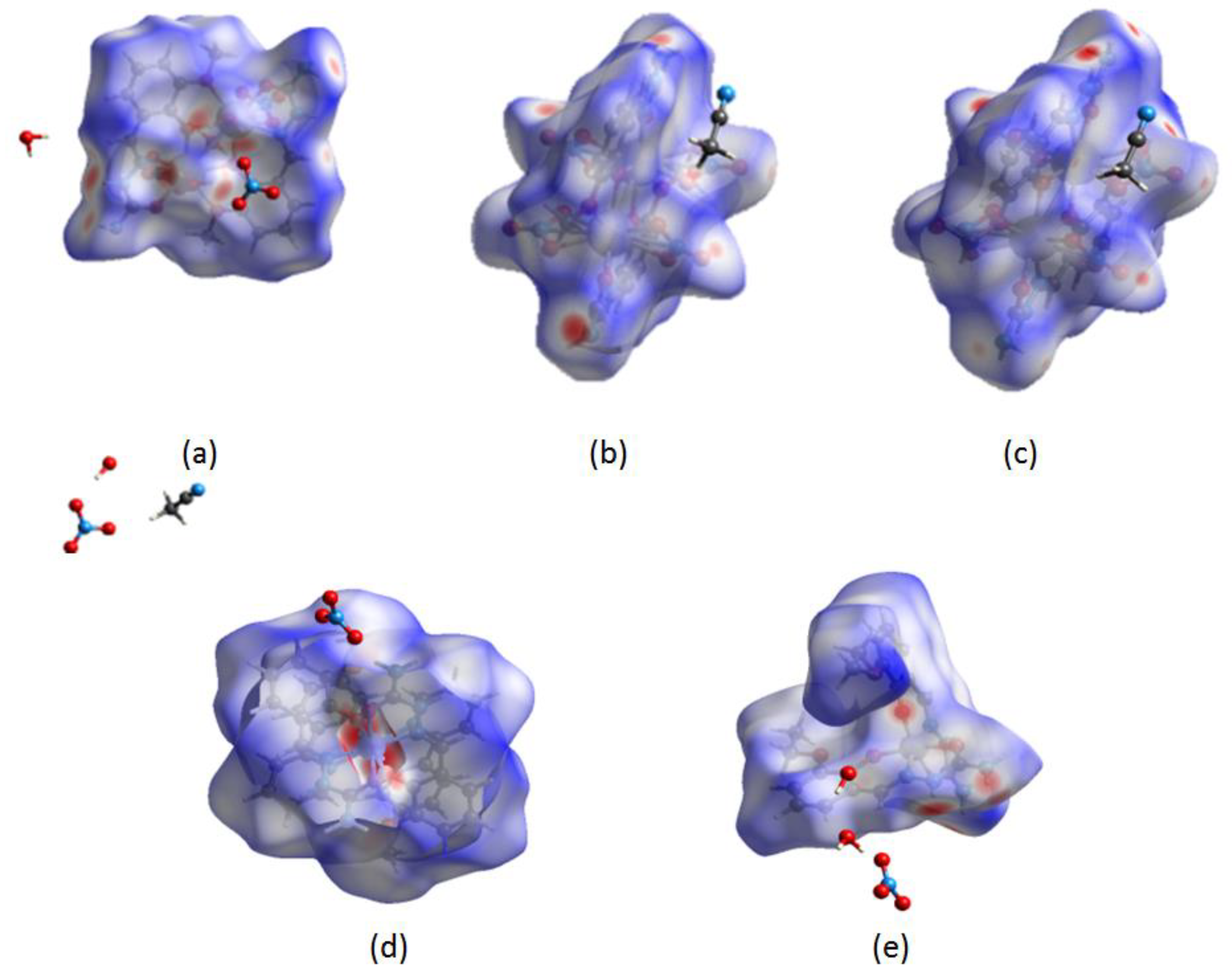
| REFCODE | Notations | Metal Type | Oxidation State | Complex Charge | dM-O (Å) | dM-N (Å) | Space Group |
|---|---|---|---|---|---|---|---|
| BAKTOZ [24] | Dy-HMBS-1 | Dy | +3 | 2+ | 2.278–2.293 | 2.506 | P21/n |
| BAKTUF [24] | Dy-HMBS-2 | Dy | +3 | 0 | 2.303–2.325 | 2.460 | P-1 |
| BAKVAN [24] | Er-HMBS | Er | +3 | 0 | 2.284–2.316 | 2.442 | P-1 |
| BEGSEO03 [18] | Ni-HMBS | Ni | +2 | 2+ | 2.032–2.090 | 2.015 | Pbcn |
| GILQAV [6] | V-HMBS | V | +5 | 0 | 1.888–2.000 | 2.173 | P21/c |
| Notation % Interactions | Dy-HMBS-1 | Dy-HMBS-2 | Er-HMBS | Ni-HMBS | V-HMBS |
|---|---|---|---|---|---|
| H···H | 25.1 | 22.9 | 21.3 | 43.5 | 30.5 |
| H···O | 30.6 | 16.3 | 17.3 | 24.2 | 17.3 |
| O···H | 17.2 | 25.0 | 26.4 | 4.2 | 28.0 |
| O···N | 0.6 | 2.3 | 1.5 | / | 0.1 |
| O···C | 0.7 | 1.9 | 1.5 | / | 1.0 |
| O···O | 1.9 | 4.8 | 3.9 | / | 0.8 |
| N···N | 0.3 | 1.0 | 2.5 | / | / |
| N···C | 0.3 | 1.8 | 0.7 | 0.7 | 1.6 |
| C···C | 0.2 | 1.4 | 0.9 | 1.6 | 4.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jevtovic, V.; Živković, J.M.; Rakić, A.A.; Alrashidi, A.A.; Alreshidi, M.A.; Alzahrani, E.A.; Alshammari, O.A.O.; Hussien, M.A.; Dimić, D. The Effect of Central Metal Ions (Dy, Er, Ni, and V) on the Structural and HSA-Binding Properties of 2-Hydroxy-3-methoxybenzaldehyde Semicarbazone Complexes. Inorganics 2025, 13, 95. https://doi.org/10.3390/inorganics13030095
Jevtovic V, Živković JM, Rakić AA, Alrashidi AA, Alreshidi MA, Alzahrani EA, Alshammari OAO, Hussien MA, Dimić D. The Effect of Central Metal Ions (Dy, Er, Ni, and V) on the Structural and HSA-Binding Properties of 2-Hydroxy-3-methoxybenzaldehyde Semicarbazone Complexes. Inorganics. 2025; 13(3):95. https://doi.org/10.3390/inorganics13030095
Chicago/Turabian StyleJevtovic, Violeta, Jelena M. Živković, Aleksandra A. Rakić, Aljazi Abdullah Alrashidi, Maha Awjan Alreshidi, Elham A. Alzahrani, Odeh A. O. Alshammari, Mostafa Aly Hussien, and Dušan Dimić. 2025. "The Effect of Central Metal Ions (Dy, Er, Ni, and V) on the Structural and HSA-Binding Properties of 2-Hydroxy-3-methoxybenzaldehyde Semicarbazone Complexes" Inorganics 13, no. 3: 95. https://doi.org/10.3390/inorganics13030095
APA StyleJevtovic, V., Živković, J. M., Rakić, A. A., Alrashidi, A. A., Alreshidi, M. A., Alzahrani, E. A., Alshammari, O. A. O., Hussien, M. A., & Dimić, D. (2025). The Effect of Central Metal Ions (Dy, Er, Ni, and V) on the Structural and HSA-Binding Properties of 2-Hydroxy-3-methoxybenzaldehyde Semicarbazone Complexes. Inorganics, 13(3), 95. https://doi.org/10.3390/inorganics13030095