
DFT Modeling of Coordination Polymerization of Polar Olefin Monomers by Molecular Metal Complexes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
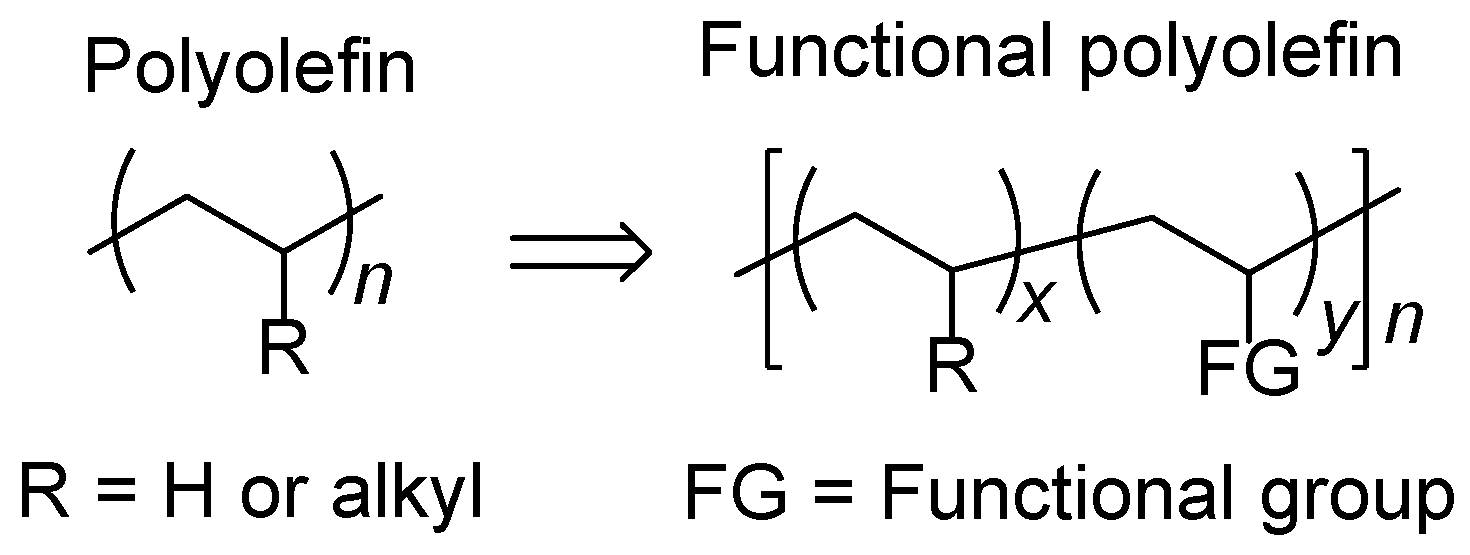
1. Introduction
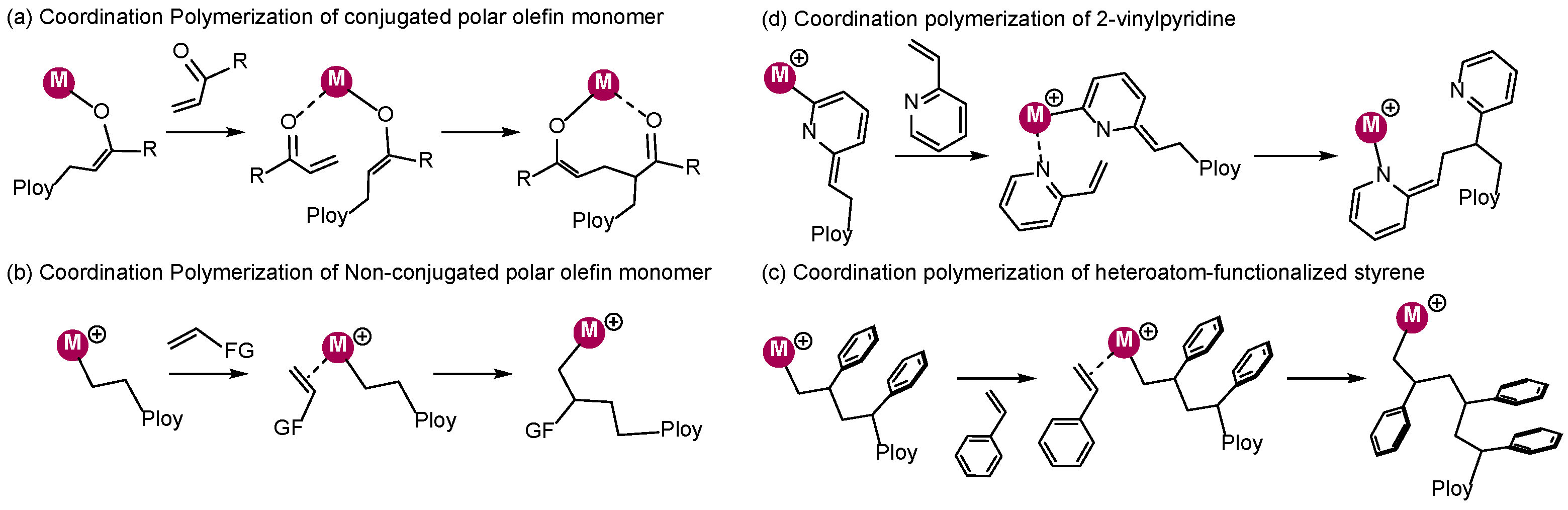
2. Coordination Polymerization of Conjugated Vinyl Polar Monomer
3. Coordination Polymerization of Non-Conjugated Polar Olefin Monomer
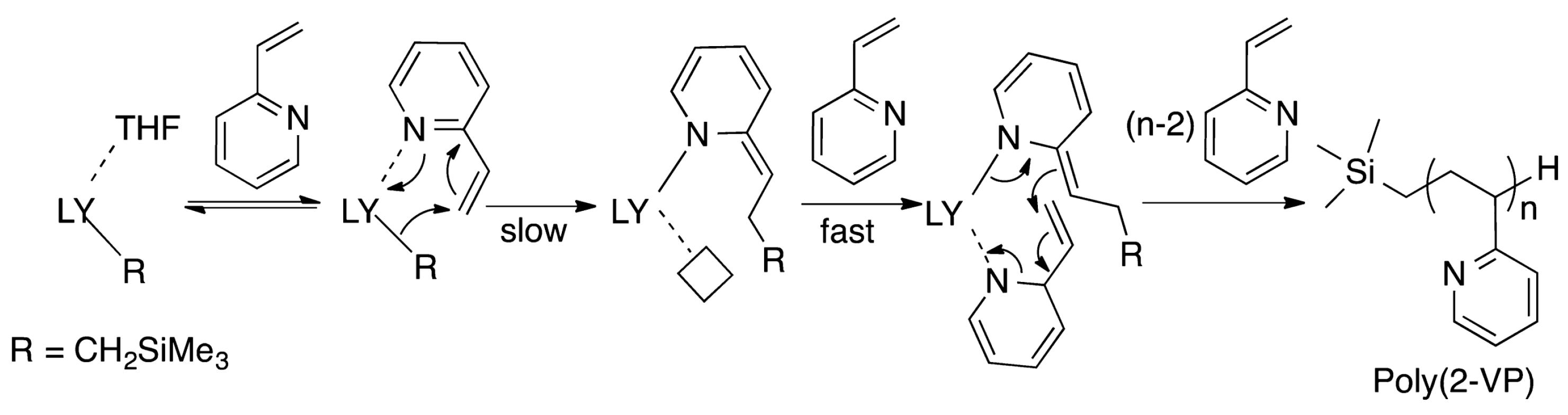
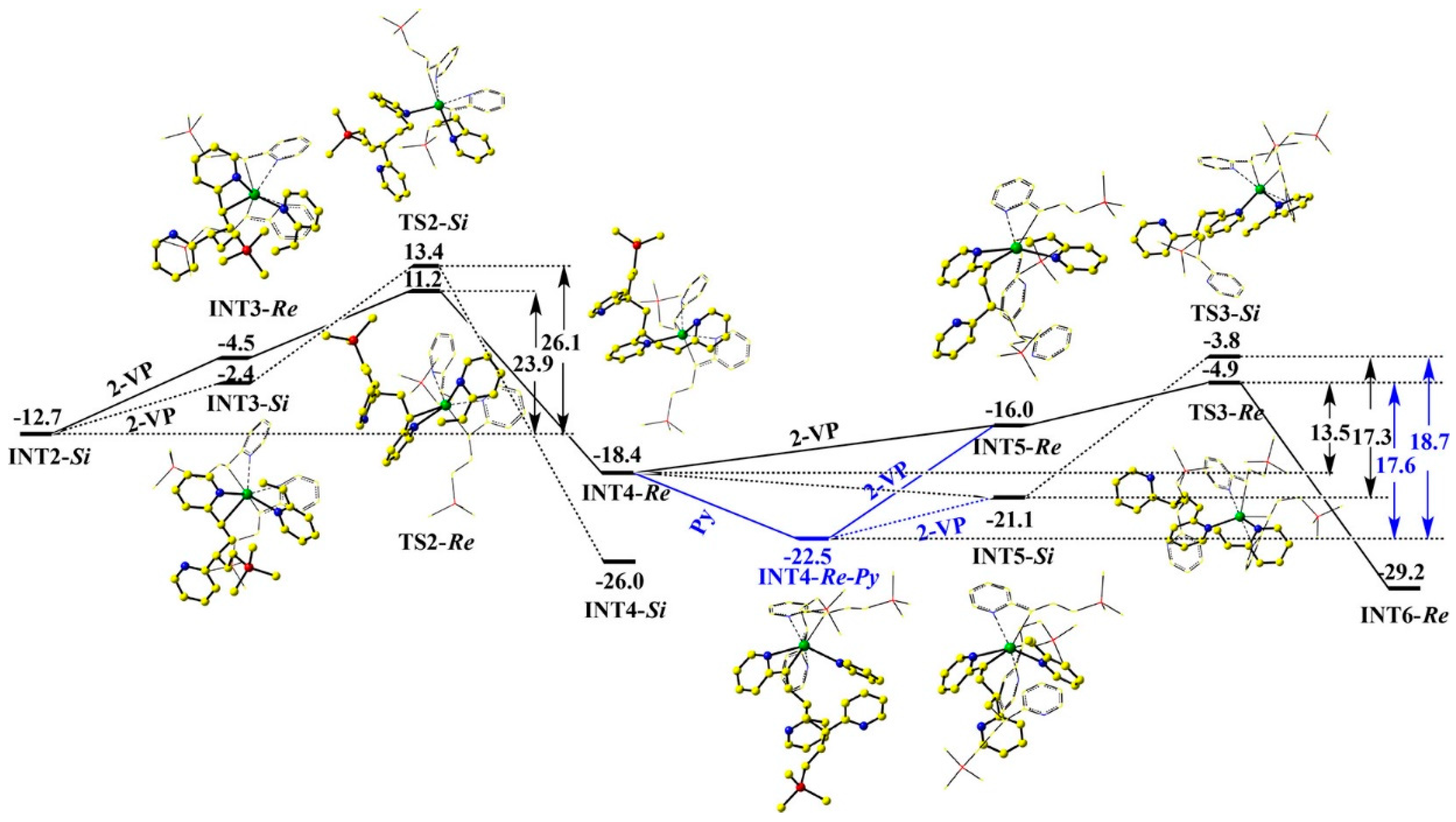
4. Coordination Polymerization of 2-Vinylpyridine
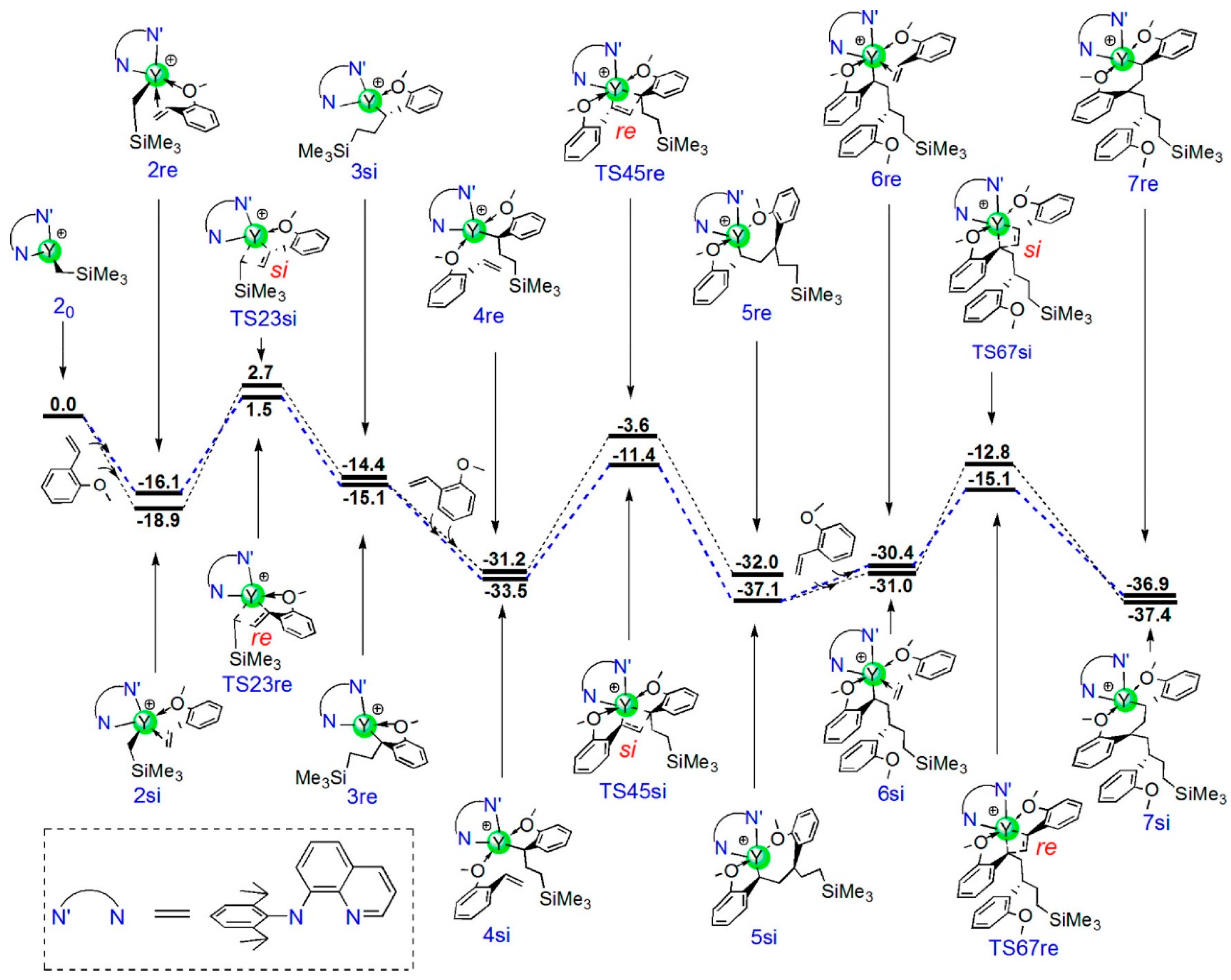
5. Stereoselective Polymerization of 4-Vinylpyridine
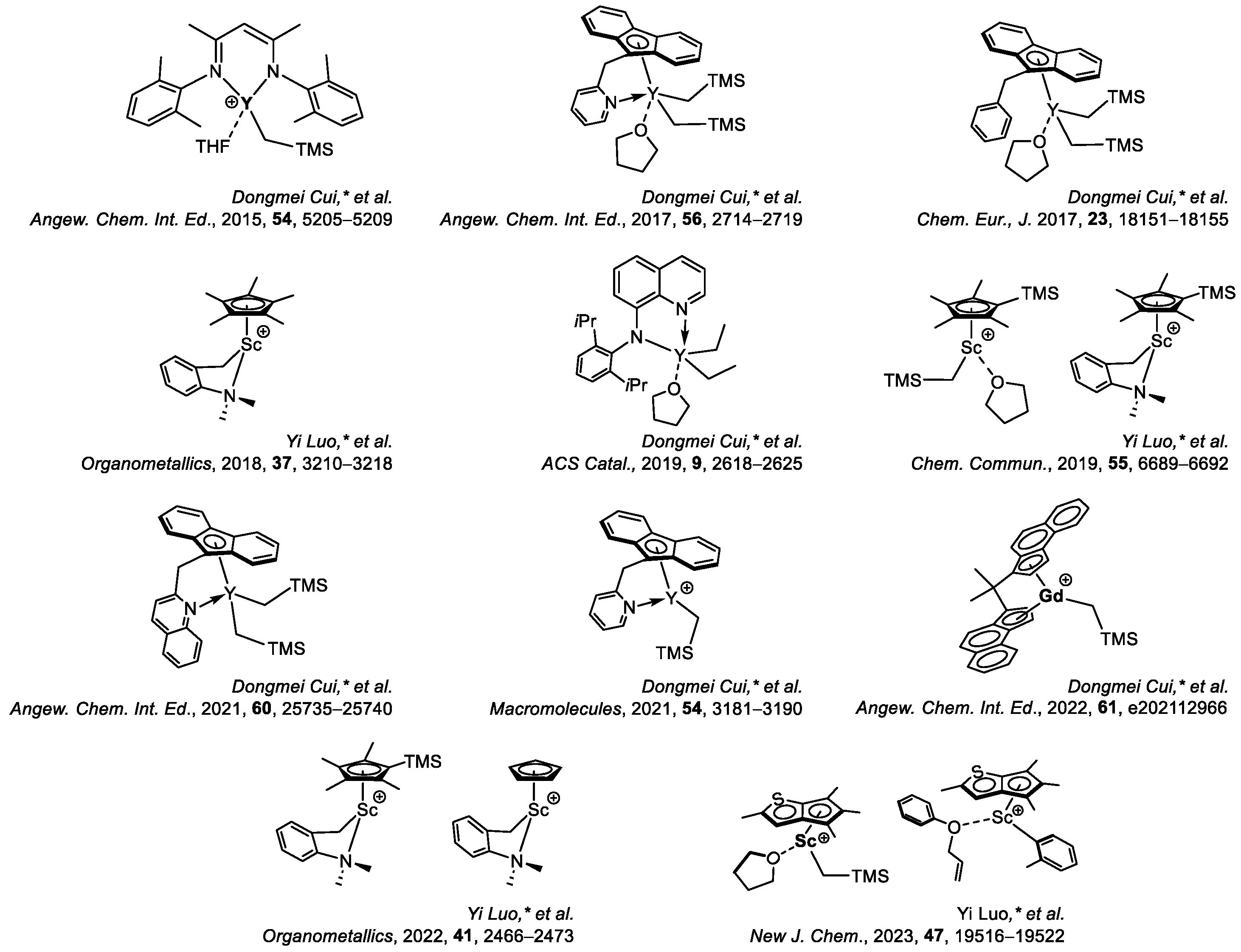
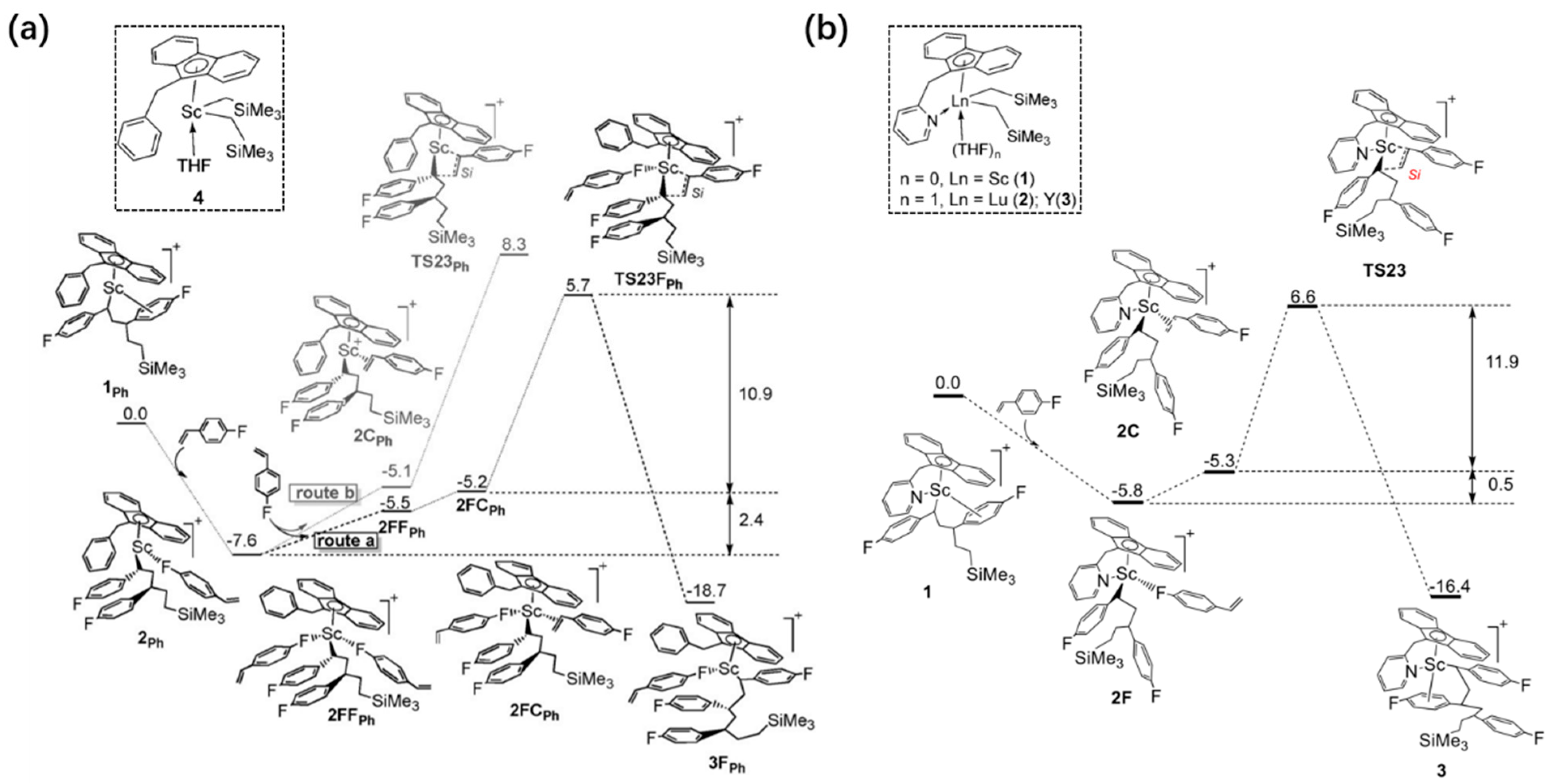
6. Stereoselective Polymerization of Heteroatom-Functionalized Styrene
7. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Reetz, M.T. One Hundred Years of the Max-Planck-Institut für Kohlenforschung. Angew. Chem. Int. Ed. 2014, 53, 8562–8586. [Google Scholar] [CrossRef] [PubMed]
- Stürzel, M.; Mihan, S.; Mülhaupt, R. From Multisite Polymerization Catalysis to Sustainable Materials and All-Polyolefin Composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef]
- Kaminsky, W. (Ed.) Polyolefins 50 Years after Ziegler and Natta I Polyethylene and Polypropylene; Springer: New York, NY, USA, 2013; Volume 257. [Google Scholar]
- Chadwick, J.C. Polyolefins—Catalyst and Process Innovations and their Impact on Polymer Properties. Macromol. React. Eng. 2009, 3, 428–432. [Google Scholar] [CrossRef]
- Busico, V. Ziegler-Natta catalysis: Forever young. MRS Bull. 2013, 38, 224–228. [Google Scholar] [CrossRef]
- Ziegler, K.; Natta, G. Paving the way to polythene. Chem. World 2013, 10, 50–53. [Google Scholar]
- McDaniel, M.P. A Review of the Phillips Supported Chromium Catalyst and Its Commercial Use for Ethylene Polymerization. Angew. Chem. Int. Ed. 2003, 42, 5010–5030. [Google Scholar]
- Baier, M.C.; Zuideveld, M.A.; Mecking, S. Post-Metallocenes in the Industrial Production of Polyolefins. Angew. Chem. Int. Ed. 2014, 53, 9722–9744. [Google Scholar] [CrossRef]
- Mülhaupt, R. Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality. Macromol. Chem. Phys. 2013, 214, 159–174. [Google Scholar] [CrossRef]
- Dubé, M.; Salehpour, S. Applying the Principles of Green Chemistry to Polymer Production Technology. Macromol. React. Eng. 2014, 8, 7–28. [Google Scholar] [CrossRef]
- Gahleitner, M.; Resconi, L.; Doshev, P. Heterogeneous Ziegler-Natta, Metallocene, and Post-Metallocene Catalysis: Successes and Challenges in Industrial Application. MRS Bull. 2013, 38, 229–233. [Google Scholar]
- Qiao, J.; Guo, M.; Wang, L.; Liu, D.; Zhang, X.; Yu, L.; Song, W.; Liu, Y. Recent Advances in Polyolefin Technology. Polym. Chem. 2011, 2, 1611–1623. [Google Scholar] [CrossRef]
- Chum, P.; Swogger, K.W. Olefin Polymer Technologies—History and Recent Progress at the Dow Chemical Company. Prog. Polym. Sci. 2008, 33, 797–819. [Google Scholar] [CrossRef]
- Jian, Z. Synthesis of Functionalized Polyolefins: Design from Catalysts to Polar Monomers. Acta Polym. Sin. 2018, 11, 1359–1371. [Google Scholar]
- Dong, J.; Hu, Y. Design and Synthesis of Structurally Well-Defined Functional Polyolefins via Transition Metal-Mediated Olefin Polymerization Chemistry. Coord Chem. Rev. 2006, 250, 47–65. [Google Scholar] [CrossRef]
- Boaen, N.K.; Hillmyer, M.A. Post-Polymerization Functionalization of Polyolefins. Chem. Soc. Rev. 2005, 34, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Franssen, N.M.G.; Reeka, J.N.H.; Bruin, B. Synthesis of Functional ‘Polyolefins’: State of the Art and Remaining Challenges. Chem. Soc. Rev. 2013, 42, 5809–5832. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. Designing Catalysts for olefin Polymerization and Copolymerization: Beyond Electronic and Steric Tuning. Nat. Rev. Chem. 2018, 2, 6–14. [Google Scholar] [CrossRef]
- Gladysz, J.A.; Ball, Z.T.; Bertrand, G.; Blum, S.A.; Dong, V.M.; Dorta, R.; Hahn, F.E.; Humphrey, M.G.; Jones, W.D.; Klosin, J.; et al. Organometallics Roundtable 2011. Organometallics 2012, 31, 1–18. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C. Polar and Functionalized Polyolefins: New Catalysts, New Modulation Strategies and New Materials. Acta Polym. Sin. 2018, 11, 1372–1384. [Google Scholar]
- Nakamura, A.; Anselment, T.; Claverie, J.; Goodall, B.; Jordan, R.; Mecking, S.; Rieger, B.; Sen, A.; Leeuwen, P.; Nozaki, K. Ortho-Phosphonebenzenesulfonate: A Superb Ligand for Palladium-Catalyzed Coordination-Insertion Copolymerization of Polar Vinyl Monomers. Acc. Chem. Res. 2013, 46, 1438–1449. [Google Scholar] [CrossRef]
- Kesti, M.R.; Coates, G.W.; Waymouth, R.M. Homogeneous Ziegler-Natta Polymerization of Functionalized Monomers Catalyzed by Cationic Group IV Metallocenes. J. Am. Chem. Soc. 1992, 114, 9679. [Google Scholar] [CrossRef]
- Kim, K.H.; Jo, W.H.; Kwak, S.; Kim, K.U.; Hwang, S.S.; Kim, J. Synthesis and Characterization of Poly(styrene-co-4-[(tert-butyldimethylsilyl)oxy]styrene) as a Precursor of Hydroxyl-Functionalized Syndiotactic Polystyrene. Macromolecules 1999, 32, 8703. [Google Scholar] [CrossRef]
- Xu, G.; Chung, T.C. Synthesis of Syndiotactic Polystyrene Derivatives Containing Amino Groups. Macromolecules 2000, 33, 5803. [Google Scholar] [CrossRef]
- Kawabe, M.; Murata, M. Syndiospecific Living Polymerization of Silyl-Protected Hydroxystyrene Derivatives and Preparation of Syndiotactic Poly(4-hydroxystyrene) with Narrow Molecular Weight Distribution. Macromol. Chem. Phys. 2001, 202, 3157–3164. [Google Scholar]
- Saito, Y.; Nakata, N.; Ishii, A. Copolymerization of Ethylene withiPr3Si-Protected 5-Hexen-1-ol with an [OSSO]-Type Bis(phenolato) Dichloro Zirconium(IV) Complex. Bull. Chem. Soc. Jpn. 2016, 89, 666. [Google Scholar] [CrossRef]
- Saito, Y.; Nakata, N.; Ishii, A. Highly Isospecifi c Polymerization of Silyl-Protected ω-Alkenols Using an [OSSO]-Type Bis(phenolato) Dichloro Zirconium(IV) Precatalyst. Macromol. Rapid Commun. 2016, 37, 969. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, S.; Liang, H.; Xie, X.; Zhu, F. Titanium Complex with an [OSSO]-Type Bis(Phenolate) Ligand for Ethylene Copolymerization with Vinyl Polar Monomer Based on Group Protection. RSC Adv. 2019, 9, 26582–26587. [Google Scholar] [CrossRef]
- Luckham, S.; Nozaki, K. Toward the Copolymerization of Propylene with Polar Comonomers. Acc. Chem. Res. 2021, 54, 344–355. [Google Scholar] [CrossRef]
- Chen, Z.; Brookhart, M. Exploring Ethylene/Polar Vinyl Monomer Copolymerizations Using Ni and Pd α-Diimine Catalysts. Acc. Chem. Res. 2018, 51, 1831–1839. [Google Scholar] [CrossRef]
- Tan, C.; Chen, C. Emerging Palladium and Nickel Catalysts for Copolymerization of Olefins with Polar Monomers. Angew. Chem. Int. Ed. 2019, 58, 7192–7200. [Google Scholar] [CrossRef]
- Ito, S. Palladium-Catalyzed Homo- and Copolymerization of Polar Monomers: Synthesis of Aliphatic and Aromatic Polymers. Bull. Chem. Soc. Jpn. 2018, 91, 251–261. [Google Scholar] [CrossRef]
- Nakamura, A.; Ito, S.; Nozaki, K. Coordination-Insertion Copolymerization of Fundamental Polar Monomers. Chem. Rev. 2009, 109, 5215–5244. [Google Scholar] [CrossRef]
- Mu, H.; Pan, L.; Song, D.; Li, Y. Neutral Nickel Catalysts for Olefin Homo- and Copolymerization: Relationships between Catalyst Structures and Catalytic Properties. Chem. Rev. 2015, 115, 12091–12137. [Google Scholar] [CrossRef] [PubMed]
- Delferro, M.; Marks, T. Multinuclear Olefin Polymerization Catalysts. Chem. Rev. 2011, 111, 2450–2485. [Google Scholar] [CrossRef]
- Mu, H.; Zhou, G.; Hu, X.; Jian, Z. Recent Advances in Nickel Mediated Copolymerization of Olefin with Polar Monomers. Coord. Chem. Rev. 2021, 435, 213802–213819. [Google Scholar] [CrossRef]
- Birajdar, R.S.; Chikkali, S. Insertion Copolymerization of Functional Olefins: Quo Vadis? Eur. Polym. J. 2021, 143, 110183–110221. [Google Scholar] [CrossRef]
- Tan, C.; Zou, C.; Chen, C. Material Properties of Functional Polyethylenes from Transition-Metal-Catalyzed Ethylene−Polar Monomer Copolymerization. Macromolecules 2022, 55, 1910–1922. [Google Scholar] [CrossRef]
- Mua, H.; Jian, Z. Stereoselective Copolymerization of Olefin with Polar Monomers to Access Stereoregular Functionalized Polyolefin. Org. Mater. 2022, 4, 178–189. [Google Scholar] [CrossRef]
- Boffa, L.; Novak, B. Copolymerization of Polar Monomers with Olefins Using Transition-Metal Complexes. Chem. Rev. 2000, 100, 1479–1493. [Google Scholar] [CrossRef]
- Chen, E. Coordination Polymerization of Polar Vinyl Monomers by Single-Site Metal Catalysts. Chem. Rev. 2009, 109, 5157–5214. [Google Scholar] [CrossRef]
- Soller, B.S.; Salzinger, S.; Rieger, B. Rare Earth Metal-Mediated Precision Polymerization of Vinylphosphonates and Conjugated Nitrogen-Containing Vinyl Monomers. Chem. Rev. 2016, 116, 1993–2022. [Google Scholar] [CrossRef]
- Xu, T. Catalysts for Stereoselective Polymerization of Polar Vinyl Monomer. Prog. Chem. 2017, 29, 285–292. [Google Scholar]
- Adams, F.; Pahl, P.; Rieger, B. Metal-Catalyzed Group-Transfer Polymerization: A Versatile Tool for Tailor-Made Functional (Co)Polymers. Chem. Eur. J. 2018, 24, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Teator, A.J.; Varner, T.P.; Knutson, P.C.; Sorensen, C.C.; Leibfarth, F.A. 100th Anniversary of Macromolecular Science Viewpoint: The Past, Present, and Future of Stereocontrolled Vinyl Polymerization. ACS Macro Lett. 2020, 9, 1638–1654. [Google Scholar] [CrossRef] [PubMed]
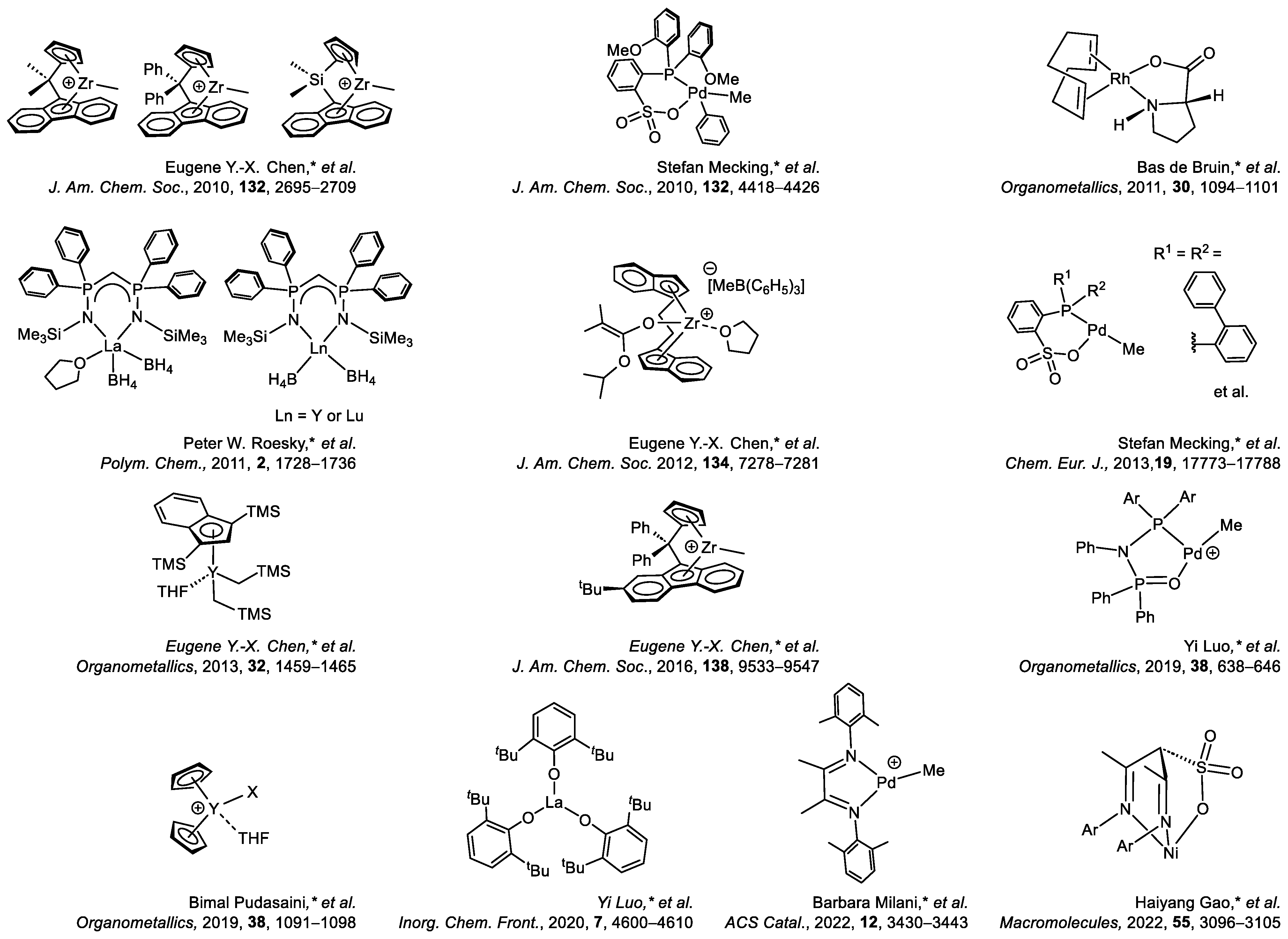
- Zhang, Y.; Ning, Y.; Caporaso, L.; Cavallo, L.; Chen, E. Catalyst-Site-Controlled Coordination Polymerization of Polar Vinyl Monomers to Highly Syndiotactic Polymers. J. Am. Chem. Soc. 2010, 132, 2695–2709. [Google Scholar] [CrossRef]
- Guironnet, D.; Caporaso, L.; Neuwald, B.; Göttker-Schnetmann, I.; Cavallo, L.; Mecking, S.J. Mechanistic Insights on Acrylate Insertion Polymerization. Am. Chem. Soc. 2010, 132, 4418–4426. [Google Scholar] [CrossRef]
- Finger, M.; Reek, J.; Bruin, B. Mechanistic Insights on Acrylate Insertion Polymerization. Organometallics 2011, 30, 1094–1101. [Google Scholar] [CrossRef]
- Guillaume, S.; Brignou, P.; Susperregui, N.; Maron, L.; Kuzdrowskac, M.; Roesky, P. Bis(Phosphinimino)Methanide Borohydride Complexes of the Rare-Earth Elements as Initiators for the Polymerization of Methyl Methacrylate: Combined Experimental and Computational Investigations. Polym. Chem. 2011, 2, 1728–1736. [Google Scholar] [CrossRef]
- Chen, X.; Caporaso, L.; Cavallo, L.; Chen, E. Stereoselectivity in Metallocene-Catalyzed Coordination Polymerization of Renewable Methylene Butyrolactones: From Stereo-Random to Stereo-perfect Polymers. J. Am. Chem. Soc. 2012, 134, 7278–7281. [Google Scholar] [CrossRef]
- Neuwald, B.; Falivene, L.; Caporaso, L.; Cavallo, L.; Mecking, S. Exploring Electronic and Steric Effects on the Insertion and Polymerization Reactivity of Phosphinesulfonato PdII Catalysts. Chem. Eur. J. 2013, 19, 17773–17788. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, X.; Chen, Y.; Caporaso, L.; Cavallo, L.; Chen, E. Rare-Earth Half-Sandwich Dialkyl and Homoleptic Trialkyl Complexes for Rapid and Stereoselective Polymerization of a Conjugated Polar Olefin. Organometallics 2013, 32, 1459–1465. [Google Scholar] [CrossRef]
- Vidal, F.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E. Robust Cross-Linked Stereocomplexes and C60 Inclusion Complexes of Vinyl-Functionalized Stereoregular Polymers Derived from Chemo/Stereoselective Coordination Polymerization. J. Am. Chem. Soc. 2016, 138, 9533–9547. [Google Scholar] [CrossRef]
- Sun, J.; Chen, M.; Luo, G.; Chen, C.; Luo, Y. Diphosphazane-monoxide and Phosphine-sulfonate Palladium Catalyzed Ethylene Copolymerization with Polar Monomers: A Computational Study. Organometallics 2019, 38, 638–646. [Google Scholar] [CrossRef]
- Pudasaini, B. Yttrium Catalyzed Dialkyl Vinyl Phosphonate Polymerization: Mechanistic Insights on the Precision Polymerization from DFT. Organometallics 2019, 38, 1091–1098. [Google Scholar] [CrossRef]
- Zhao, Y.; Luo, G.; Xu, X.; Hou, Z.; Luo, Y. A Computational Study of the Reactivity of Rare-Earth/Phosphorus Lewis Pairs Toward Polymerization of Conjugated Polar Alkenes. Inorg. Chem. Front. 2020, 7, 4600–4610. [Google Scholar] [CrossRef]
- Alberoni, C.; D’Alterio, M.; Balducci, G.; Immirzi, B.; Polentarutti, M.; Pellecchia, C.; Milani, B. Tunable “In-Chain” and “At the End of the Branches” Methyl Acrylate Incorporation in the Polyolefin Skeleton through Pd(II) Catalysis. ACS Catal. 2022, 12, 3430–3443. [Google Scholar] [CrossRef]
- Du, W.; Zheng, H.; Li, Y.; Cheung, C.; Li, D.; Gao, H.; Deng, H.; Gao, H. Neutral Tridentate α-Sulfonato-β-diimine Nickel Catalyst for (Co)polymerizations of Ethylene and Acrylates. Macromolecules 2022, 55, 3096–3105. [Google Scholar] [CrossRef]
- Michalak, A.; Ziegler, T. Late Transition Metals as Homo- and Co-Polymerization Catalysts. Top. Organomet. Chem. 2005, 12, 145–186. [Google Scholar]
- Nishiura, M.; Guo, F.; Hou, Z. Half-Sandwich Rare-Earth-Catalyzed Olefin Polymerization, Carbometalation, and Hydroarylation. Acc. Chem. Res. 2015, 48, 2209–2220. [Google Scholar] [CrossRef]
- Su, B.; Jia, P.; Wang, Y.; Li, Y.; He, H.; Li, Q. Copolymerization of Ethylene/Polar Monomer Catalyzed by Phosphinoarenesulfonate (PO) Metal Catalysts and the Catalytic Mechanism. Chin. J. Org. Chem. 2016, 36, 2344–2352. [Google Scholar] [CrossRef]
- Keyes, A.; Alhan, H.; Ordonez, E.; Ha, U.; Beezer, D.; Dau, H.; Liu, Y.; Tsogtgerel, E.; Jones, G.; Harth, E. Olefins and Vinyl Polar Monomers: Bridging the Gap for Next Generation Materials. Angew. Chem. Int. Ed. 2019, 58, 12370–12391. [Google Scholar] [CrossRef]
- Cui, D. Studies on Homo- and Co-polymerizations of Polar and Non-polar Monomers Using Rare-earth Metal Catalysts. Acta Polym. Sin. 2020, 51, 12–29. [Google Scholar]
- Chen, J.; Gao, Y.; Marks, T. Early Transition Metal Catalysis for Olefin–Polar Monomer Copolymerization. Angew. Chem. Int. Ed. 2020, 59, 14726–14735. [Google Scholar] [CrossRef]
- Zhou, G.; Cui, L.; Mu, H.; Jian, Z. Custom-Made Polar Monomers Utilized in Nickel and Palladium Promoted Olefin Copolymerization. Polym. Chem. 2021, 12, 3878–3892. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Cao, Y. Late Transition Metal Complexes for Olefin Copolymerization with Polar Monomers. Chin. J. Org. Chem. 2021, 41, 1396–1433. [Google Scholar] [CrossRef]
- Jian, Z.; Falivene, L.; Boffa, G.; Sánchez, S.; Caporaso, L.; Grassi, A.; Mecking, S. Direct Synthesis of Telechelic Polyethylene by Selective Insertion Polymerization. Angew. Chem. Int. Ed. 2016, 55, 14378–14383. [Google Scholar] [CrossRef]
- Wang, C.; Luo, G.; Nishiura, M.; Song, G.; Yamamoto, A.; Luo, Y.; Hou, Z. Heteroatom-Assisted Olefin Polymerization by Rare-Earth Metal Catalysts. Sci. Adv. 2017, 3, e1701011. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Motta, A.; Zhang, J.; Gao, Y.; Marks, T. Mechanism of Organoscandium-Catalyzed Ethylene Copolymerization with Amino-Olefins: A Quantum Chemical Analysis. ACS Catal. 2019, 9, 8810–8818. [Google Scholar] [CrossRef]
- You, F.; Liu, H.; Luo, G.; Shi, X. Tridentate Diarylamido-based Pincer Complexes of Nickel and Palladium: Sidearm Effects in Polymerization of Norbornene. Dalton Trans. 2019, 48, 12219–12227. [Google Scholar] [CrossRef]
- Hong, C.; Wang, X.; Chen, C. Palladium-Catalyzed Dimerization of Vinyl Ethers: Mechanism, Catalyst Optimization, and Polymerization Applications. Macromolecules 2019, 52, 7123–7129. [Google Scholar]
- Zhong, Y.; Douair, I.; Wang, T.; Wu, C.; Maron, L.; Cui, D. Access to Hydroxy-Functionalized Polypropylene through Coordination Polymerization. Angew. Chem. Int. Ed. 2020, 59, 4947–4952. [Google Scholar] [CrossRef]
- Mehmood, A.; Xu, X.; Kang, X.; Luo, Y. Origin of Different Chain-End Microstructures in Ethylene/Vinyl Halide Copolymerization Catalysed by Phosphine–Sulfonate Palladium Complexes. New J. Chem. 2020, 44, 16941–16947. [Google Scholar] [CrossRef]
- Mehmood, A.; Xu, X.; Raza, W.; Kim, K.; Luo, Y. Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers. Polymers 2020, 12, 2401–2416. [Google Scholar] [CrossRef]
- Song, C.; Chen, J.; Fu, Z.; Yan, L.; Gao, F.; Cao, Q.; Li, H.; Yan, X.; Chen, S.; Zhang, S.; et al. Syndiospecific Polymerization of o-Methoxystyrene and Its Silyloxy or Fluorine-Substituted Derivatives by HNC-Ligated Scandium Catalysts: Synthesis of Ultrahigh-Molecular-Weight Functionalized Polymers. Macromolecules 2021, 54, 10838–10849. [Google Scholar] [CrossRef]
- Ji, G.; Chen, Z.; Wang, X.; Ning, X.; Xu, C.; Zhang, X.; Tao, W.; Li, J.; Gao, Y.; Shen, Q.; et al. Direct Copolymerization of Ethylene With Protic Comonomers Enabled By Multinuclear Ni Catalyst. Nat. Commun. 2021, 12, 6283–6292. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, Z.; Zhao, Y.; Luo, Y.; He, S. Multivariate Linear Regression Models to Predict Monomer Poisoning Effect in Ethylene/Polar Monomer Copolymerization Catalyzed by Late Transition Metals. Inorganics 2022, 10, 26–36. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, H.; Xin, S.; Li, H.; Luo, Y.; He, S. DFT Studies on the Early-Transition-Metal-Catalyzed Polymerization of Polar Monomers with a Methylene Spacer between Vinyl and Functional Groups. Organometallics 2022, 41, 3514–3521. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, Z.; Jiang, H.; Wang, Q.; Li, S.; Cui, D. Polar Group-Promoted Copolymerization of Ethylene with Polar Olefins Macromolecules 2023, 56, 1547−1553.
- Zhang, N.; Xue, Z.; Shi, L.; Luo, G. Unveiling the Detailed Mechanism and Origins of Chemo-, Regio-, and Stereoselectivity of Rare-Earth Catalyzed Alternating Copolymerization of Polar and Nonpolar Olefins. Inorg. Chem. 2024, 63, 3544–3559. [Google Scholar] [CrossRef] [PubMed]
- Klinger, D.; Wang, C.; Connal, L.; Audus, D.; Jang, S.; Kraemer, S.; Killops, K.; Fredrickson, G.; Kramer, E.; Hawker, C. A Facile Synthesis of Dynamic, Shape-Changing Polymer Particles. Angew. Chem. Int. Ed. 2014, 53, 7018–7022. [Google Scholar] [CrossRef]
- Jang, S.; Audus, D.; Klinger, D.; Krogstad, D.; Kim, B.; Cameron, A.; Kim, S.; Delaney, K.; Hur, S.; Killops, K.; et al. Striped, Ellipsoidal Particles by Controlled Assembly of Diblock Copolymers. J. Am. Chem. Soc. 2013, 135, 6649–6657. [Google Scholar] [CrossRef]
- Natta, G.; Mazzanti, G.; Dall’Asta, G.; Long, P. Crystalline 2-Vinyl-Pyridine Polymers. Makromol. Chem. 1960, 37, 160–162. [Google Scholar] [CrossRef]
- Li, X.; Xu, T. Stereoselective Polymerization of Aromatic Vinyl Polar Monomers. Eur. J. Inorg. Chem. 2023, 26, e202200699. [Google Scholar] [CrossRef]
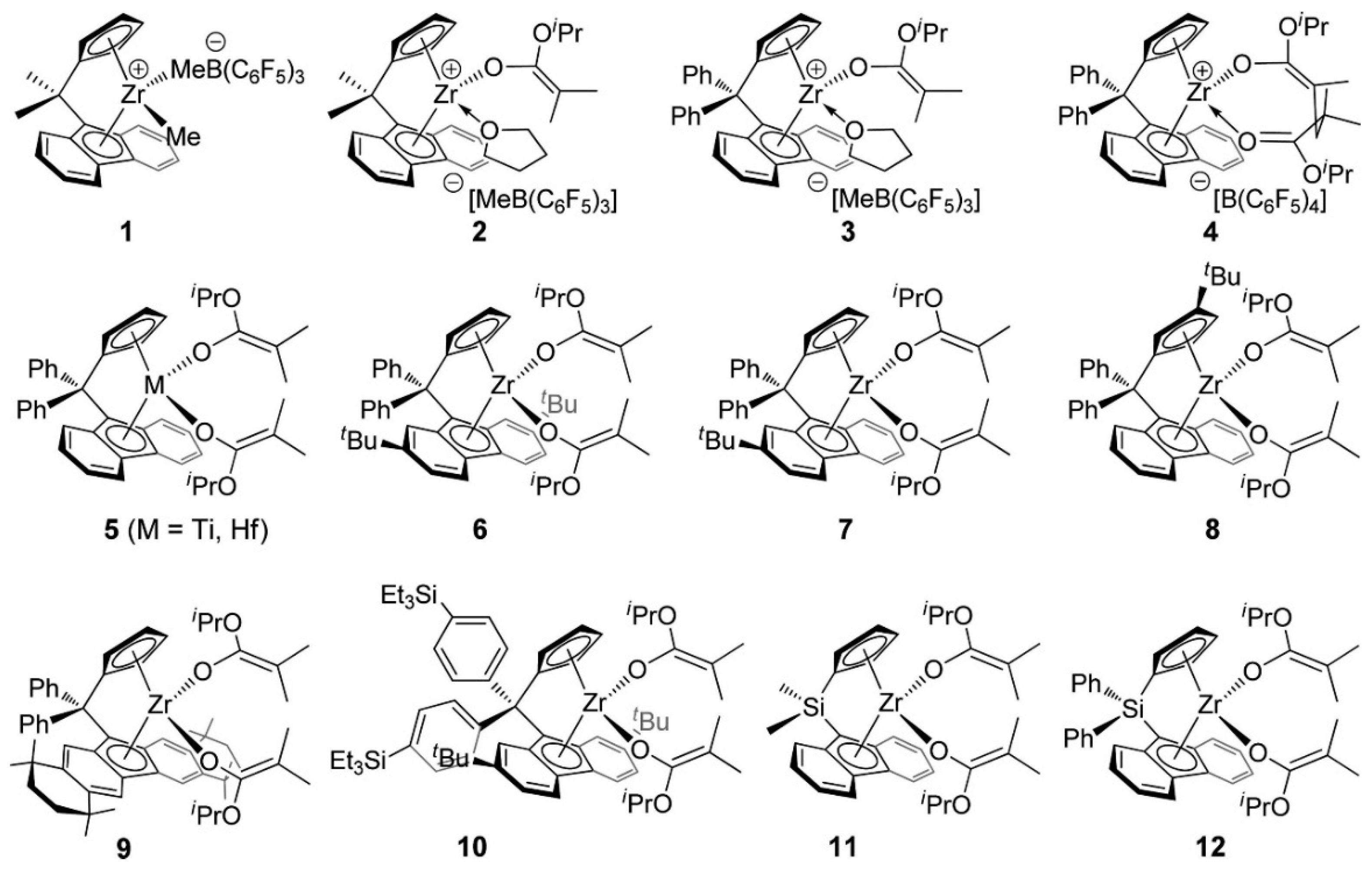
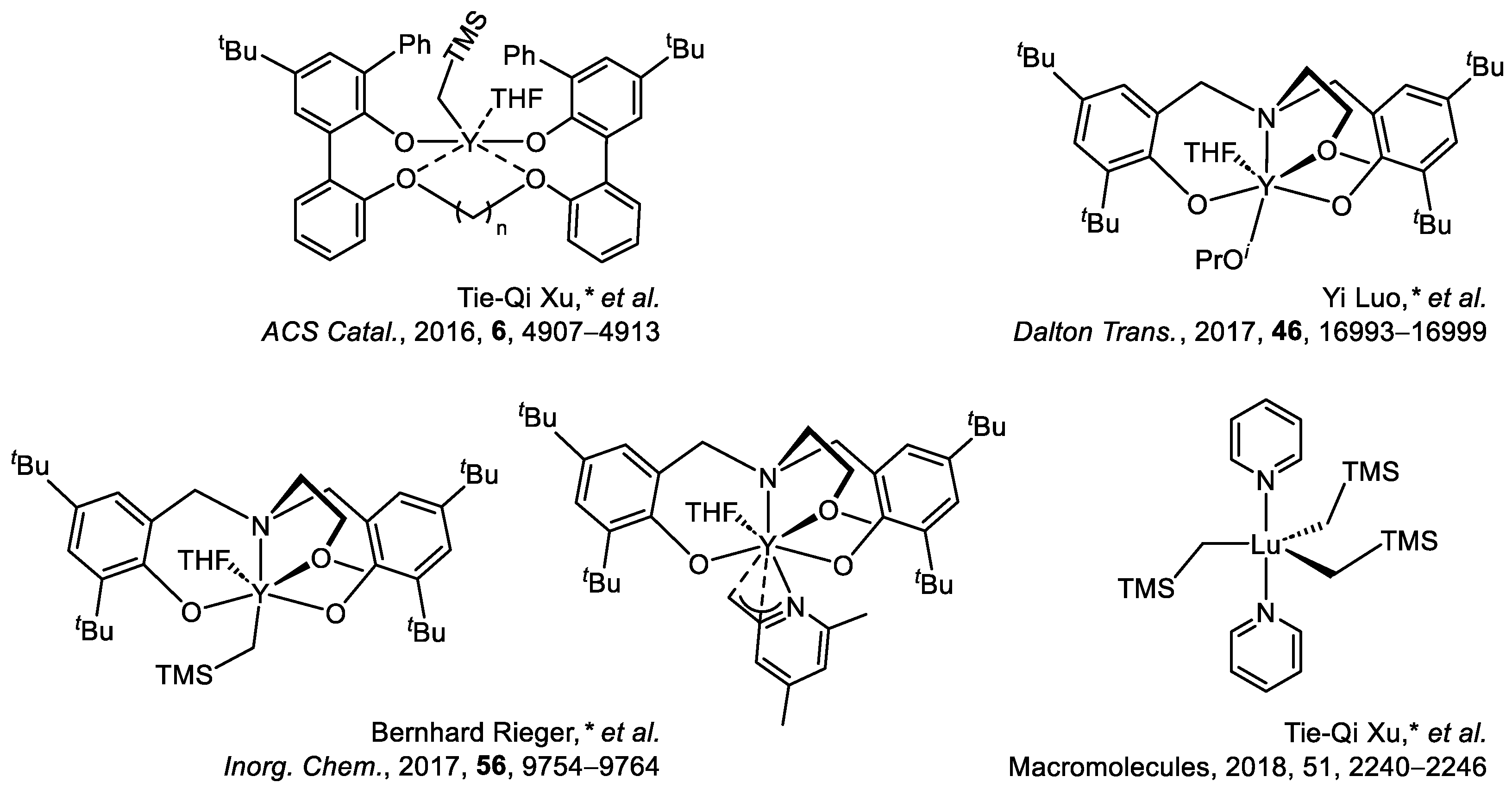
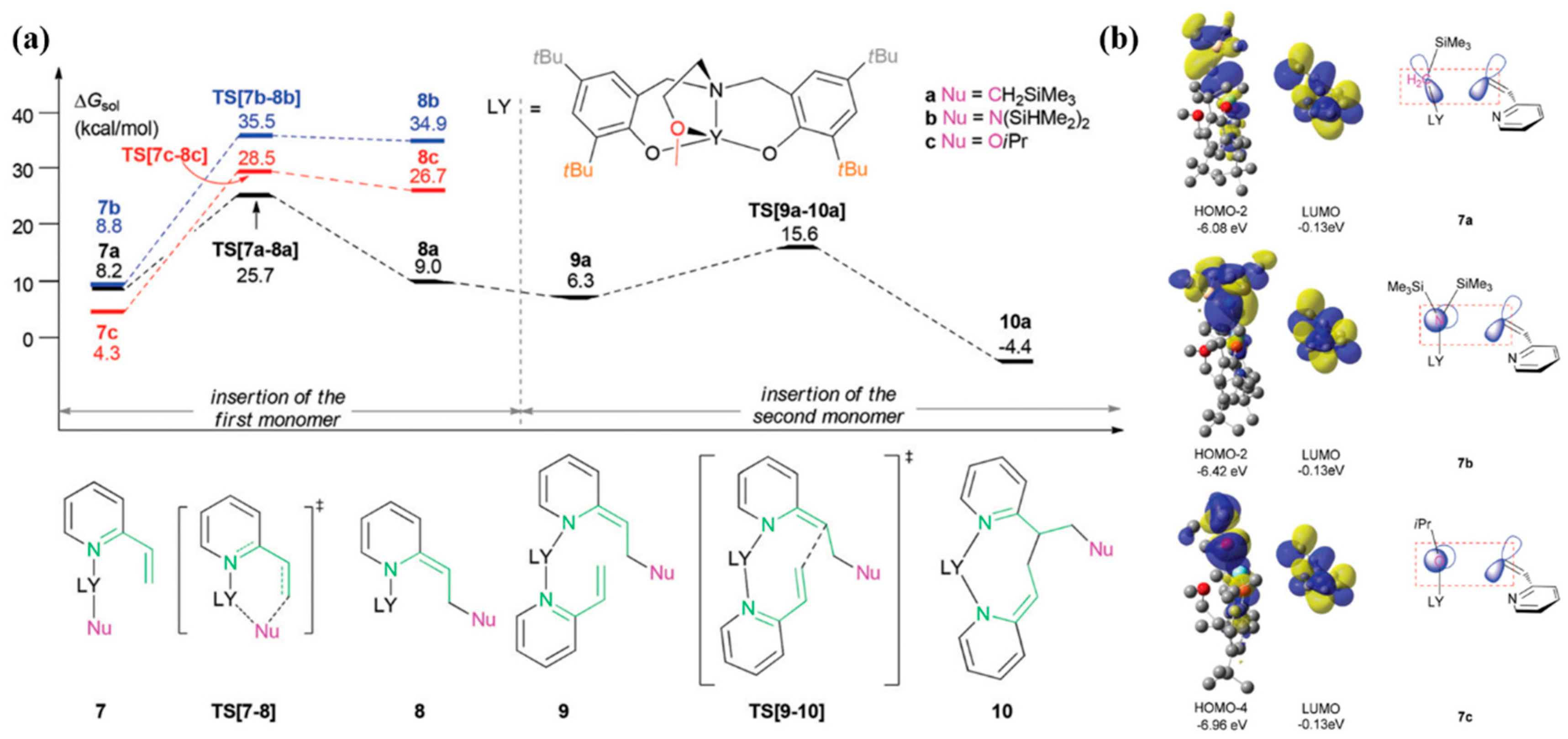
- Xu, T.; Yang, G.; Lu, X. Highly Isotactic and High-Molecular-Weight Poly(2-vinylpyridine) by Coordination Polymerization with Yttrium Bis(phenolate) Ether Catalysts. ACS Catal. 2016, 6, 4907–4913. [Google Scholar] [CrossRef]
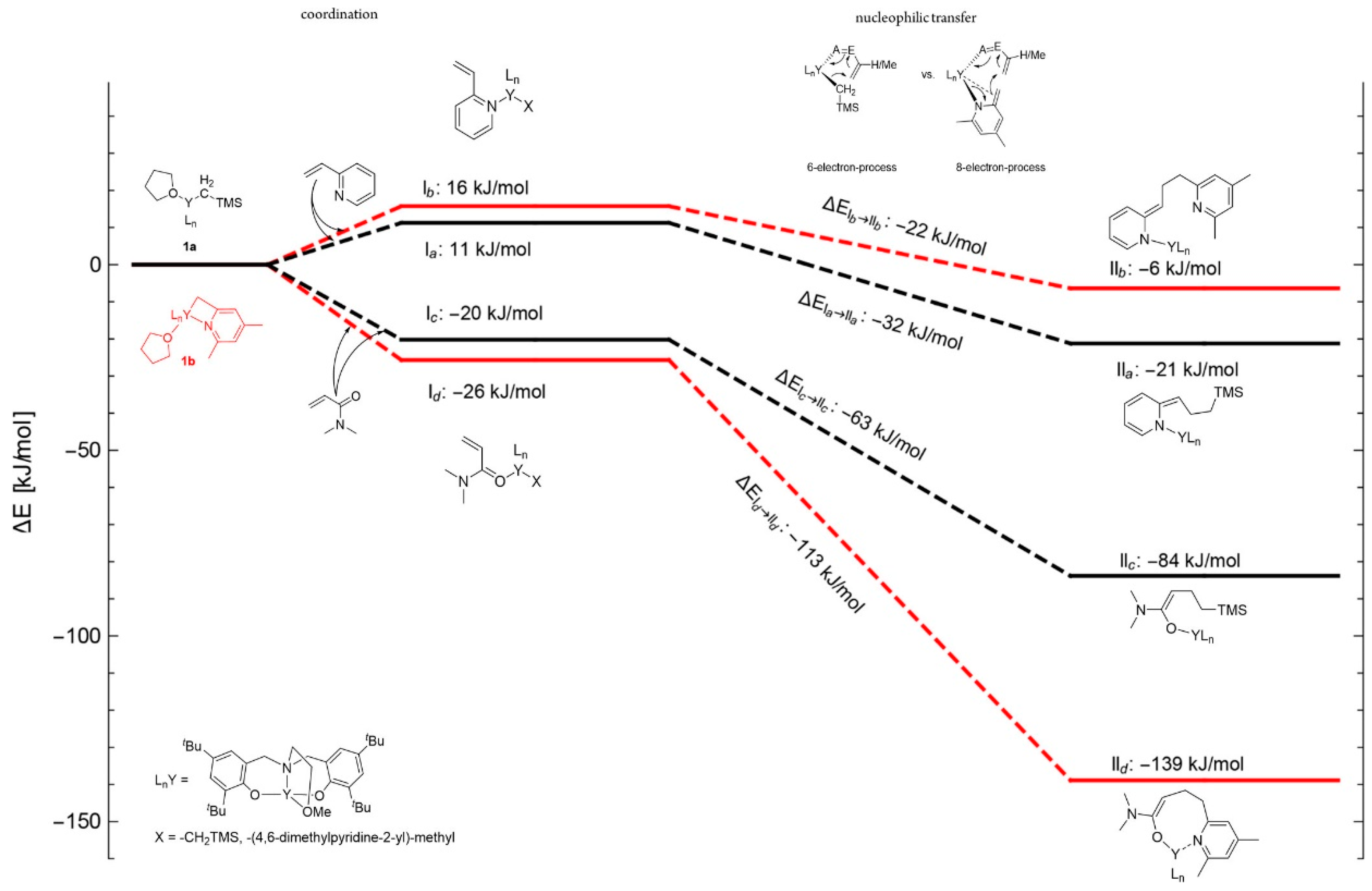
- Yang, J.; Yu, Y.; Qu, J.; Luo, Y. Effects of Nucleophilic Ligands on the Chain Initiation Efficiency of Polar Monomer Polymerizations Catalyzed by 2-Methoxyethylaminobis(Phenolate)Yttrium Complexes: A DFT Study. Dalton Trans. 2017, 46, 16993–16999. [Google Scholar] [CrossRef]
- Adams, F.; Machat, M.; Altenbuchner, P.; Ehrmaier, J.; Pöthig, A.; Karsili, T.; Rieger, B. Toolbox of Nonmetallocene Lanthanides: Multifunctional Catalysts in Group-Transfer Polymerization. Inorg. Chem. 2017, 56, 9754–9764. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Xu, T.; Lu, X. From Stereochemically Tunable Homopolymers to Stereomultiblock Copolymers: Lewis Base Regulates Stereochemistry in the Coordination Polymerization of 2-Vinylpyridine. Macromolecules 2018, 51, 2240–2246. [Google Scholar] [CrossRef]
- Weger, M.; Giuman, M.; Knaus, M.; Ackermann, M.; Drees, M.; Hornung, J.; Altmann, P.; Fischer, R.; Rieger, B. Single-Site, Organometallic Aluminum Catalysts for the Precise Group Transfer Polymerization of Michael-Type Monomers. Chem. Eur. J. 2018, 24, 14950–14957. [Google Scholar] [CrossRef]
- Su, Y.; Zhao, Y.; Zhang, H.; Luo, Y.; Xu, X. Rare-Earth Aryloxide/Ylide-Functionalized Phosphine Frustrated Lewis Pairs for the Polymerization of 4-Vinylpyridine and Its Derivatives. Macromolecules 2021, 54, 7724–7731. [Google Scholar] [CrossRef]
- Jaymand, M. Recent Progress in the Chemical Modification of Syndiotactic Polystyrene. Polym. Chem. 2014, 5, 2663–2690. [Google Scholar] [CrossRef]
- Liu, D.; Yao, C.; Wang, R.; Wang, M.; Wang, Z.; Wu, C.; Lin, F.; Li, S.; Wan, X.; Cui, D. Highly Isoselective Coordination Polymerization of ortho-Methoxystyrene with β-Diketiminato Rare-Earth-Metal Precursors. Angew. Chem. Int. Ed. 2015, 54, 5205–5209. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, M.; Wang, Z.; Wu, C.; Pan, Y.; Cui, D. Stereoselective Copolymerization of Unprotected Polar and Nonpolar Styrenes by anYttrium Precursor: Control of Polar-Group Distribution and Mechanism. Angew. Chem. Int. Ed. 2017, 56, 2714–2719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, M.; Liu, J.; Liu, D.; Cui, D. Rapid Syndiospecific (Co)Polymerization of Fluorostyrene with High Monomer Conversion. Chem. Eur. J. 2017, 23, 18151–18155. [Google Scholar] [CrossRef]
- Zhao, Y.; Luo, G.; Wang, X.; Kang, X.; Cui, D.; Hou, Z.; Luo, Y. DFT Studies on the Polymerization of Functionalized Styrenes Catalyzed by Rare-Earth-Metal Complexes: Factors Affecting C−H Activation Relevant to Step-Growth Polymerization. Organometallics 2018, 37, 3210–3218. [Google Scholar] [CrossRef]
- Liu, D.; Wang, M.; Chai, Y.; Wan, X.; Cui, D. Self-Activated Coordination Polymerization of Alkoxystyrenes by a Yttrium Precursor: Stereocontrol and Mechanism. ACS Catal. 2019, 9, 2618–2625. [Google Scholar] [CrossRef]
- Zhao, Y.; Luo, G.; Kang, X.; Guo, F.; Zhu, X.; Zheng, R.; Hou, Z.; Luo, Y. “C–H⋅⋅⋅π Interaction” Regulates the Stereoselectivity in Olefin Polymerization. Chem. Commun. 2019, 55, 6689–6692. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wu, C.; Ji, X.; Cui, D. Direct Synthesis of Functional Thermoplastic Elastomer with Excellent Mechanical Properties by Scandium-Catalyzed Copolymerization of Ethylene and Fluorostyrenes. Angew. Chem. Int. Ed. 2021, 60, 25735–25740. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, B.; Zhao, Z.; Cui, D. Chemo- and Stereoselective Polymerization of Polar Divinyl Monomers by Rare-Earth Complexes. Macromolecules 2021, 54, 3181–3190. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, Z.; Li, S.; Cui, D. Isospecific (Co)polymerization of Unmasked Polar Styrenes by Neutral Rare-Earth Metal Catalysts. Angew. Chem. Int. Ed. 2022, 61, e202112966. [Google Scholar] [CrossRef]
- Wen, X.; Xu, X.; Li, H.; Zhao, Y.; Luo, Y. Theoretical Mechanistic Studies of the Polymerization of Functionalized Styrenes Catalyzed by Rare-Earth-Metal Complexes: Stereoselectivity Regulation. Organometallics 2022, 41, 2466–2473. [Google Scholar]
- Wen, X.; Xing, X.; Zhang, W.; Lu, H.; Zhu, L.; Xu, X.; Zhao, Y.; Cui, D.; Luo, Y. DFT Studies on the Effect of Additives on Stereoselectivity in the Polymerization of Styrene Catalyzed by Rare-Earth Metal Complexes. New J. Chem. 2023, 47, 19516–19522. [Google Scholar] [CrossRef]
- Leborgne, A.; Vincens, V.; Joulgard, M.; Spassky, N. RingOpening Oligomerization Reactions Using Aluminium Complexes of Schiff’s Bases as Initiators. Makromol. Chem. Macromol. Symp. 1993, 73, 37–46. [Google Scholar] [CrossRef]
- Wisniewski, M.; Le Borgne, A.; Spassky, N. Synthesis and Properties of (D)- and (L)-Lactide Stereocopolymers Using the System Achiral Schiff’s Base/Aluminium Methoxide as Initiator. Macromol. Chem. Phys. 1997, 198, 1227–1238. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereoselective Ring-Opening Polymerization of mesoLactide: Synthesis of Syndiotactic Poly(lactic acid). J. Am. Chem. Soc. 1999, 121, 4072–4073. [Google Scholar] [CrossRef]
- Cheng, M.; Attygalle, A.B.; Lobkovsky, E.B.; Coates, G.W. Single-Site Catalysts for Ring-Opening Polymerization: Synthesis of Heterotactic Poly(lactic acid) from rac-Lactide. J. Am. Chem. Soc. 1999, 121, 11583–11584. [Google Scholar] [CrossRef]
- Chamberlain, B.M.; Cheng, M.; Moore, D.R.; Ovitt, T.M.; Lobkovsky, E.B.; Coates, G.W. Polymerization of Lactide with Zinc and Magnesium β-Diiminate Complexes: Stereocontrol and Mechanism. J. Am. Chem. Soc. 2001, 123, 3229–3238. [Google Scholar] [CrossRef] [PubMed]
- Ovitt, T.M.; Coates, G.W. Stereochemistry of Lactide Polymerization with Chiral Catalysts: New Opportunities for Stereocontrol Using Polymer Exchange Mechanisms. J. Am. Chem. Soc. 2002, 124, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Ishii, R.; Akakura, M.; Aoi, K. Stereoselective Ring-Opening Polymerization of Racemic Lactide Using Aluminum-Achiral Ligand Complexes: Exploration of a Chain End Control Mechanism. J. Am. Chem. Soc. 2002, 124, 5938–5939. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S. Enzymatic Synthesis of Polyesters via Ring-Opening Polymerization. Adv. Polym. Sci. 2006, 194, 95–132. [Google Scholar]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.; Hedrick, J.L. Organocatalytic Ring-Opening Polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef]
- Kiesewetter, M.K.; Shin, E.J.; Hedrick, J.L.; Waymouth, R.M. Organocatalysis: Opportunities and Challenges for Polymer Synthesis. Macromolecules 2010, 43, 2093–2107. [Google Scholar]
- Dove, A.P. Organic Catalysis for Ring-Opening Polymerization. ACS Macro Lett. 2012, 1, 1409–1412. [Google Scholar] [CrossRef]
- Sarazin, Y.; Carpentier, J. Discrete Cationic Complexes for Ring-Opening Polymerization Catalysis of Cyclic Esters and Epoxides. Chem. Rev. 2015, 115, 3564–3614. [Google Scholar] [CrossRef]
- Carpentier, J. Rare-Earth Complexes Supported by Tripodal Tetradentate Bis(phenolate) Ligands: A Privileged Class of Catalysts for Ring-Opening Polymerization of Cyclic Esters. Organometallics 2015, 34, 4175–4189. [Google Scholar] [CrossRef]
- Redshaw, C. Metallocalixarene catalysts: α-olefin polymerization and ROP of cyclic esters. Dalton Trans. 2016, 45, 9018–9030. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Ivchenko, P. Coordination Ring-Opening Polymerization of Cyclic Esters: A Critical Overview of DFT Modeling and Visualization of the Reaction Mechanisms. Molecules 2019, 24, 4117–4167. [Google Scholar] [CrossRef]
- Wei, J.; Diaconescu, P. Redox-Switchable Ring-Opening Polymerization with Ferrocene Derivatives. Acc. Chem. Res. 2019, 52, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Ni, X.; Ling, J. Recent Progress in Ring-opening Polymerizations Catalyzed by Rare Earth Catalysts. Acta Polym. Sin. 2021, 52, 445–455. [Google Scholar]
- Shawver, N.; Doerr, A.M.; Long, B. A perspective on redox-switchable ring-opening polymerization. J. Polym. Sci. 2023, 61, 361–371. [Google Scholar] [CrossRef]
- Guillaume, S.; Maron, L.; Roesky, P. Handbook on the Physics and Chemistry of Rare Earths; ScienceDirect: Amsterdam, The Netherlands, 2014; Volume 44, pp. 1–86. [Google Scholar]
- Sutar, A.K.; Maharana, T.; Routaray, A.; Nath, N. Ring-Opening Polymerization of Lactide. In Advanced Catalytic Materials; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 193–224. [Google Scholar]
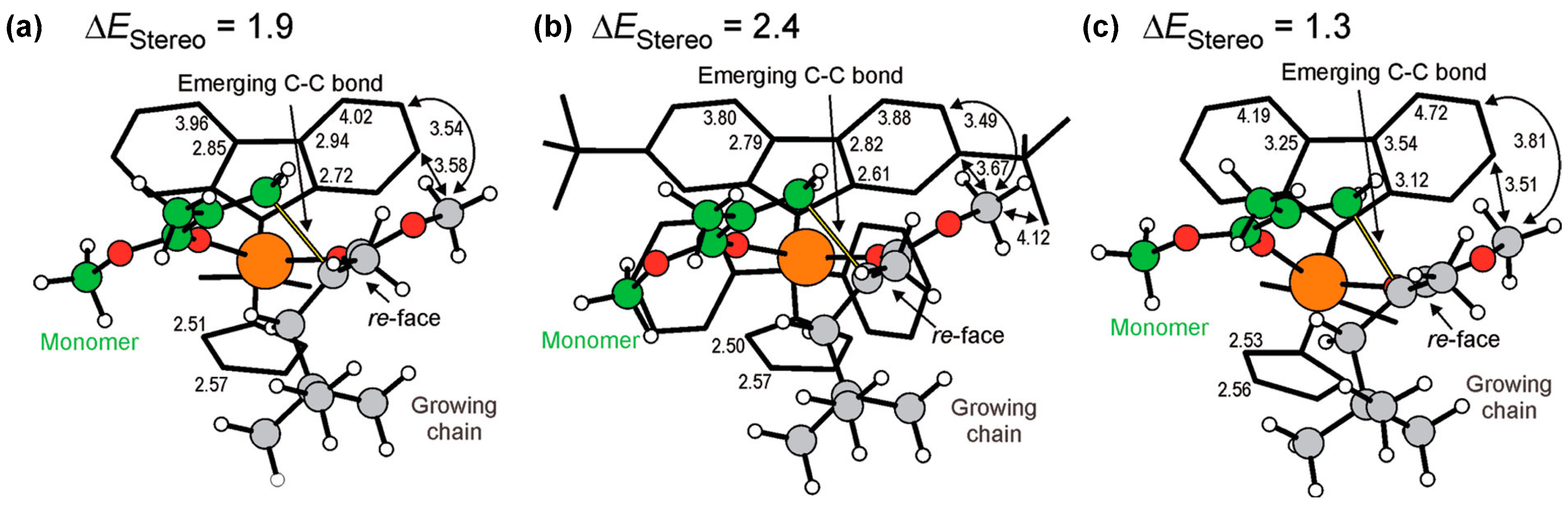
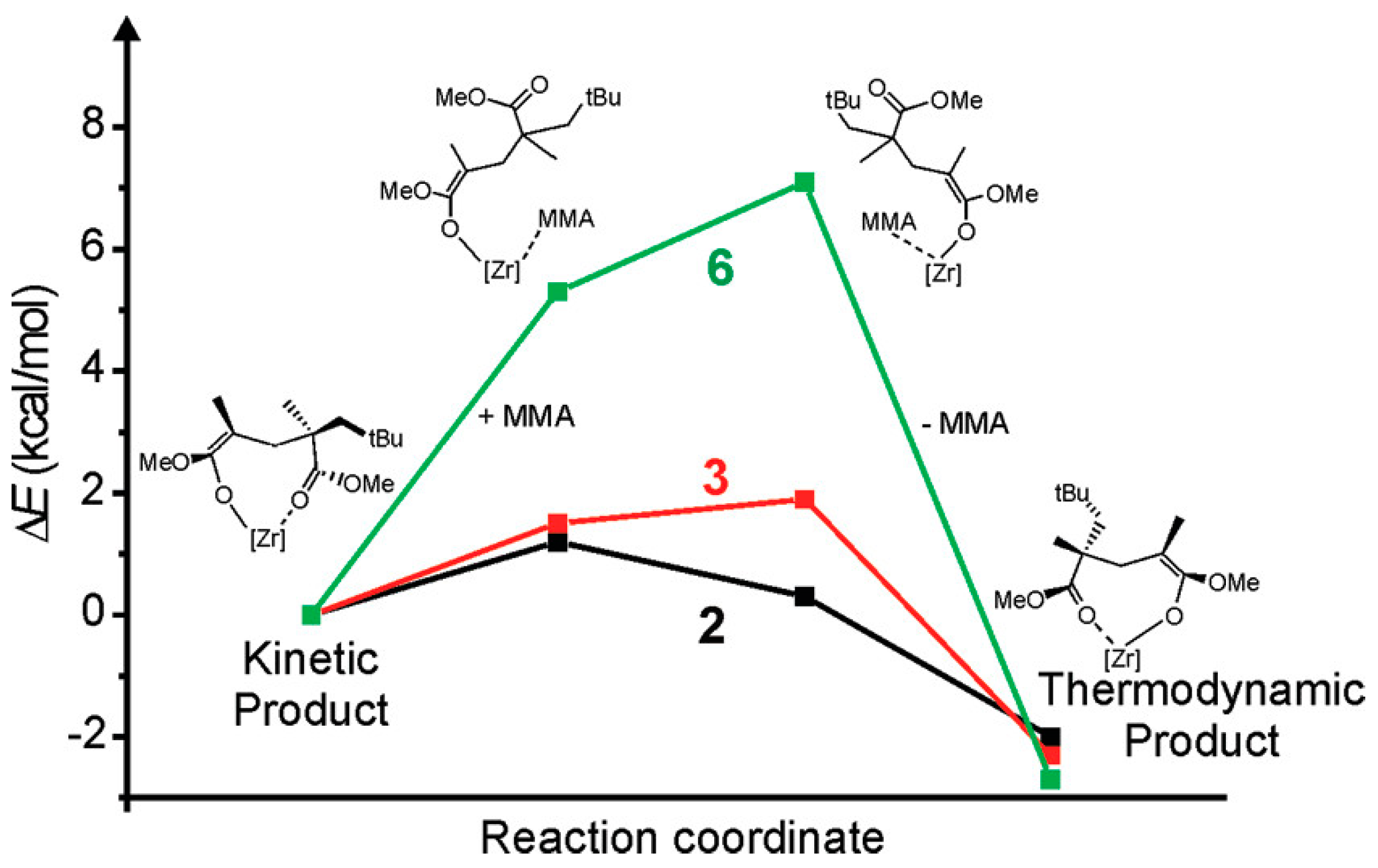

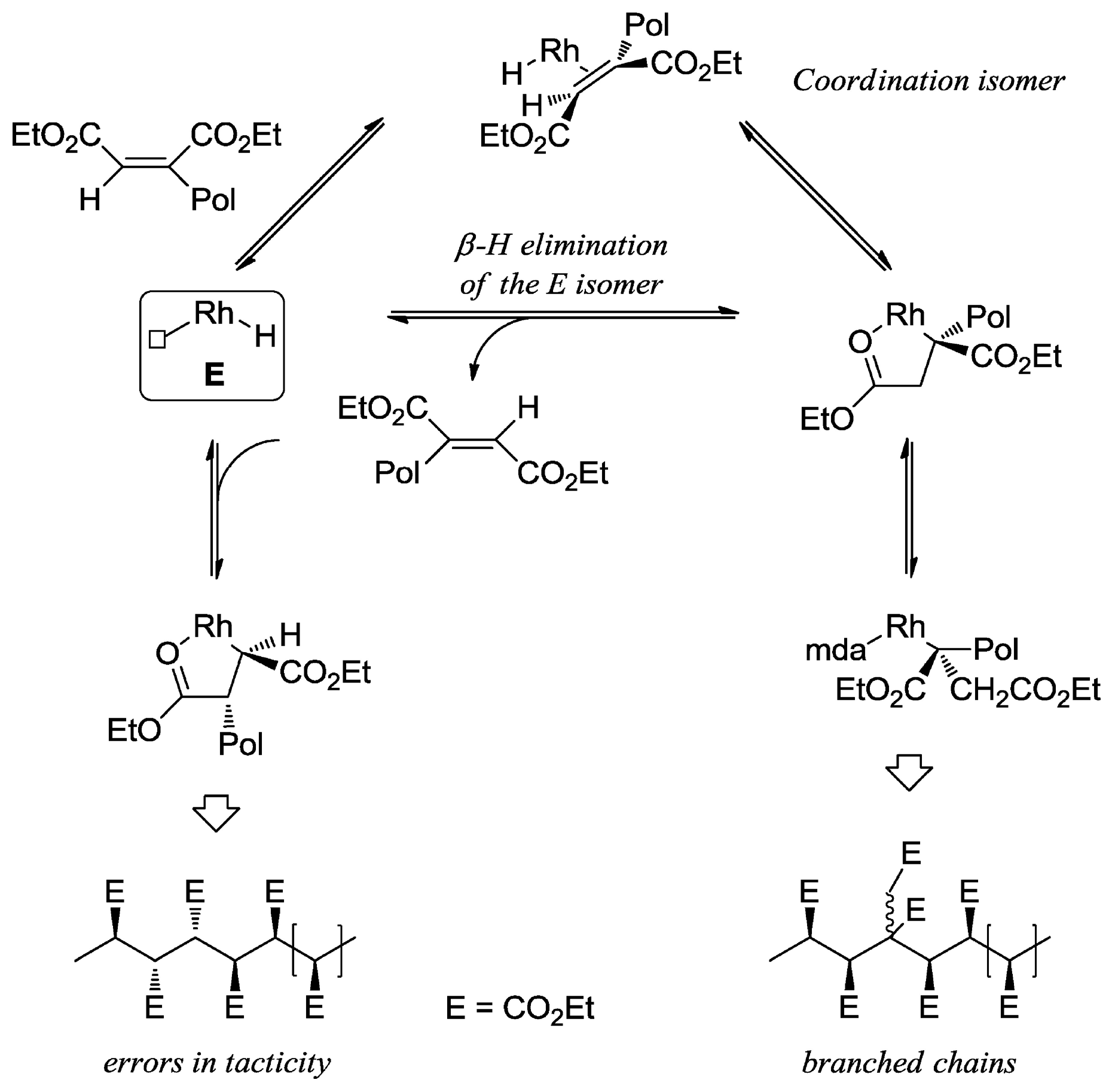
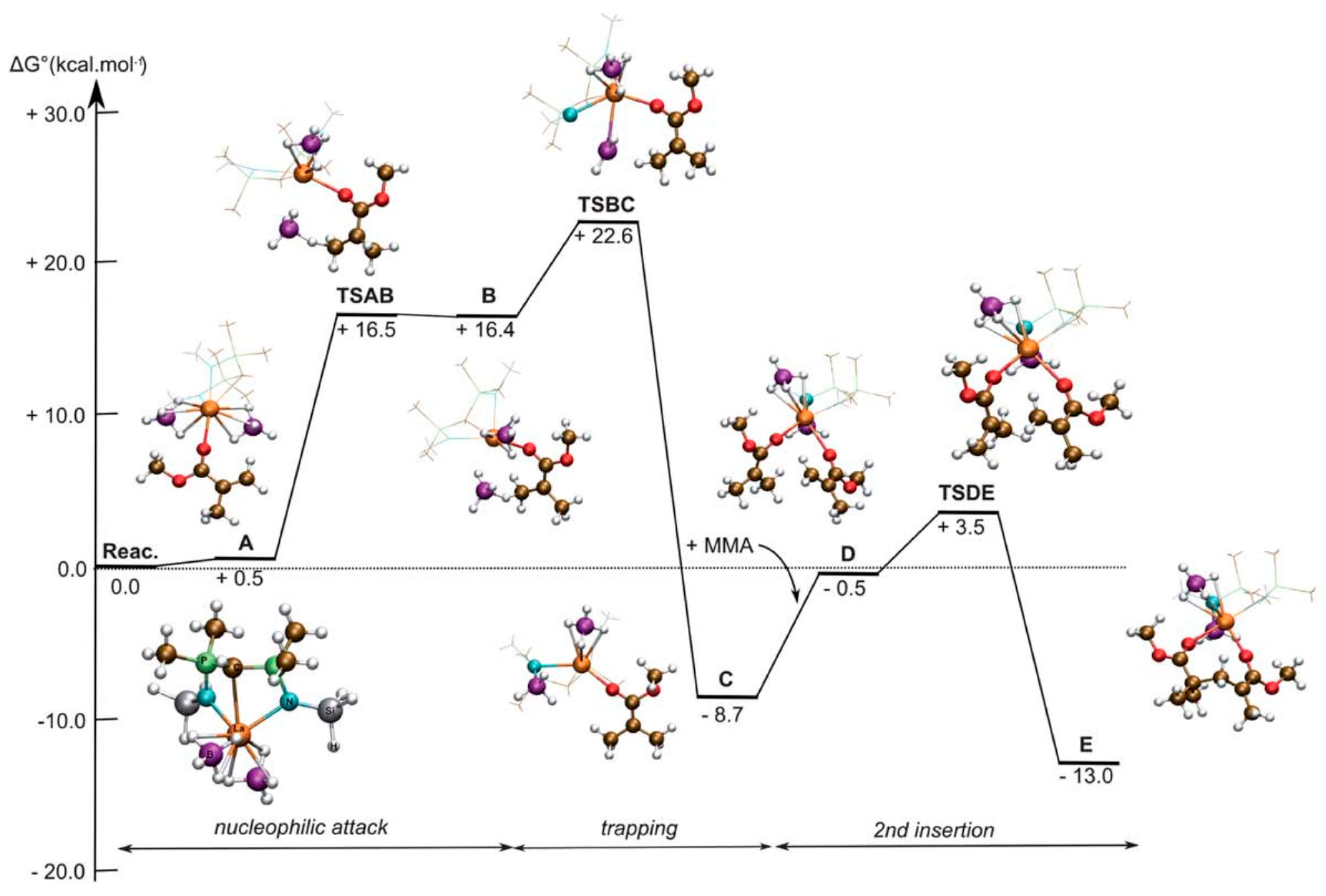
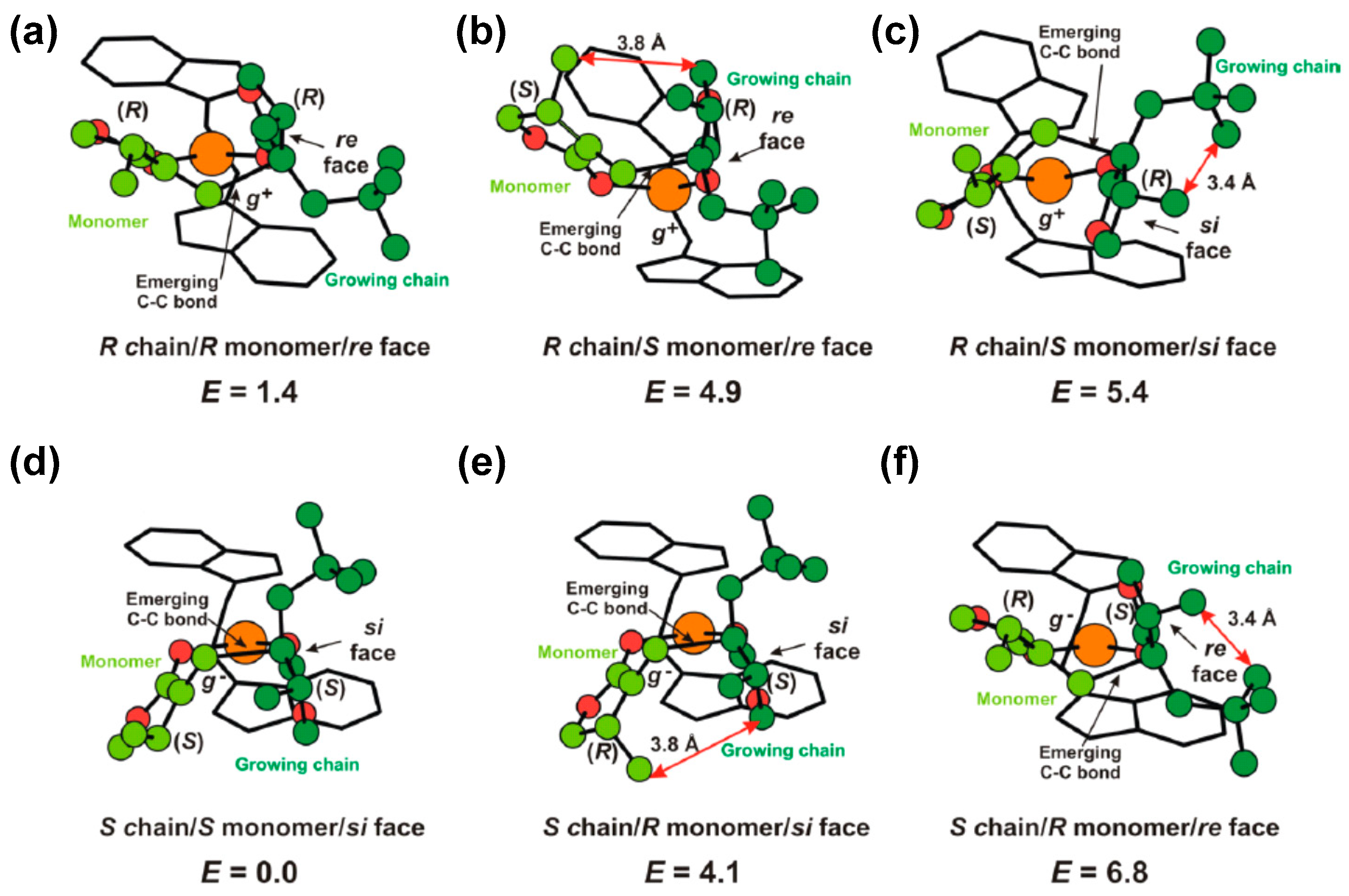
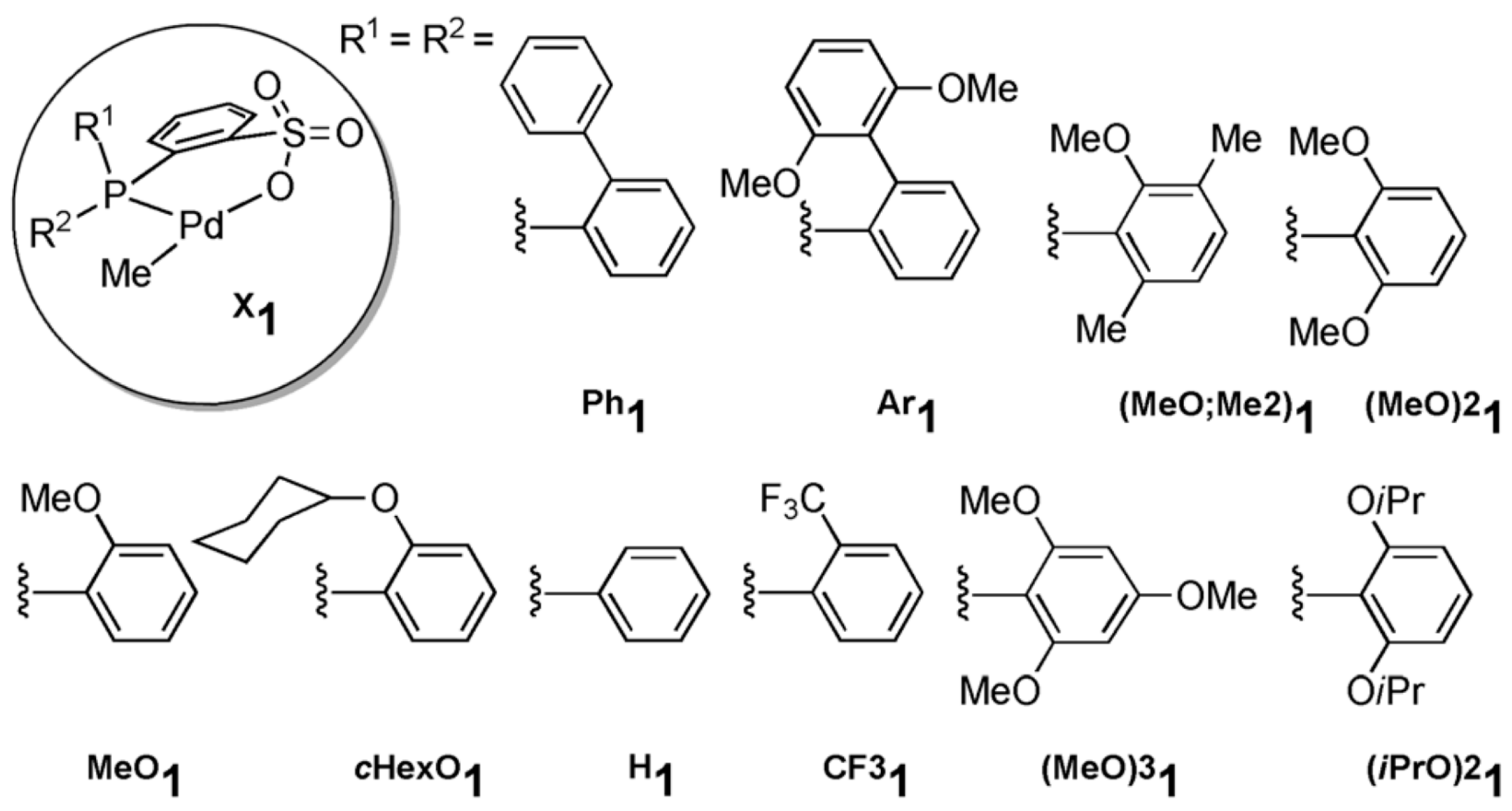
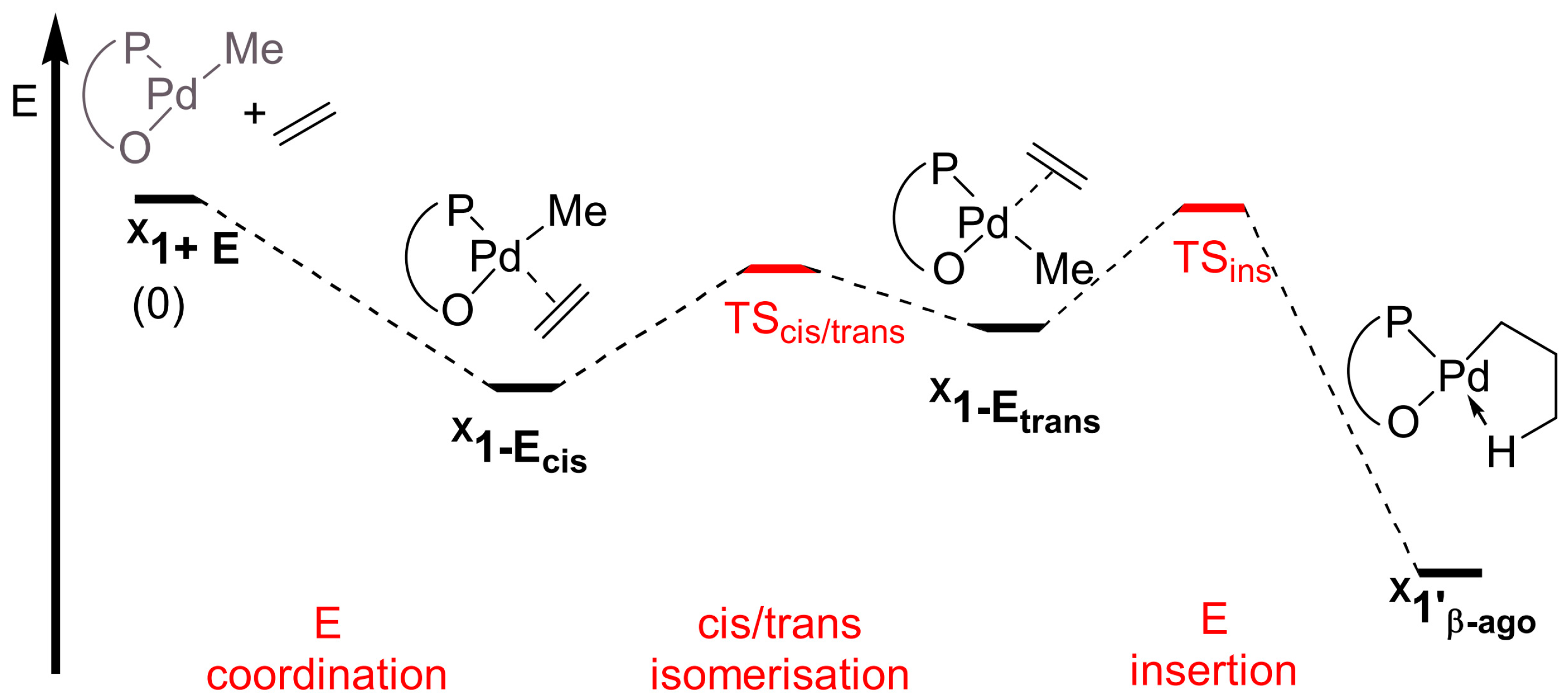
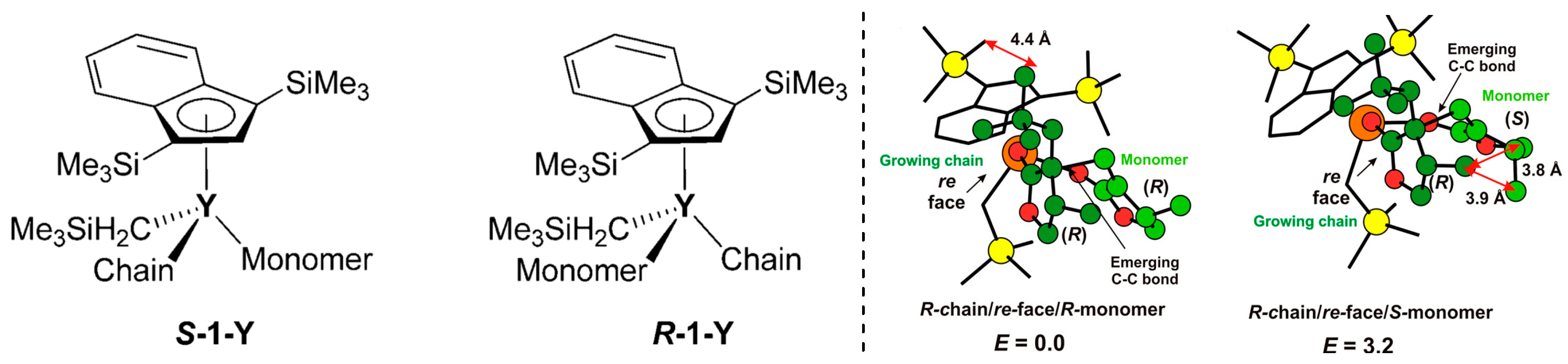
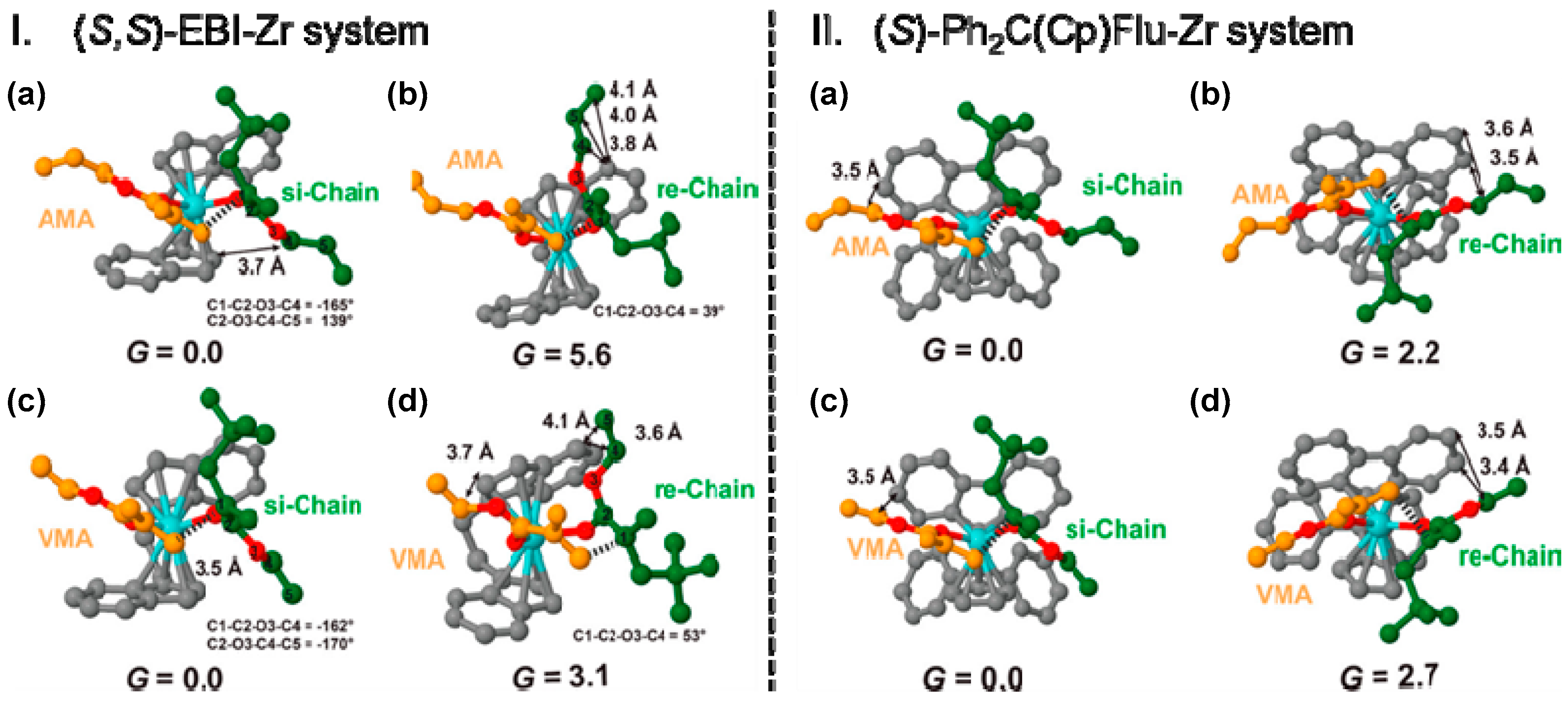
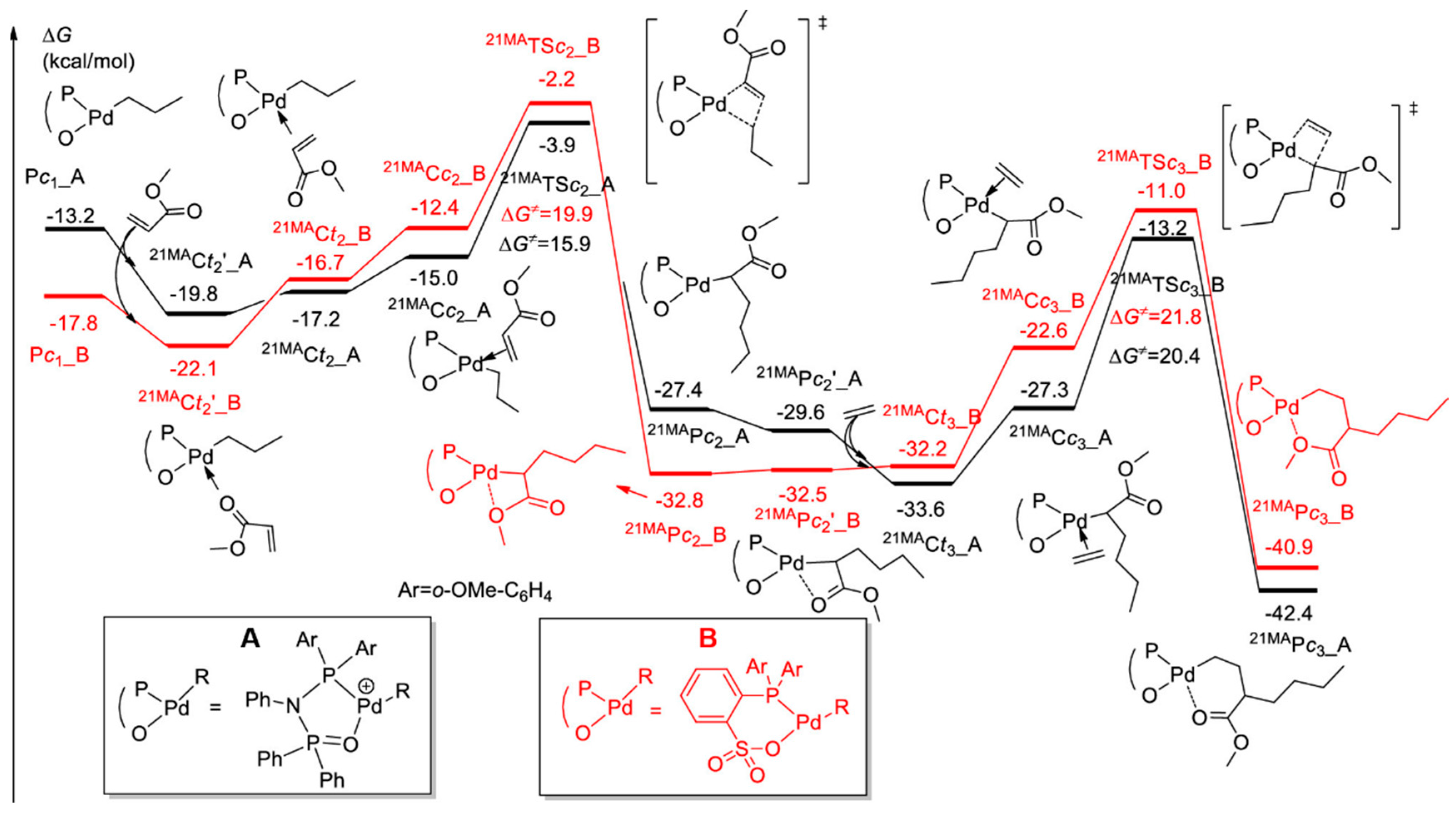
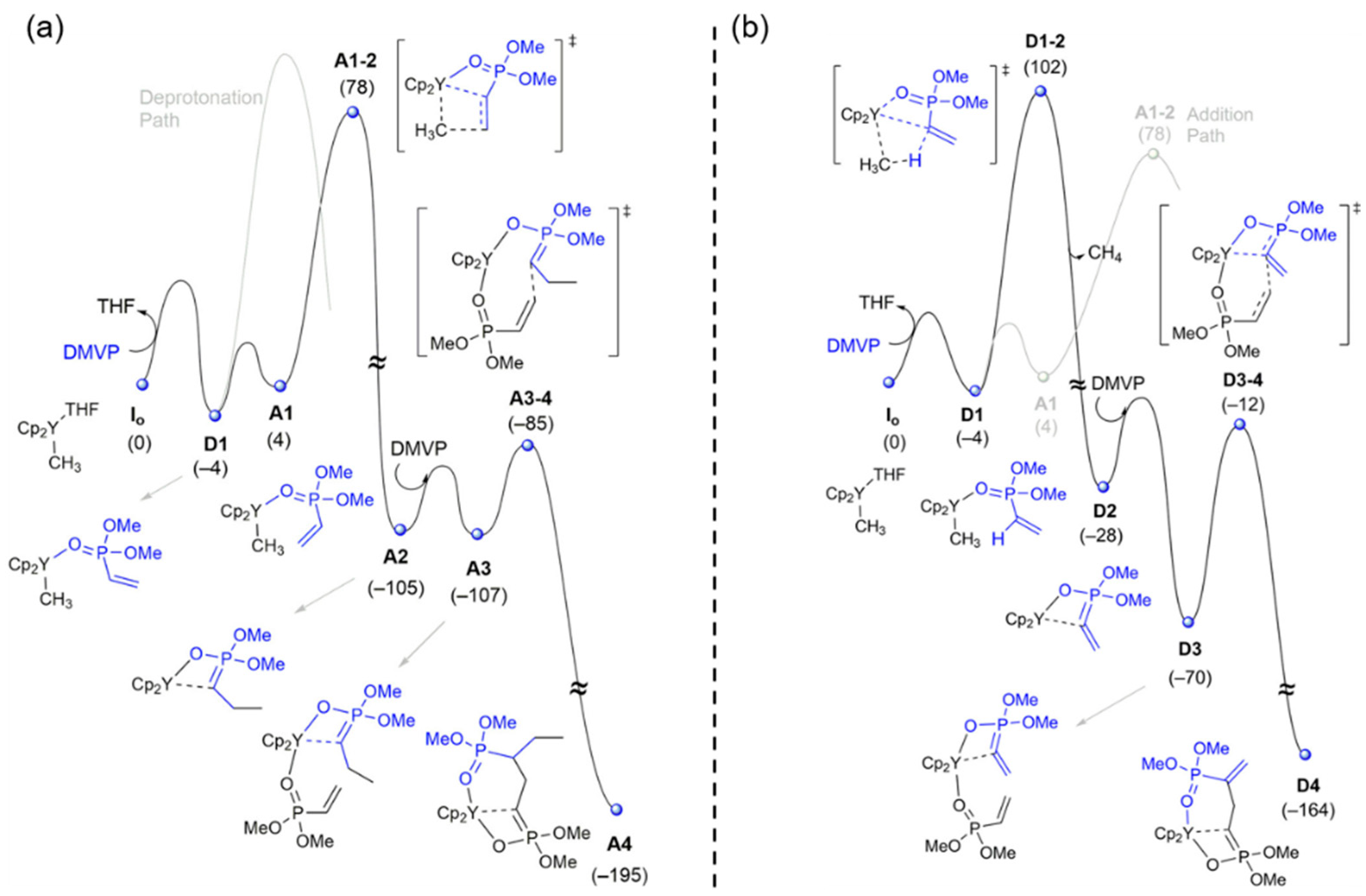
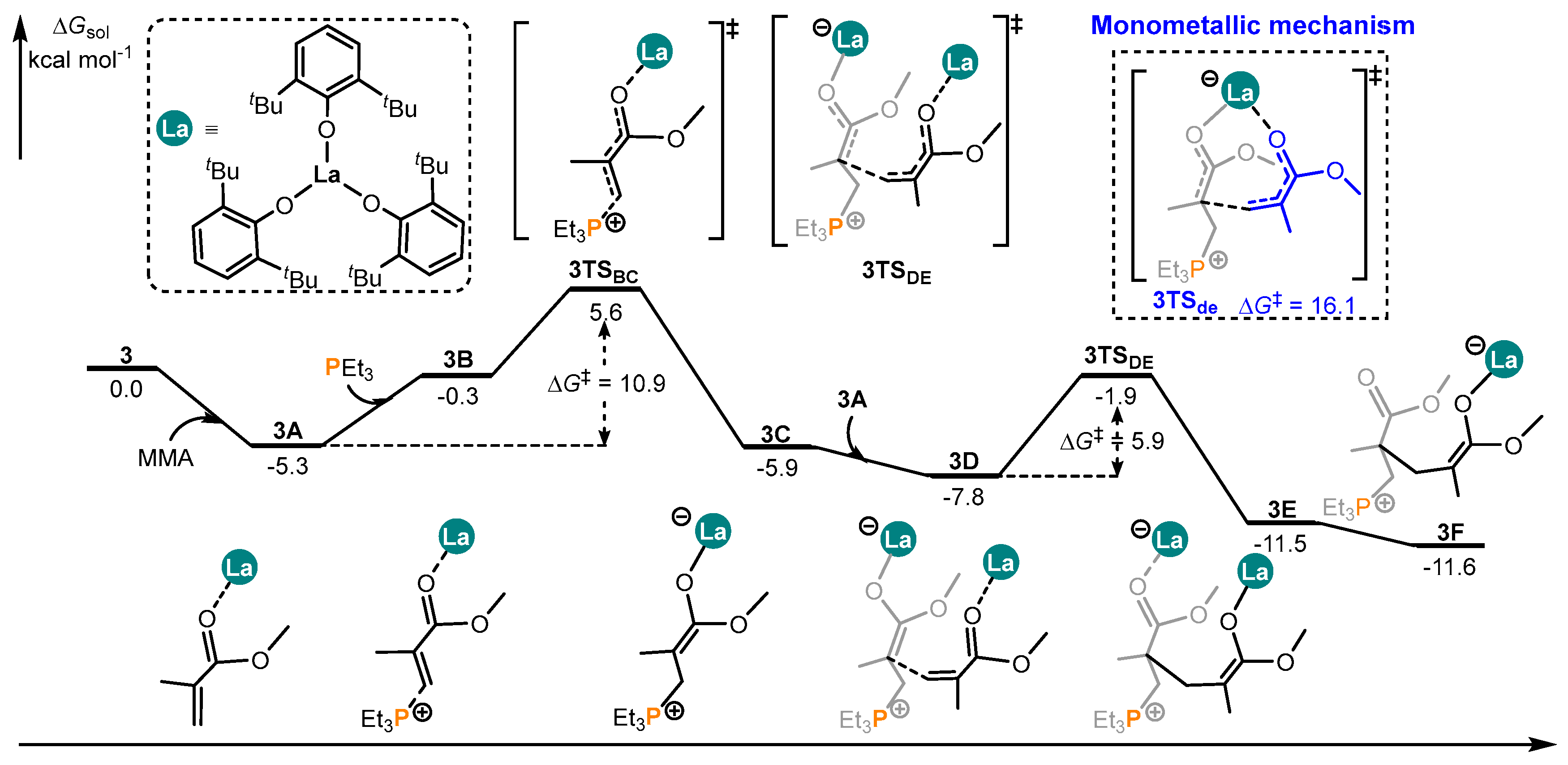

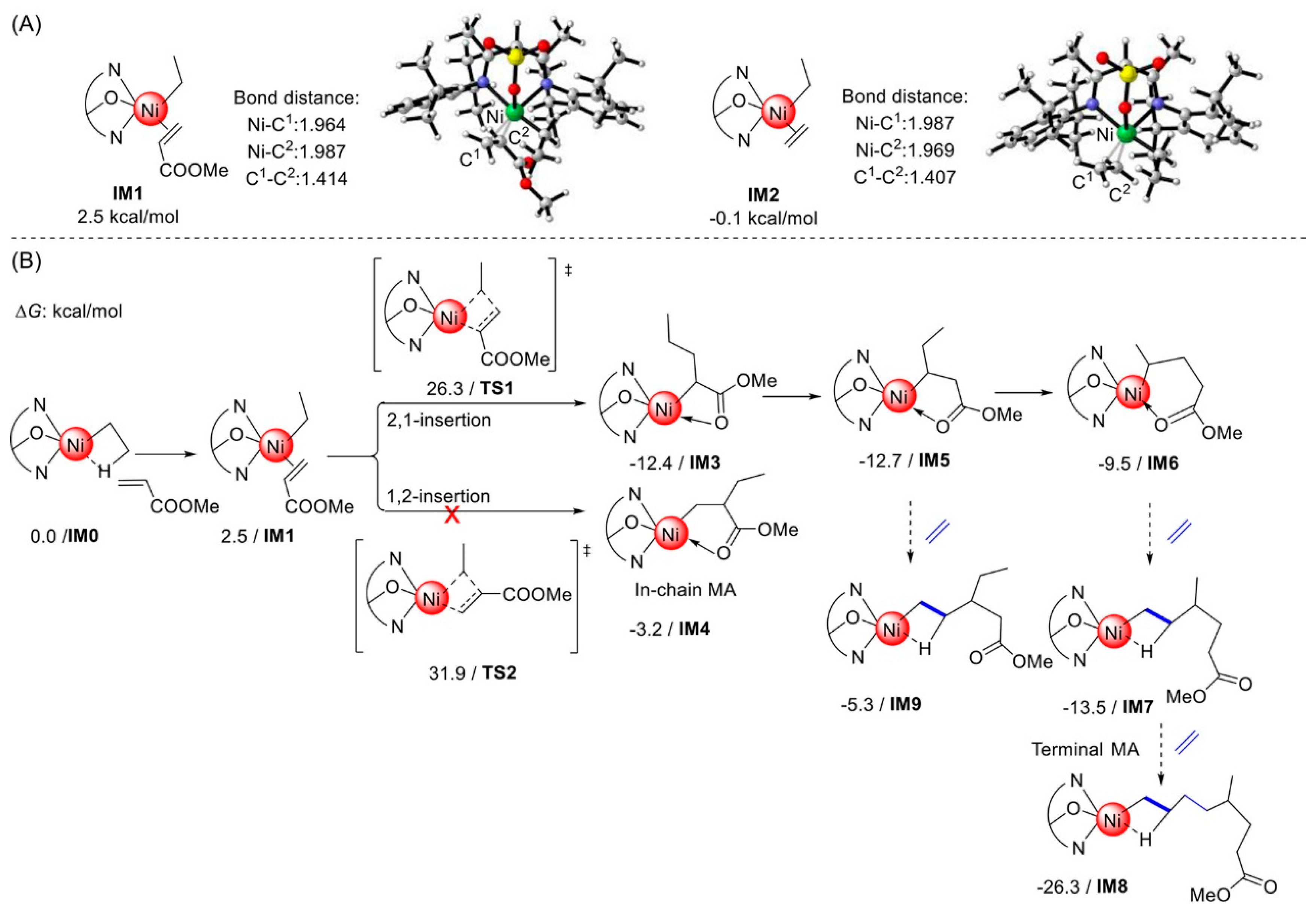
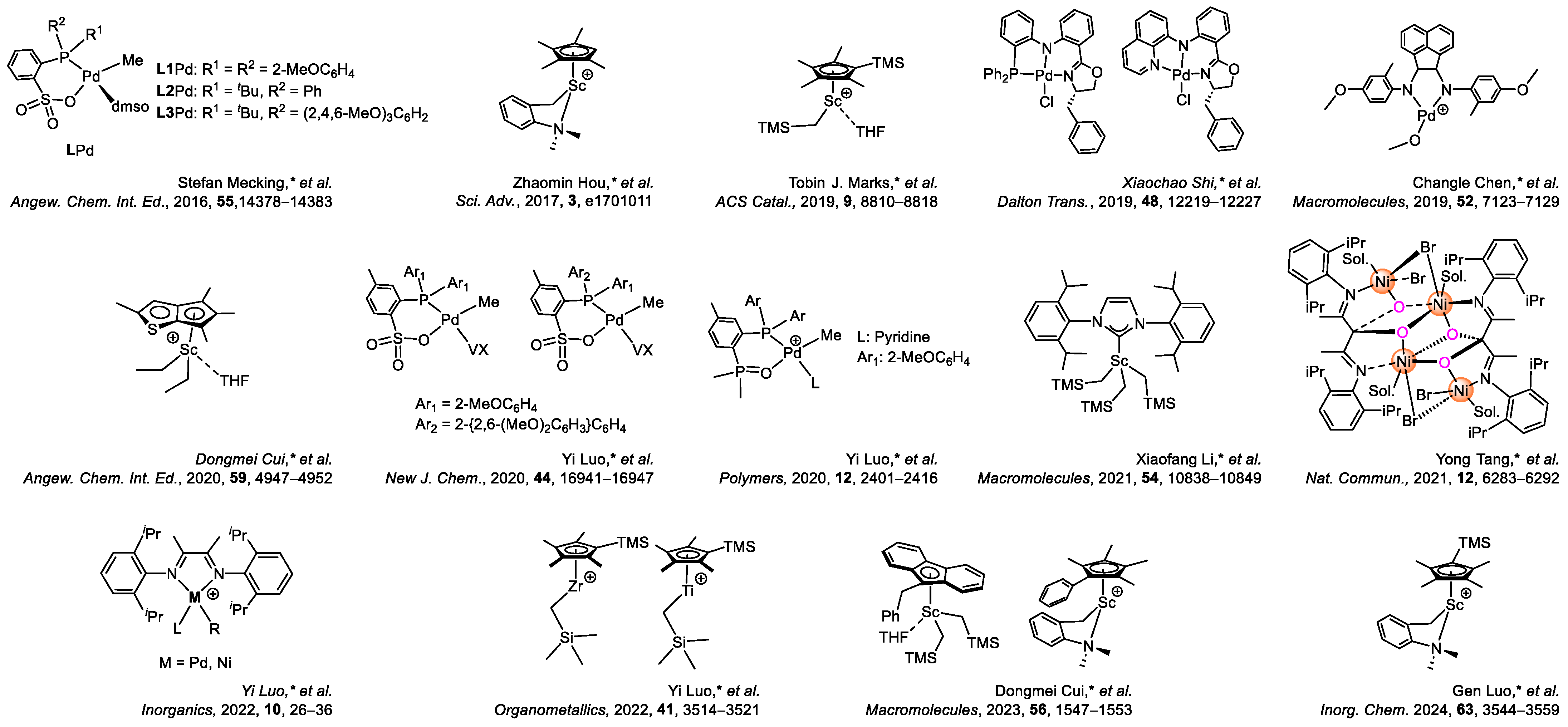

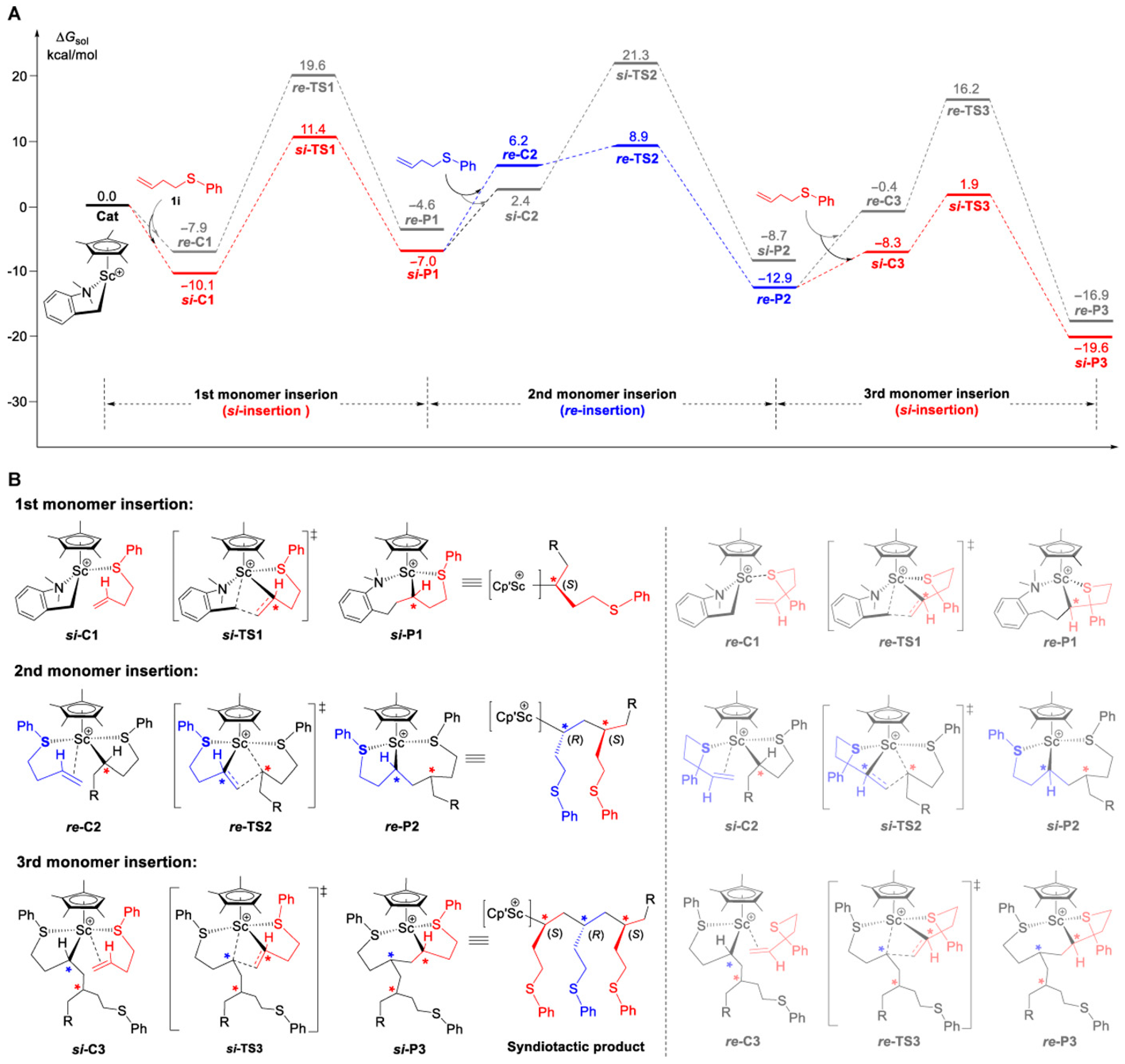
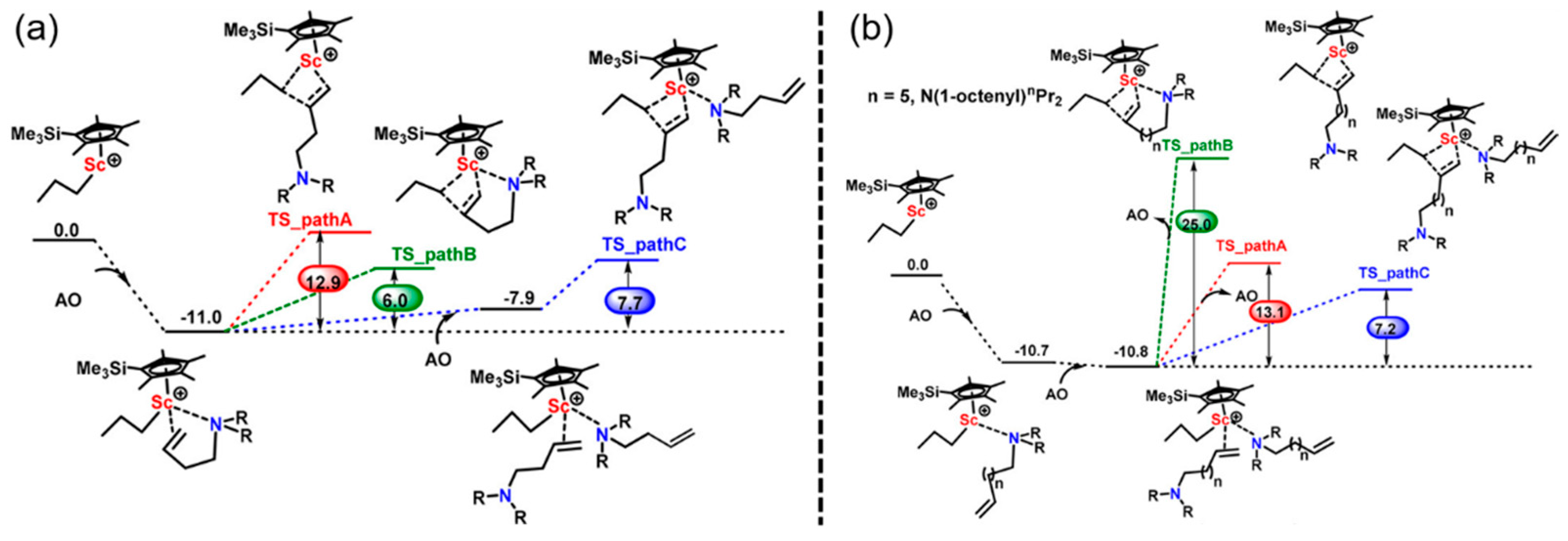
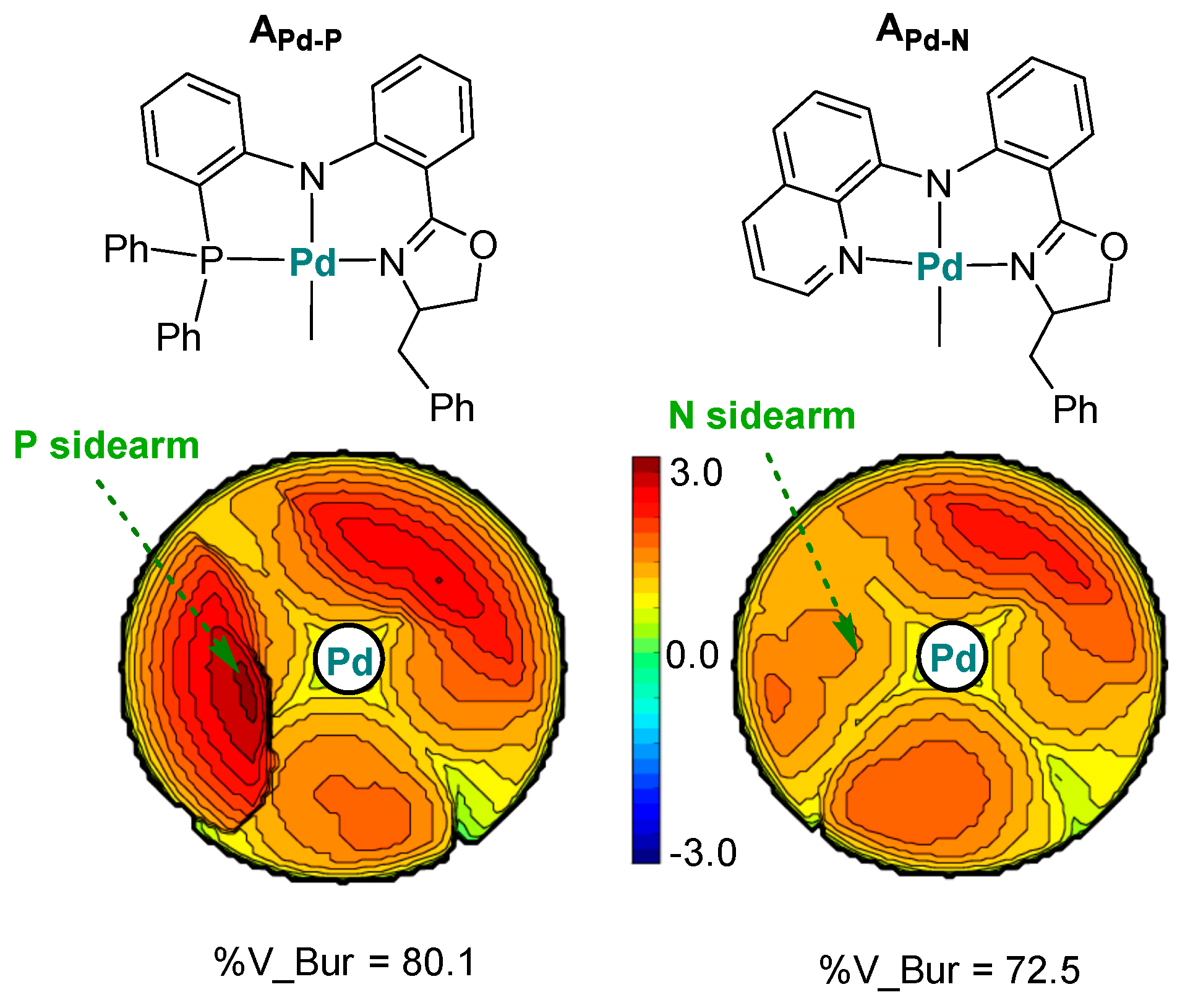
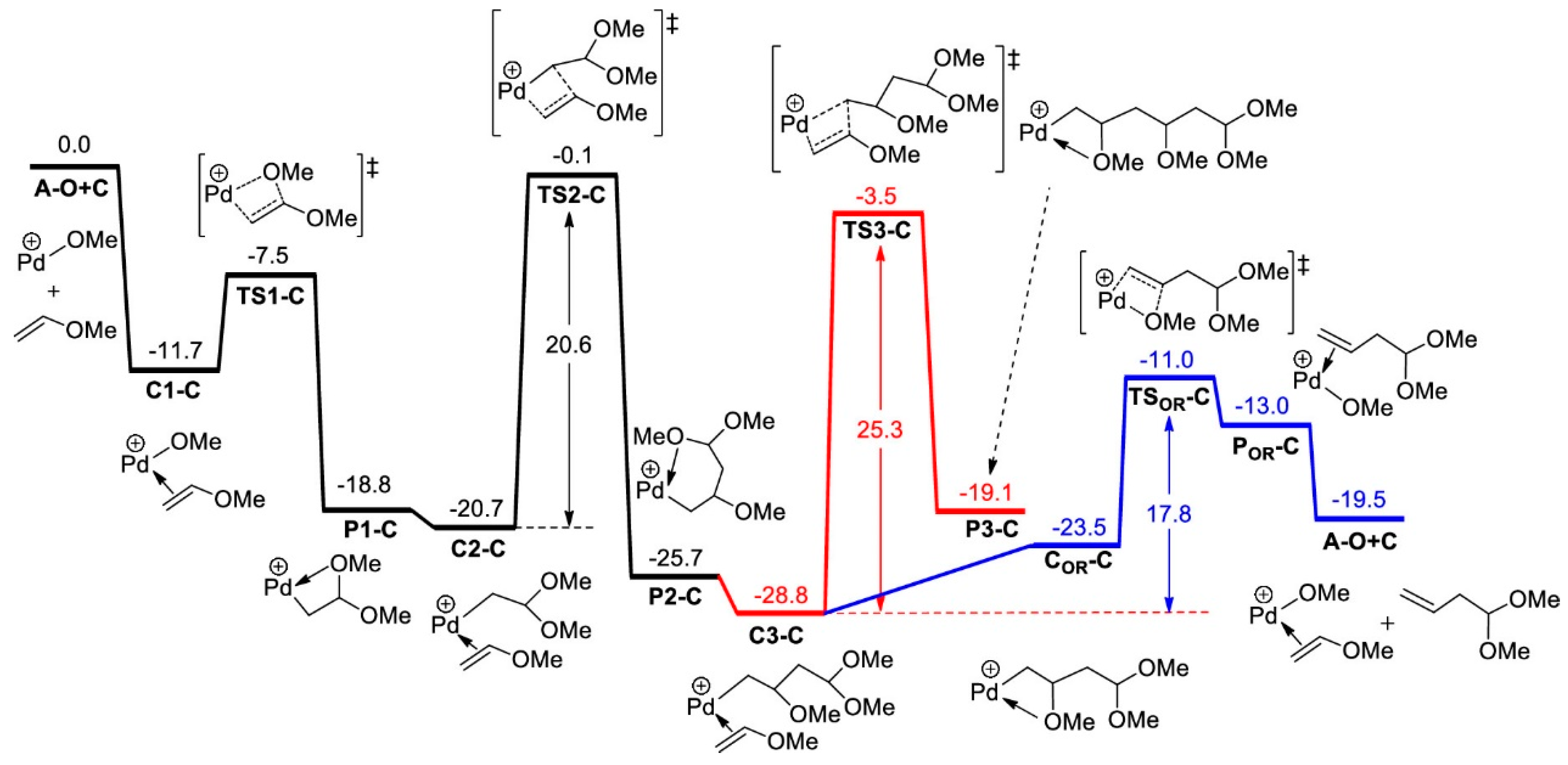
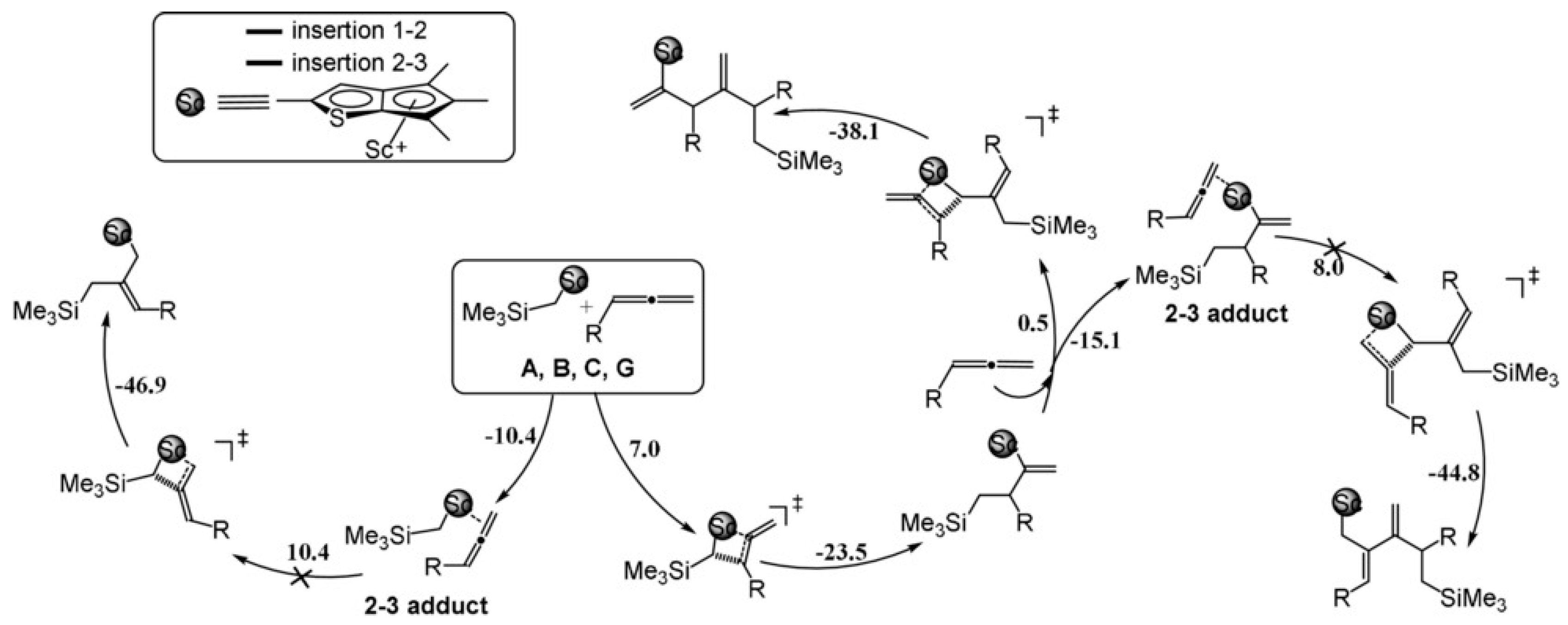
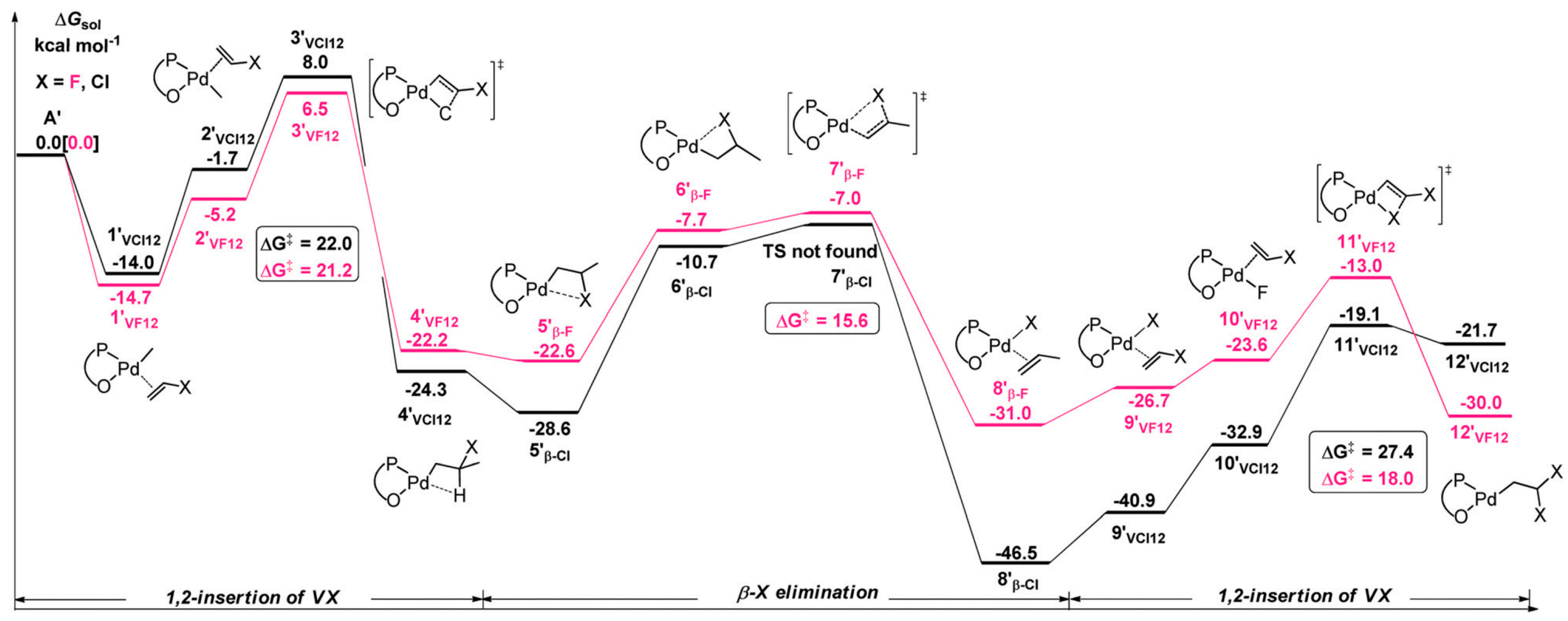
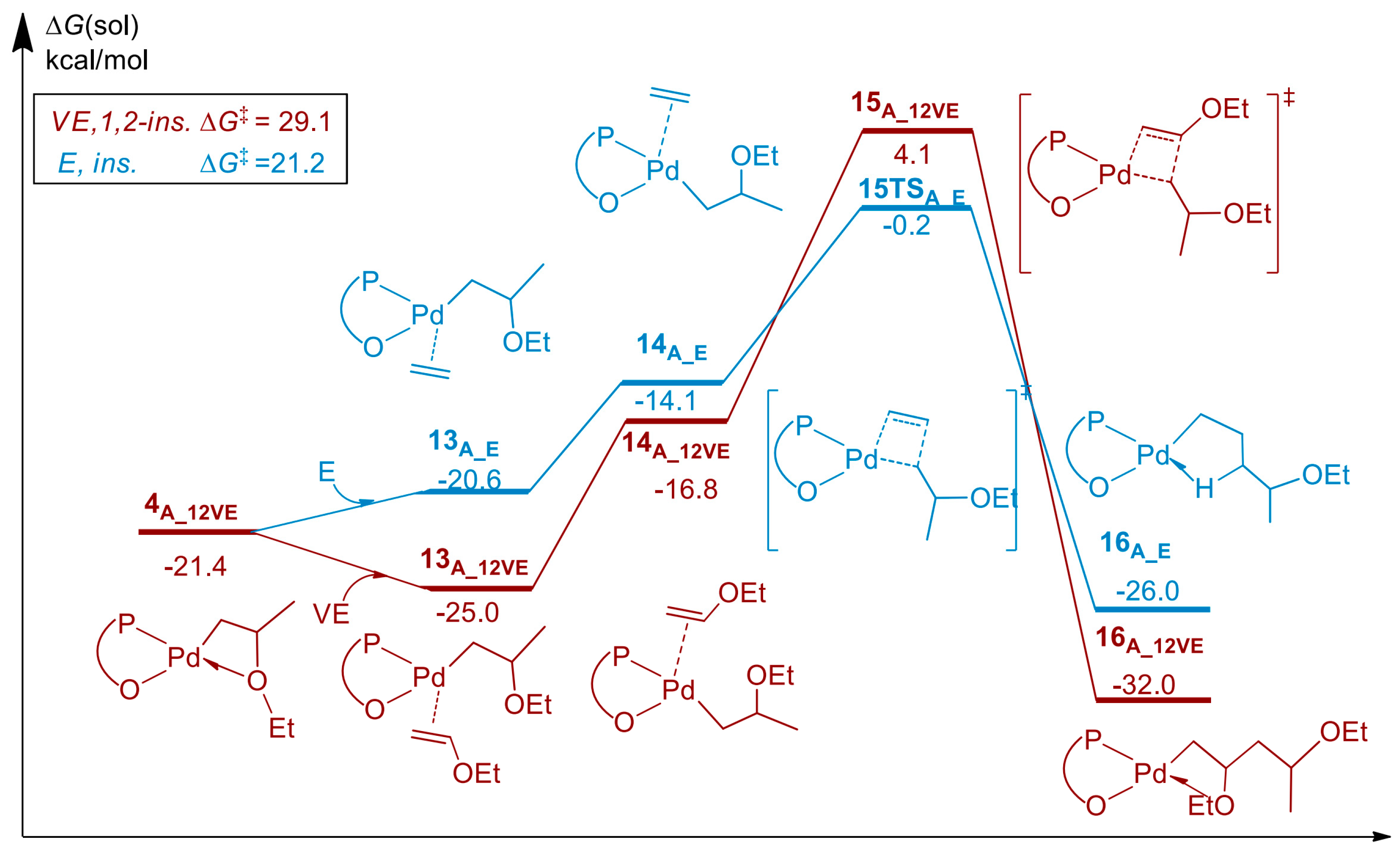

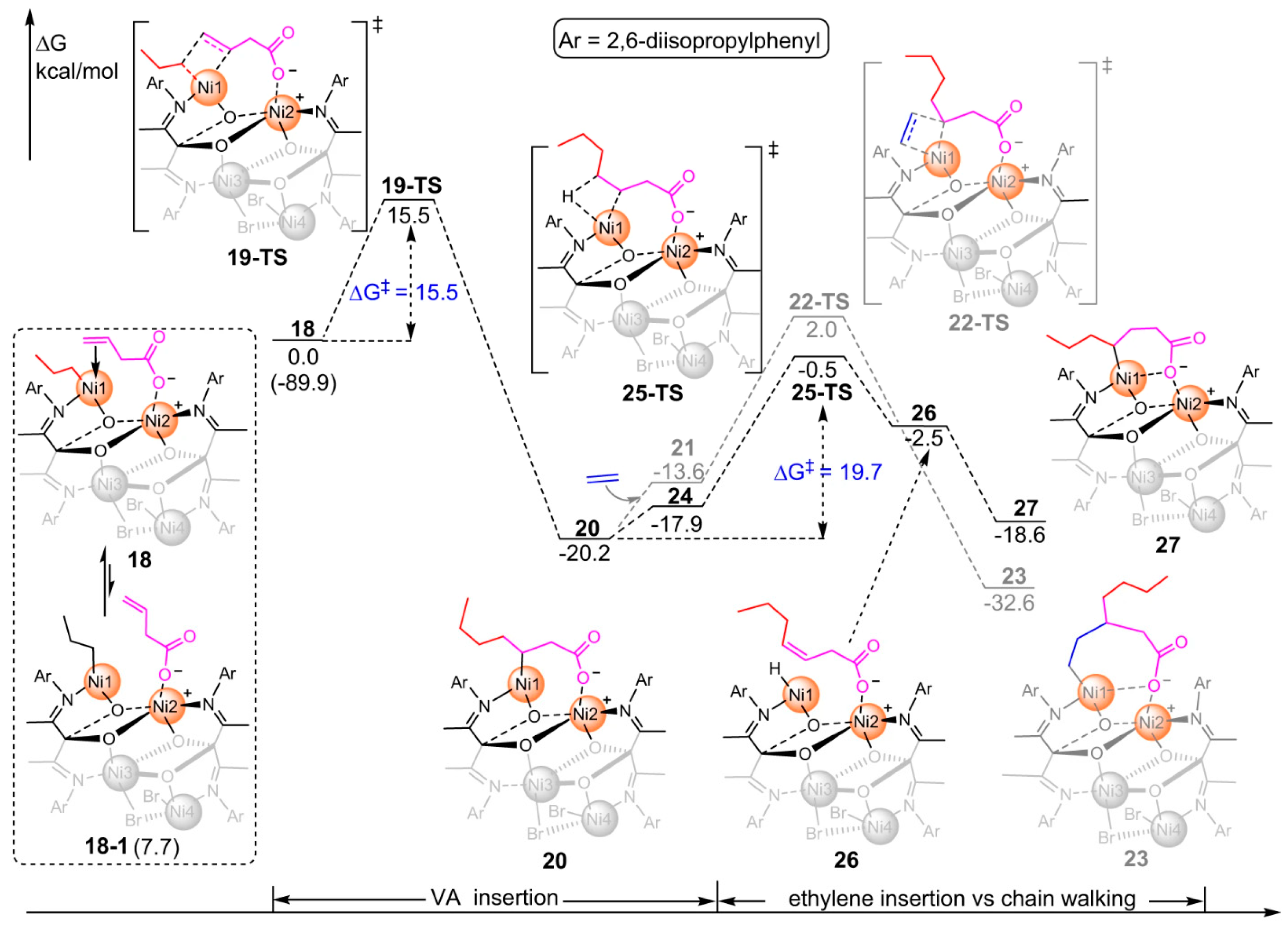
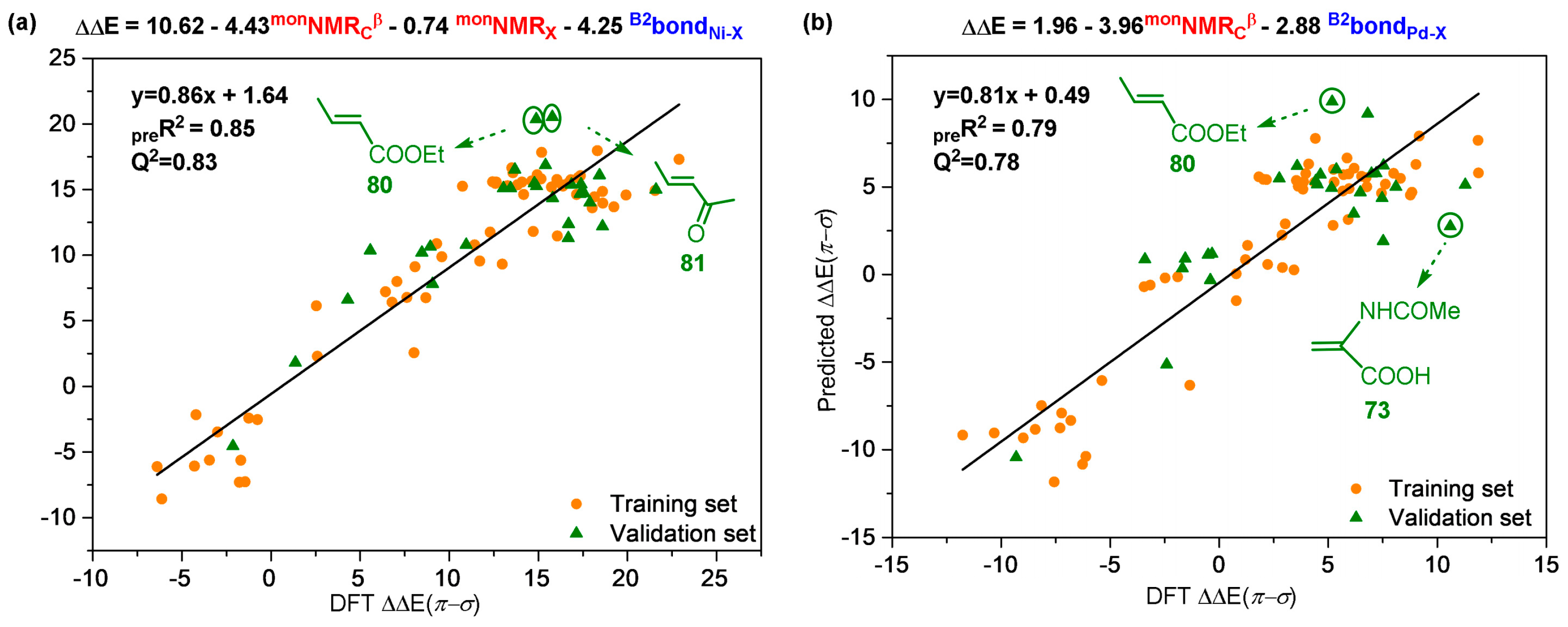
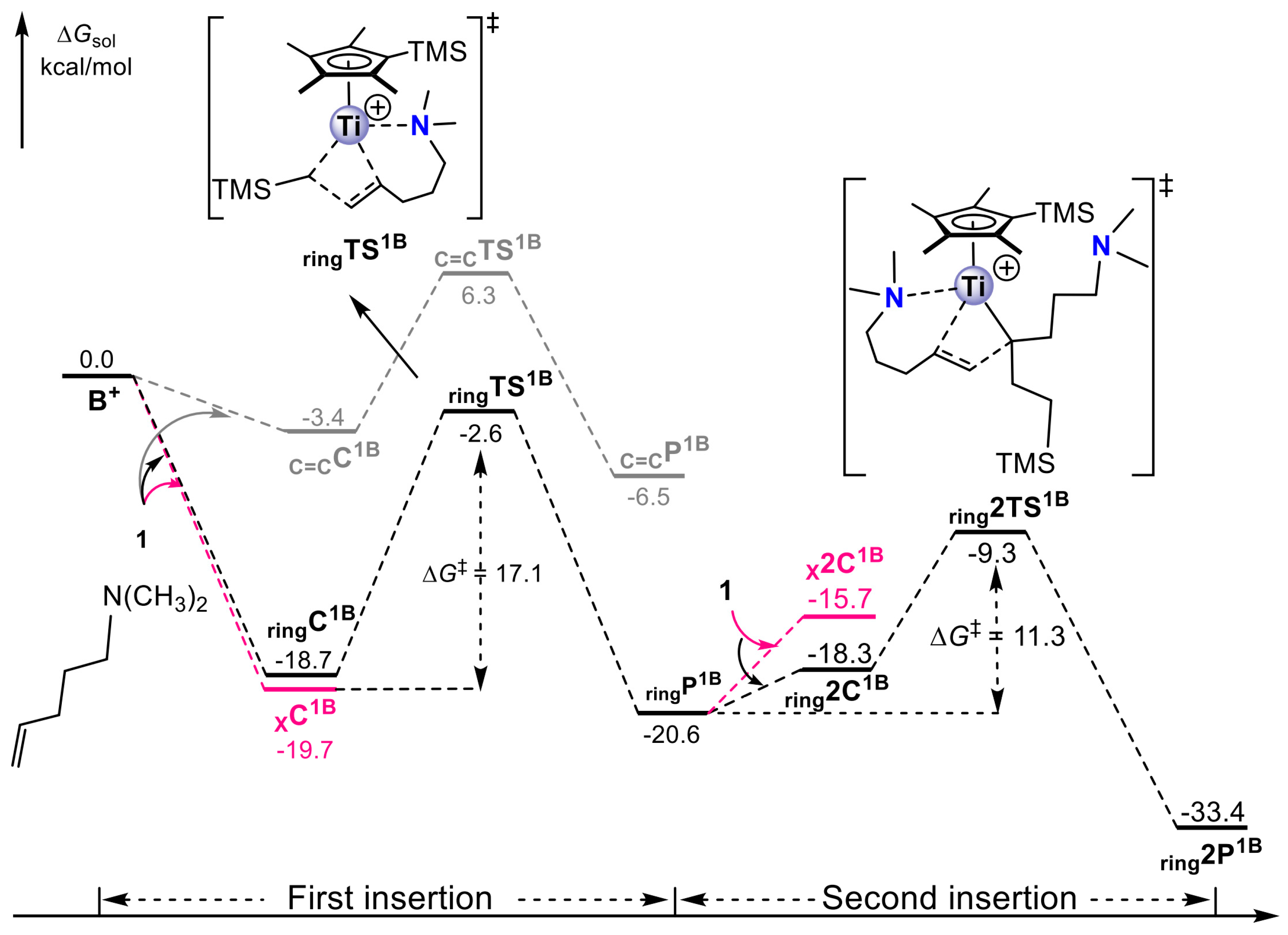






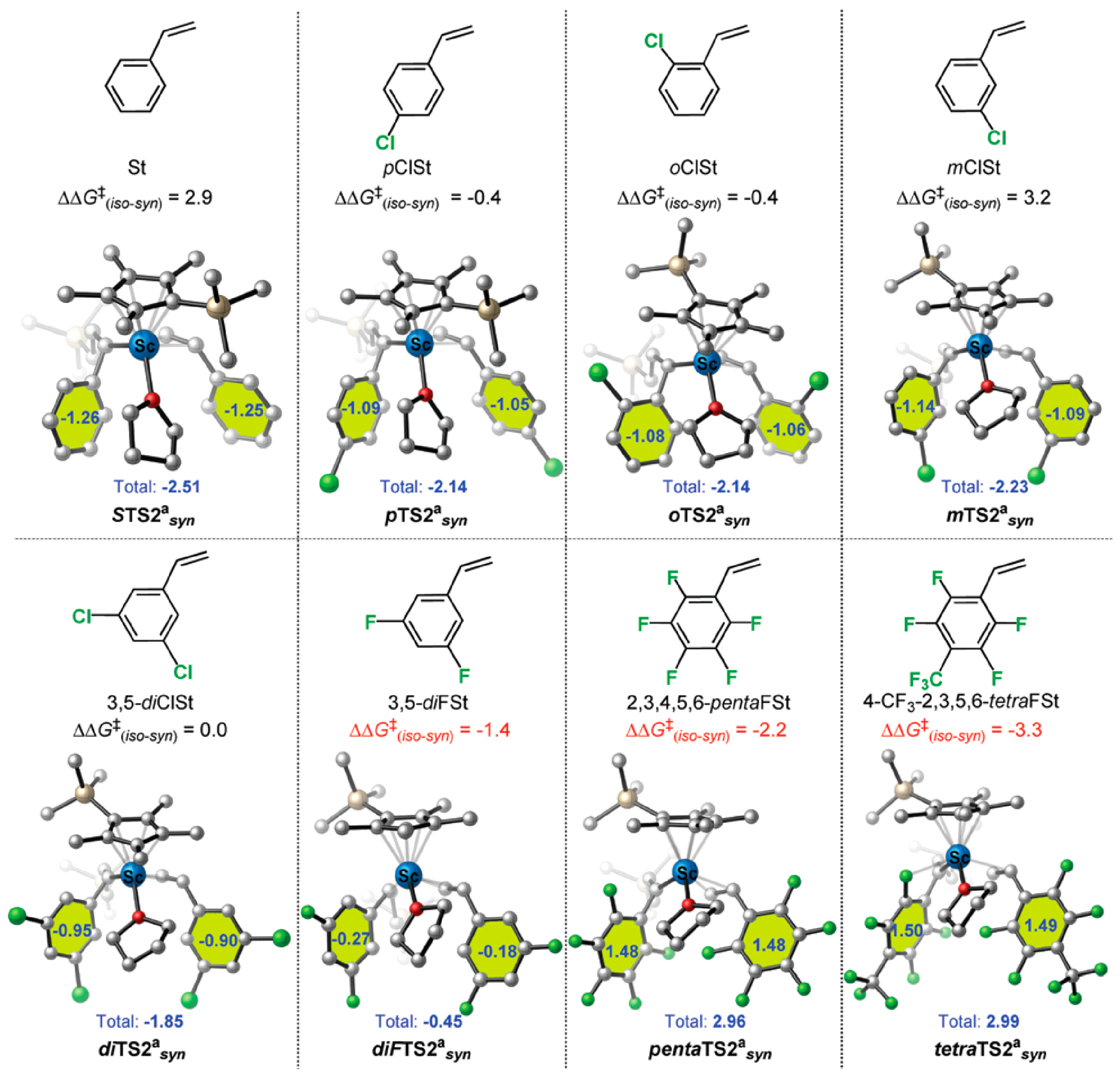
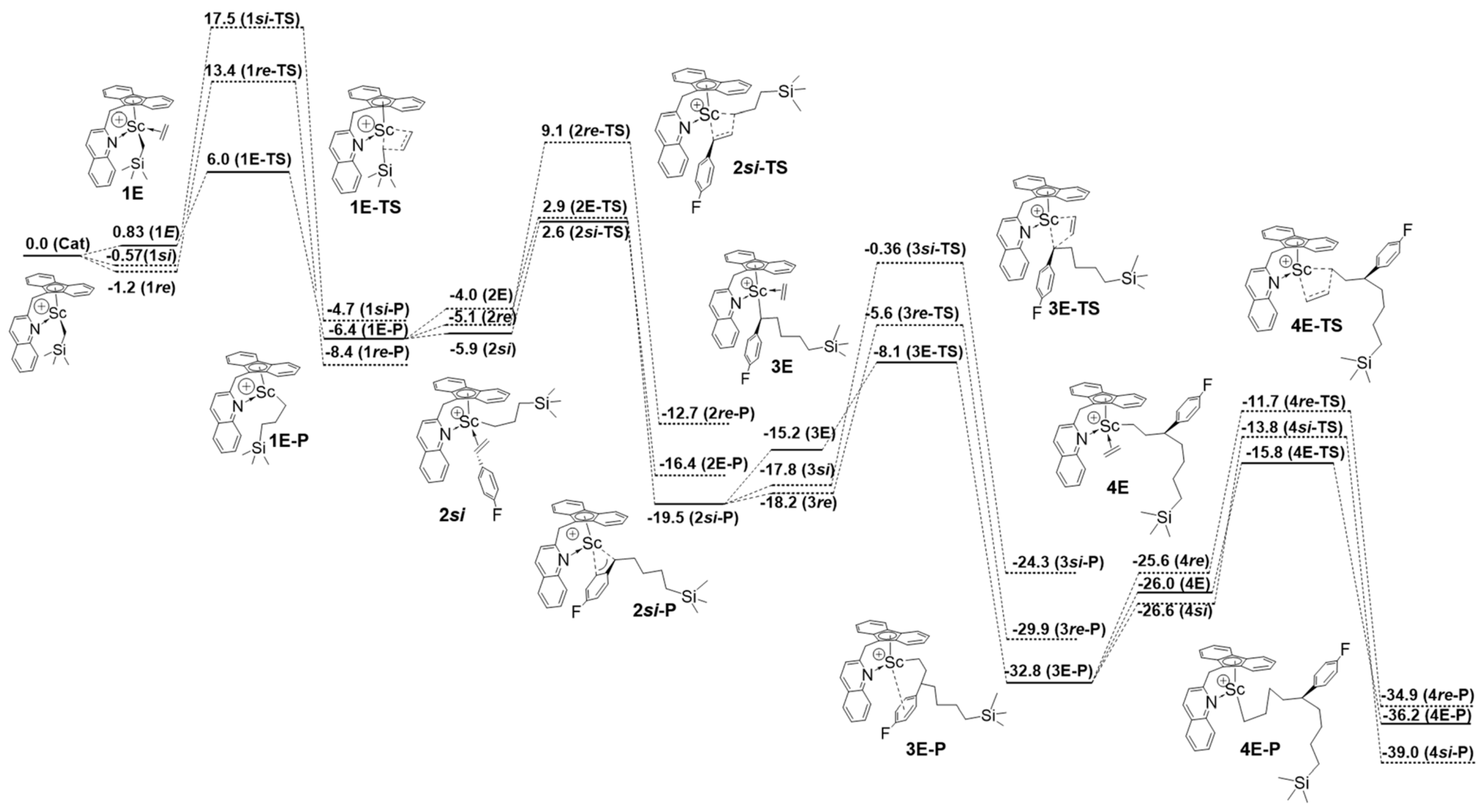
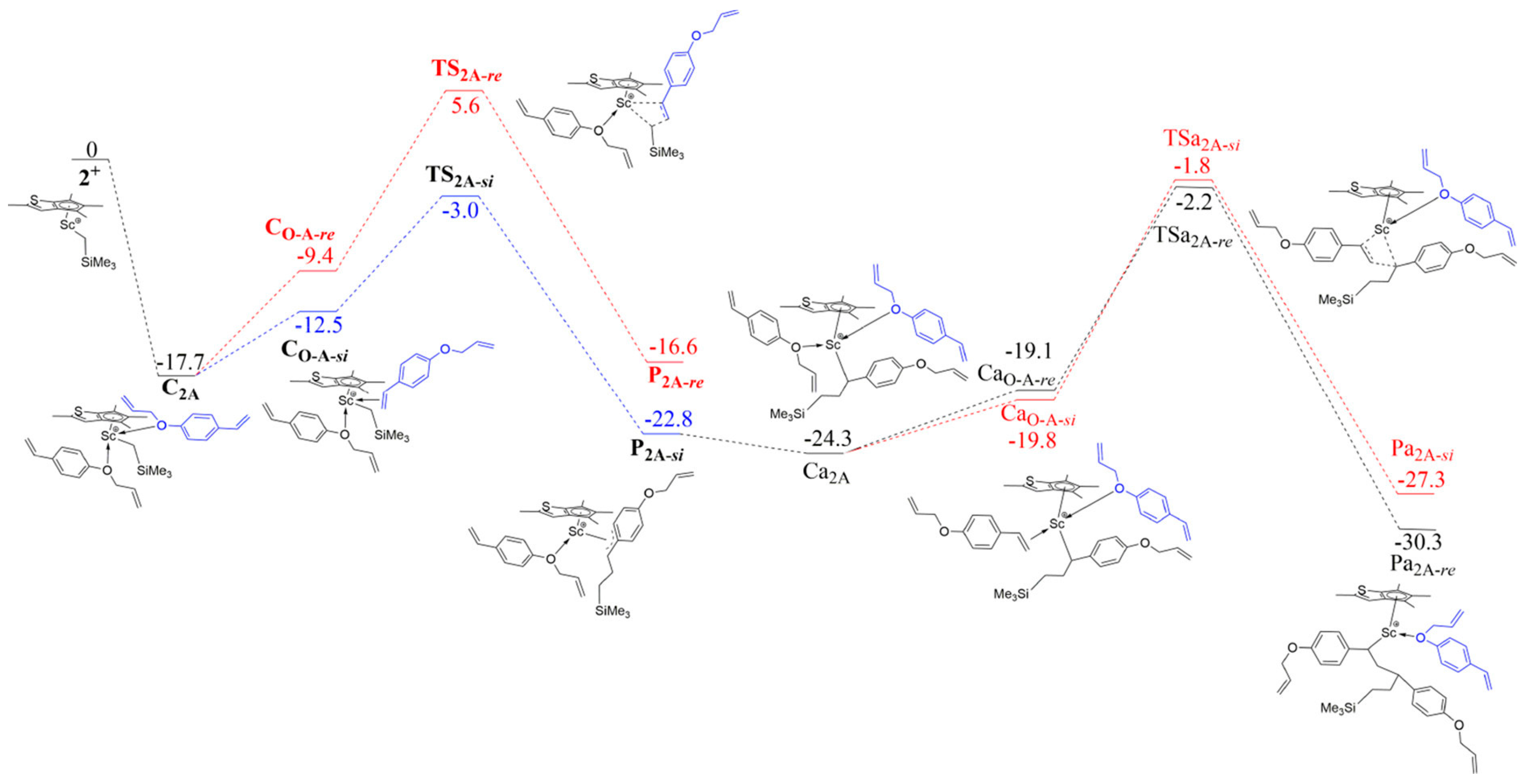
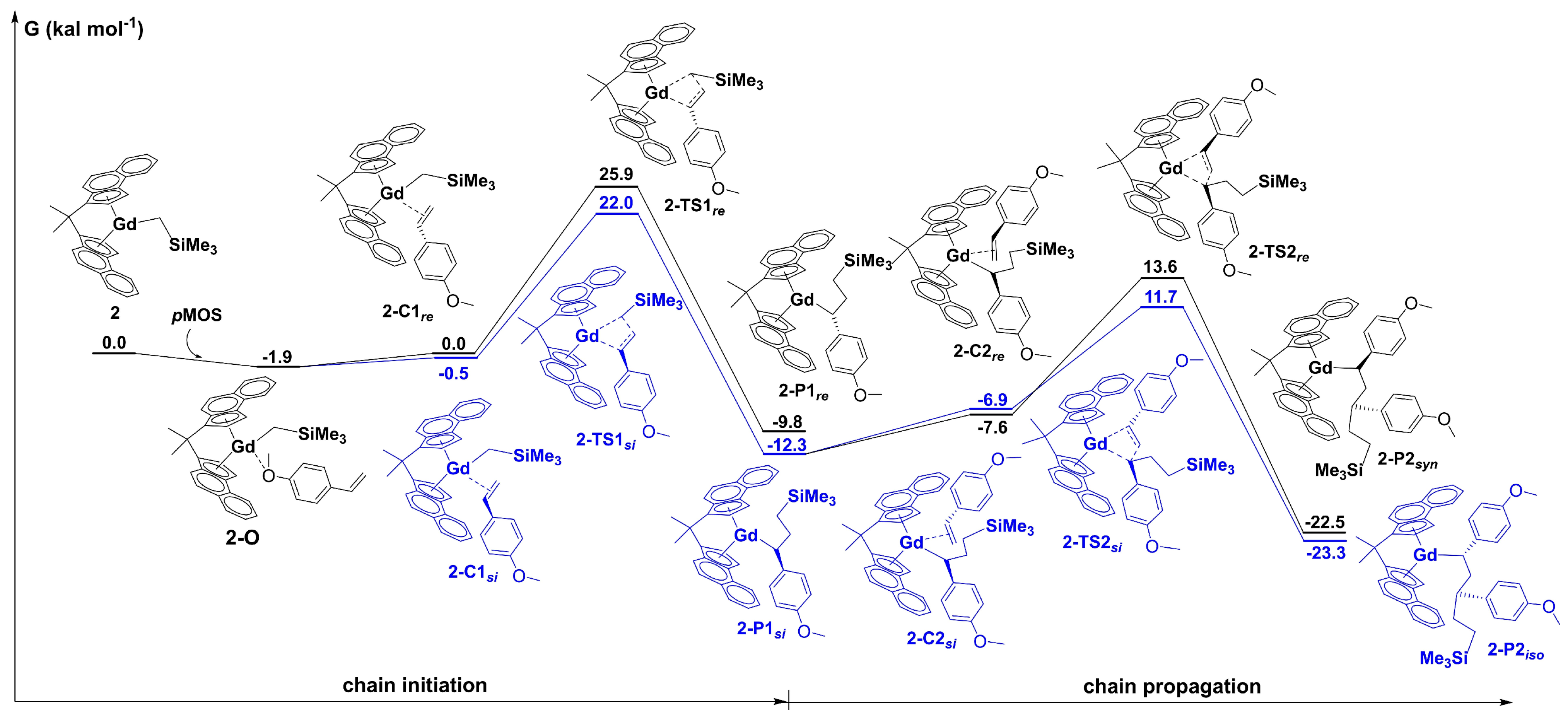
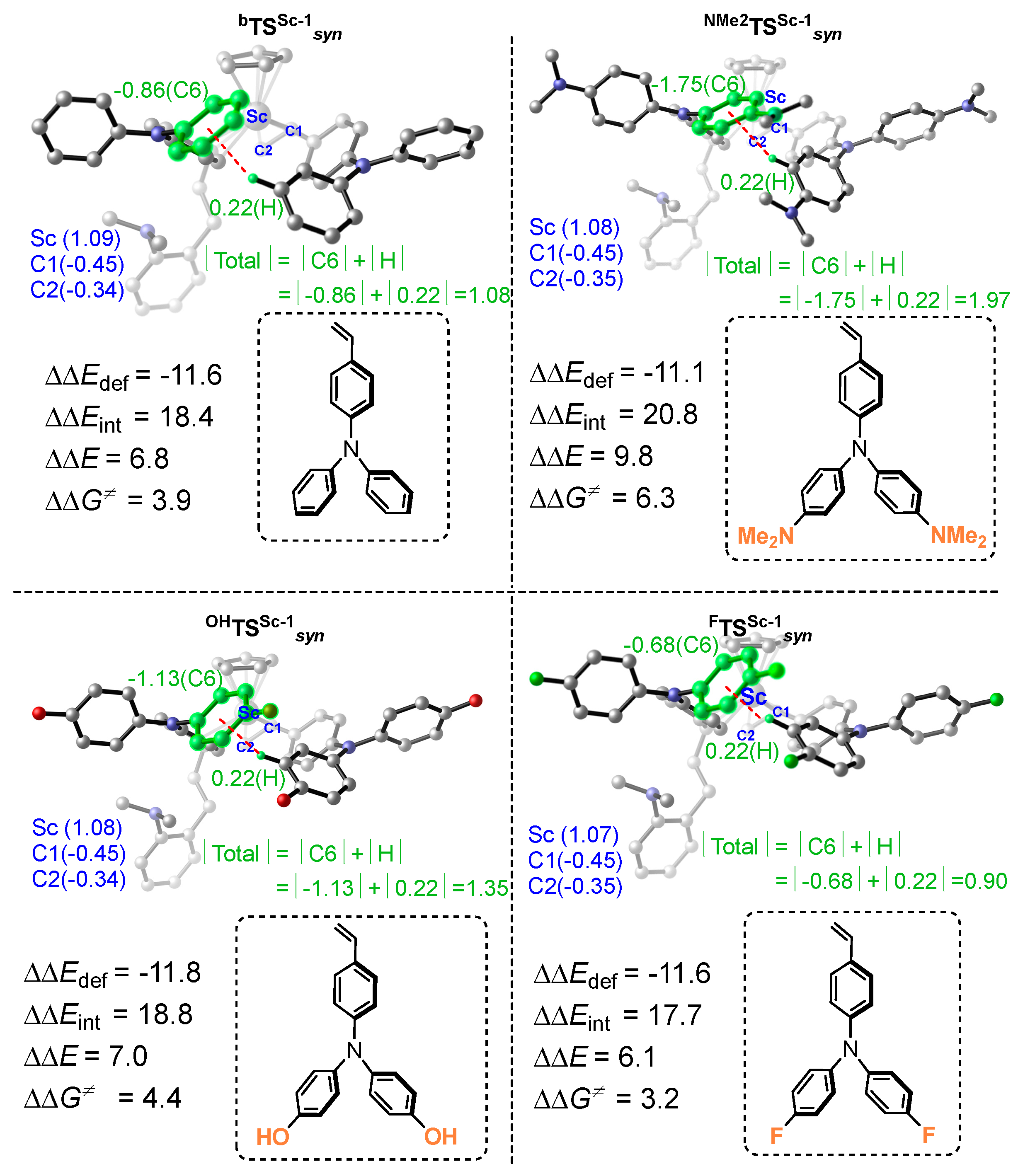
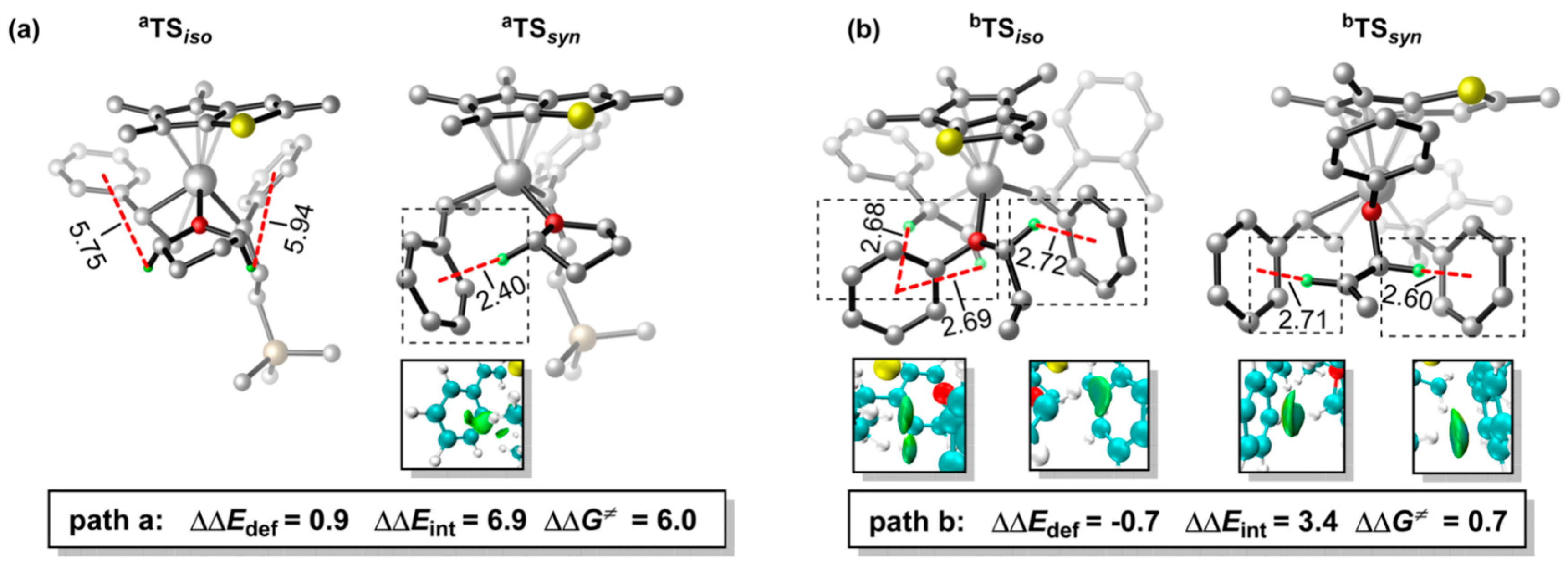
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Zhang, Z.; Luo, Y. DFT Modeling of Coordination Polymerization of Polar Olefin Monomers by Molecular Metal Complexes. Inorganics 2024, 12, 233. https://doi.org/10.3390/inorganics12090233
Zhao Y, Zhang Z, Luo Y. DFT Modeling of Coordination Polymerization of Polar Olefin Monomers by Molecular Metal Complexes. Inorganics. 2024; 12(9):233. https://doi.org/10.3390/inorganics12090233
Chicago/Turabian StyleZhao, Yanan, Zhenli Zhang, and Yi Luo. 2024. "DFT Modeling of Coordination Polymerization of Polar Olefin Monomers by Molecular Metal Complexes" Inorganics 12, no. 9: 233. https://doi.org/10.3390/inorganics12090233
APA StyleZhao, Y., Zhang, Z., & Luo, Y. (2024). DFT Modeling of Coordination Polymerization of Polar Olefin Monomers by Molecular Metal Complexes. Inorganics, 12(9), 233. https://doi.org/10.3390/inorganics12090233