

Investigations of the Influence of Two Pyridyl-Mesoionic Carbene Constitutional Isomers on the Electrochemical and Spectroelectrochemical Properties of Group 6 Metal Carbonyl Complexes


Abstract
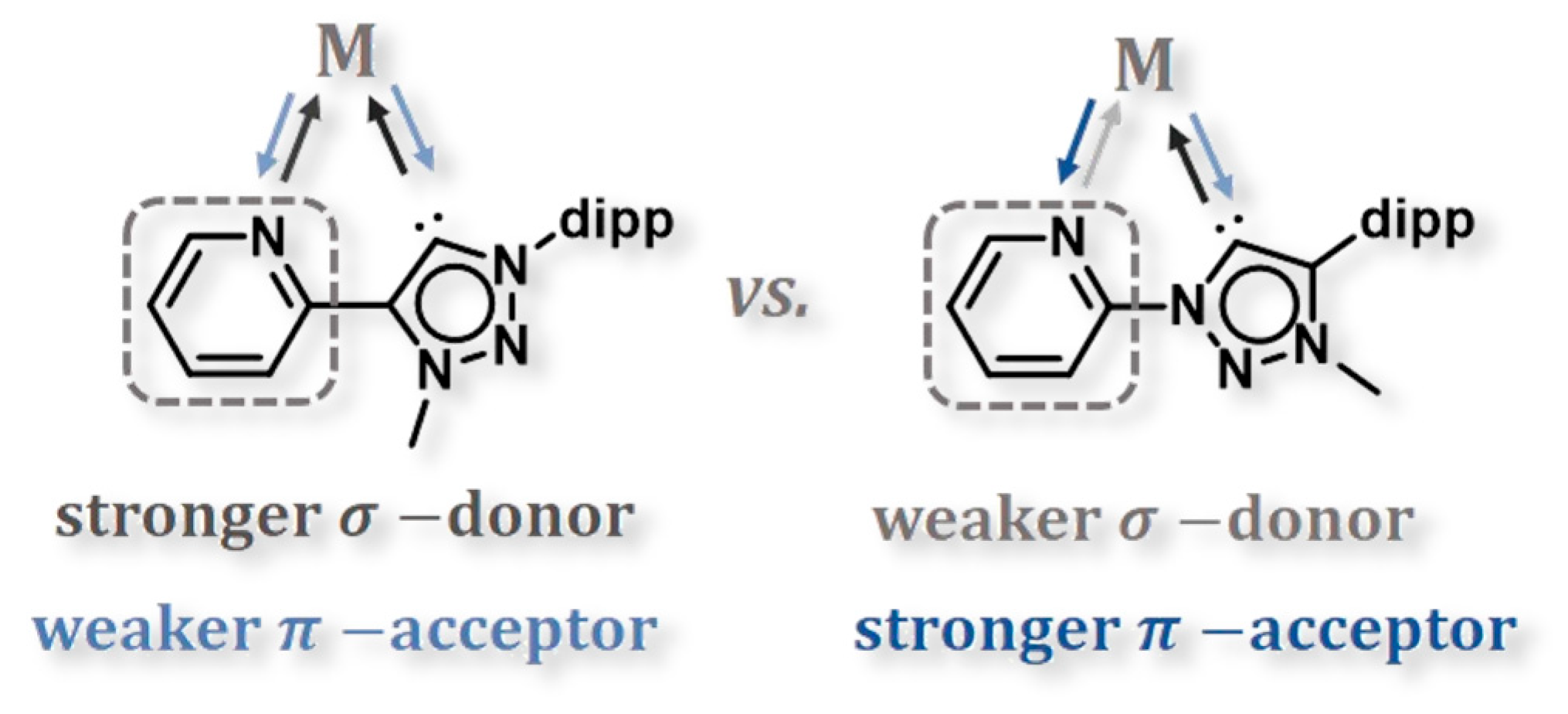
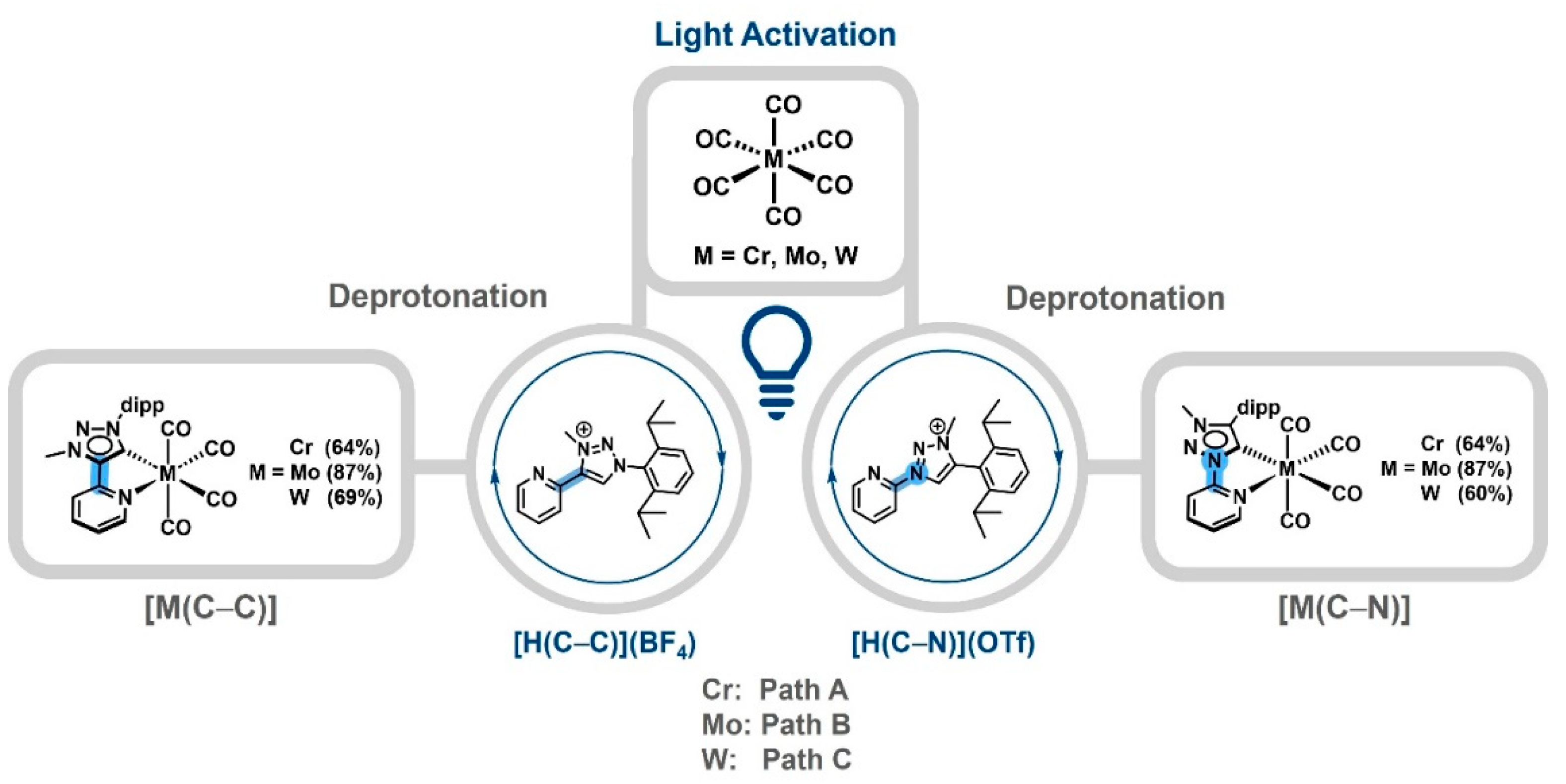
1. Introduction
2. Results and Discussion
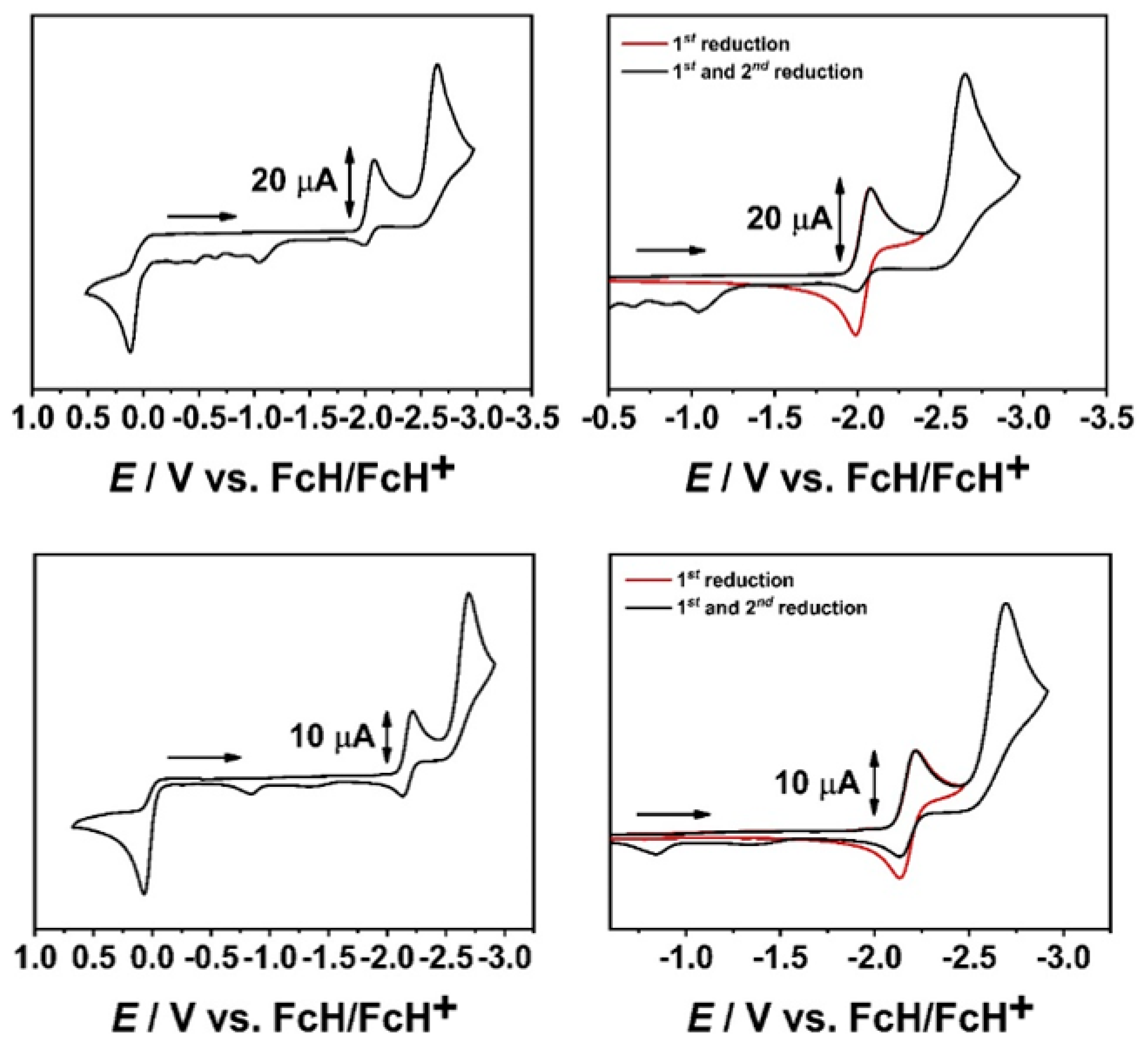
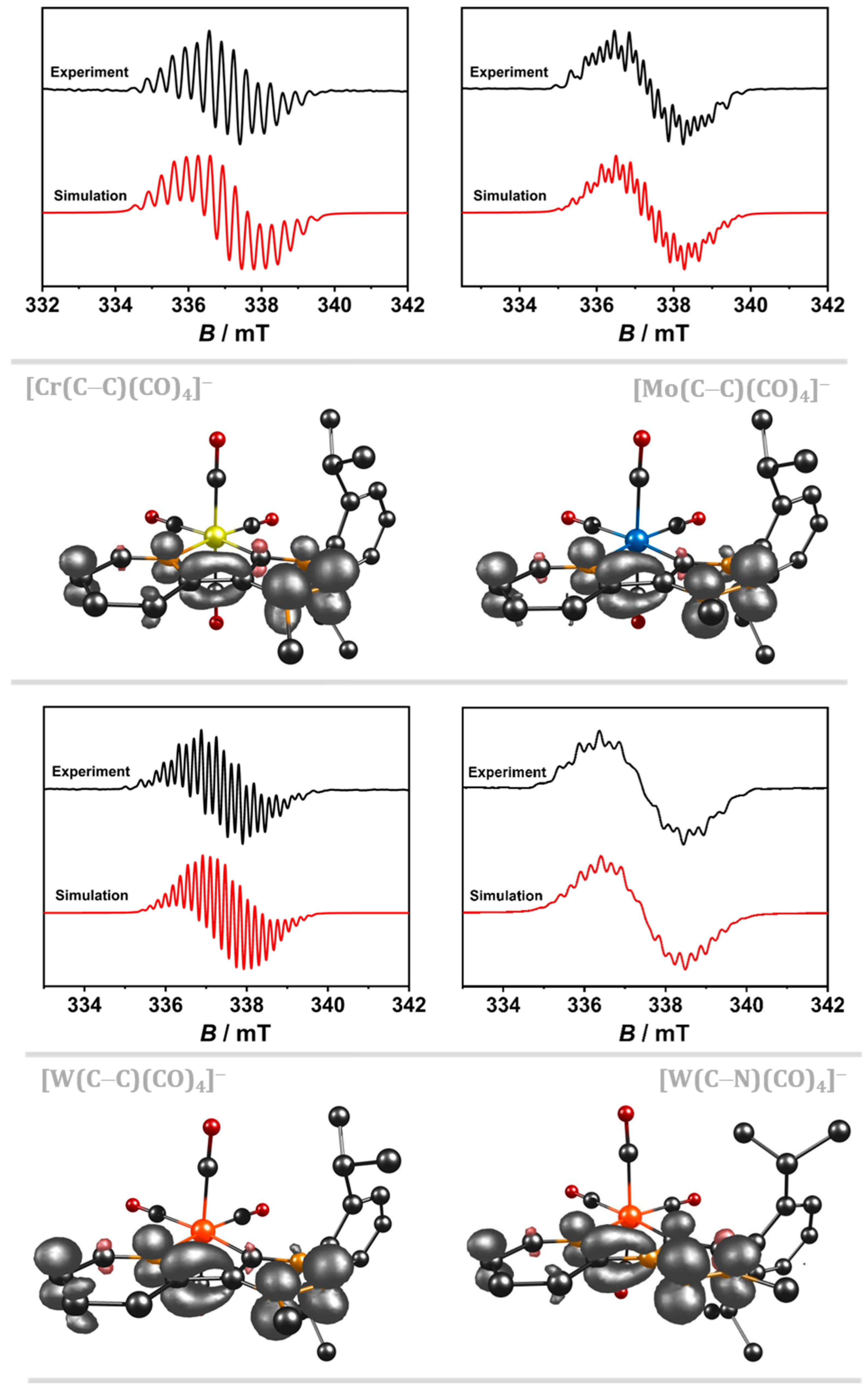
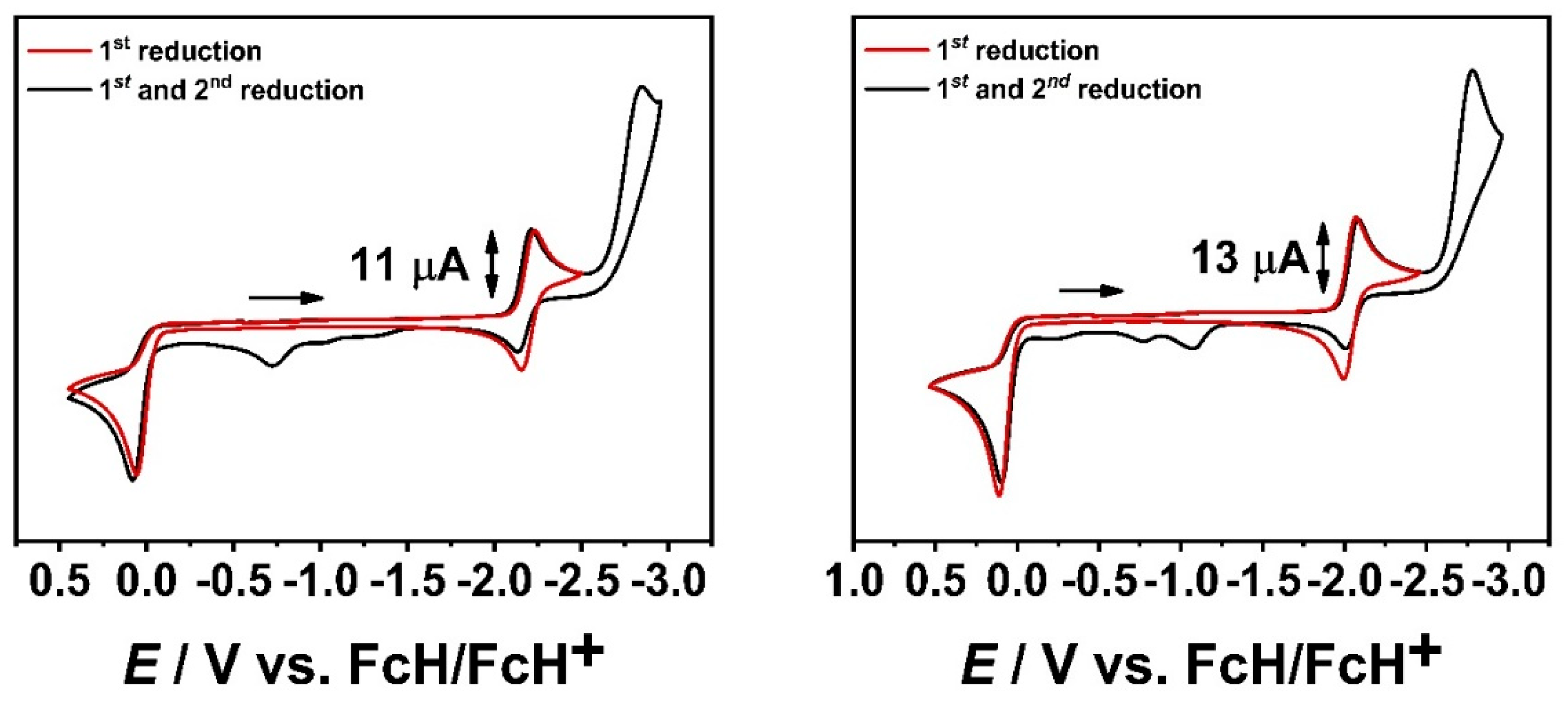
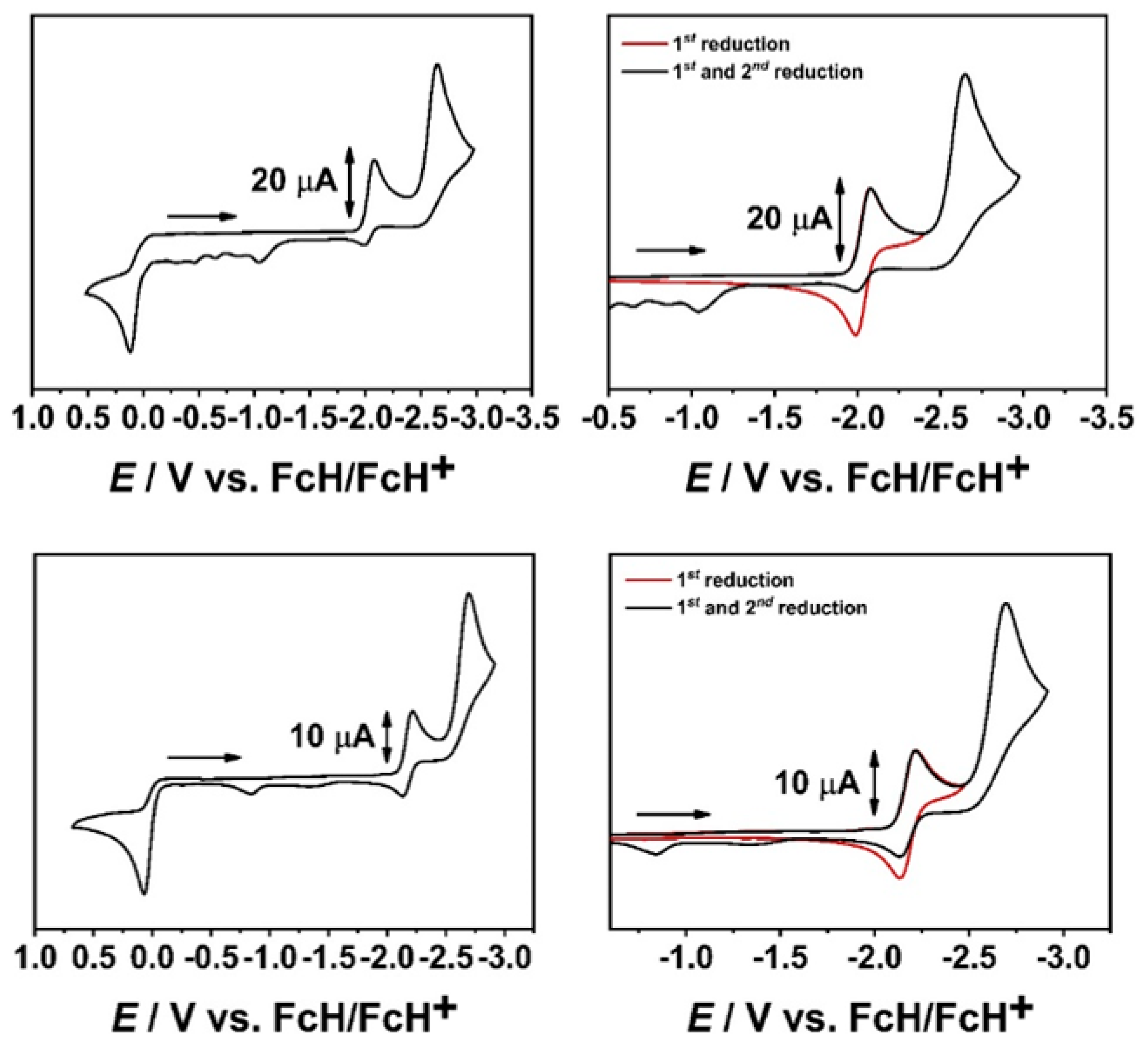
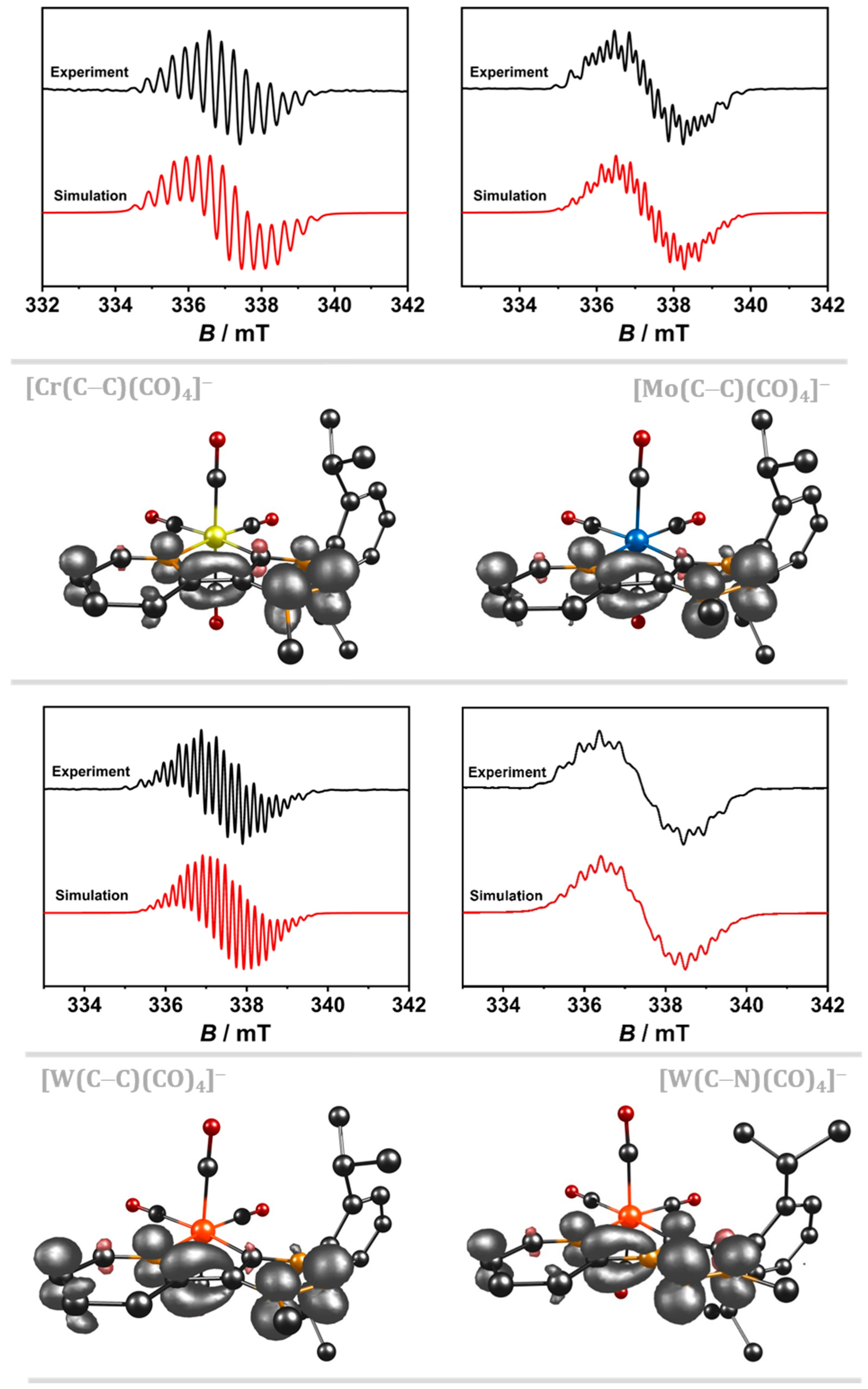
2.1. Cyclic Voltammetry with a GC WE and EPR-SEC
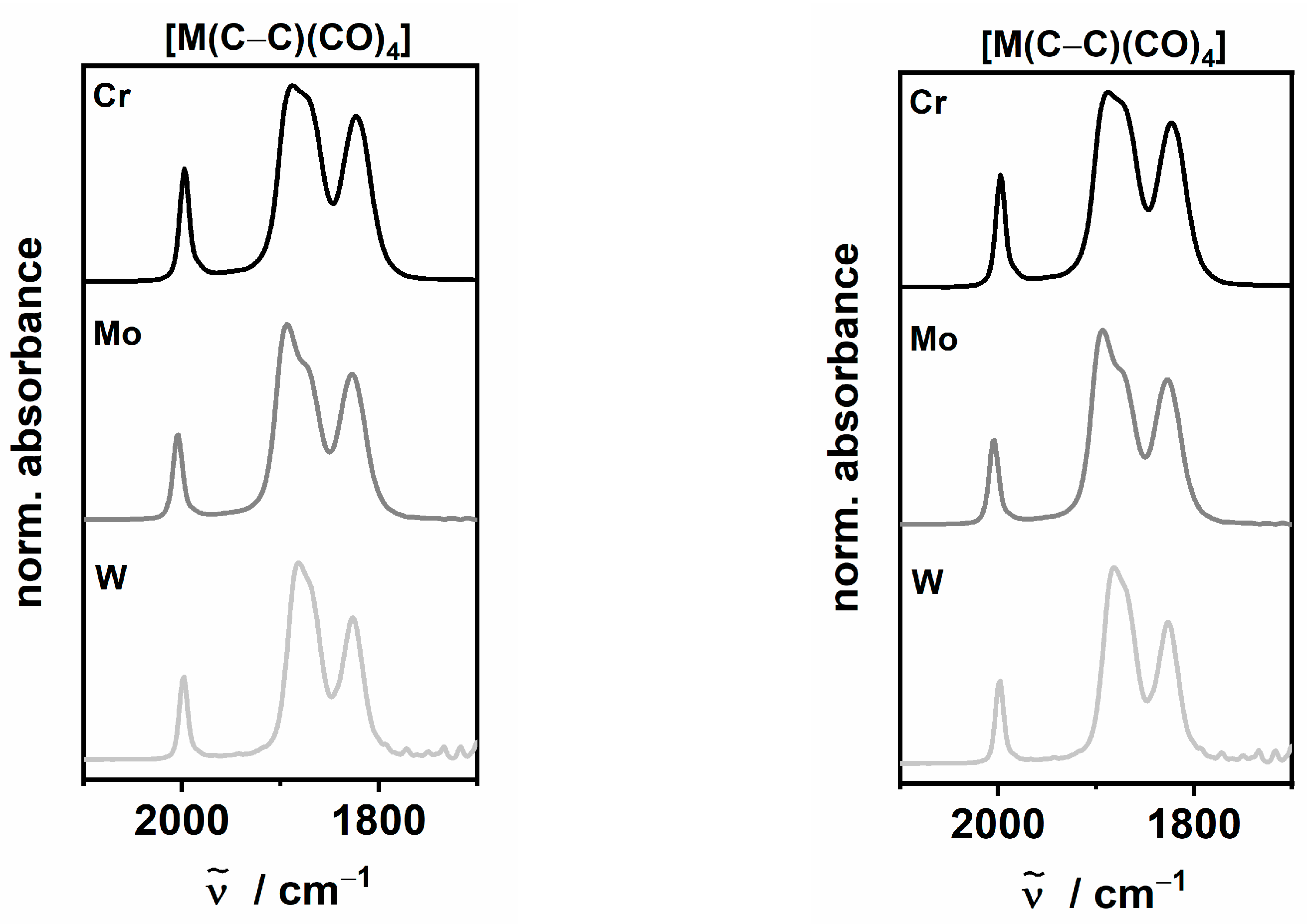
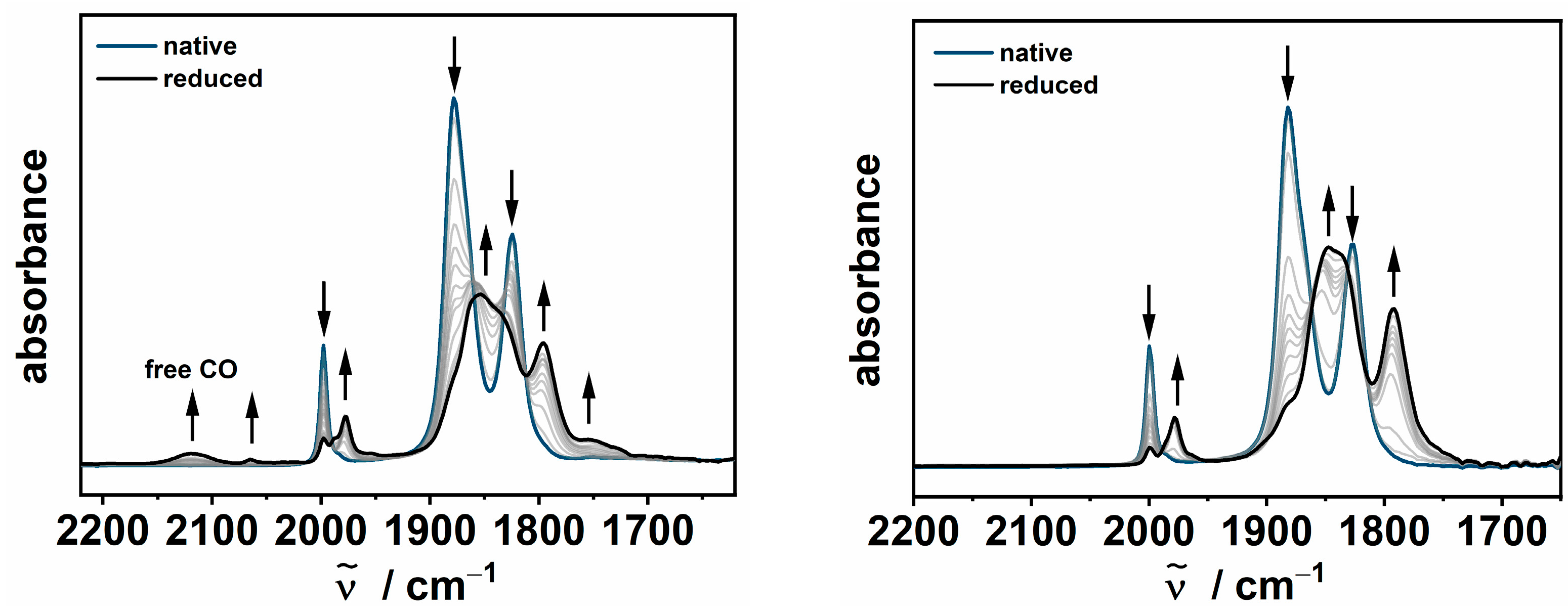
2.2. IR-Spectroelectrochemistry
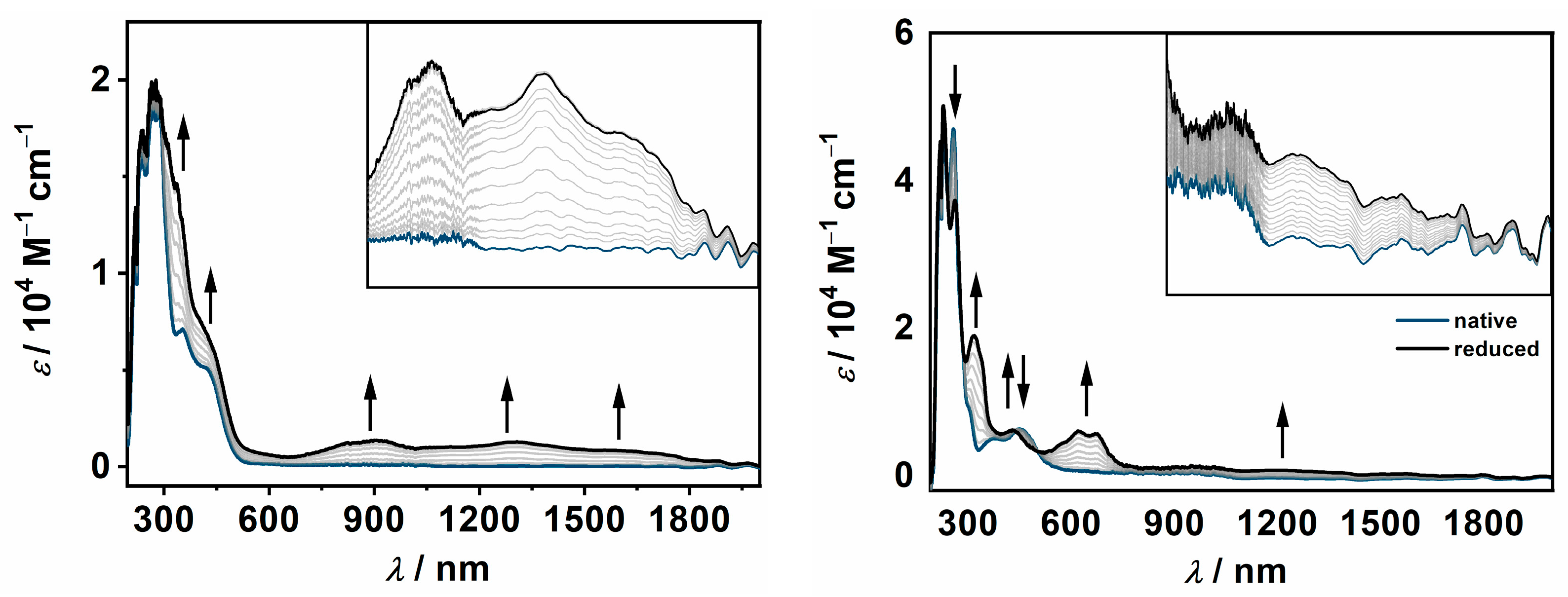
2.3. UV/vis/NIR-Spectroelectrochemistry
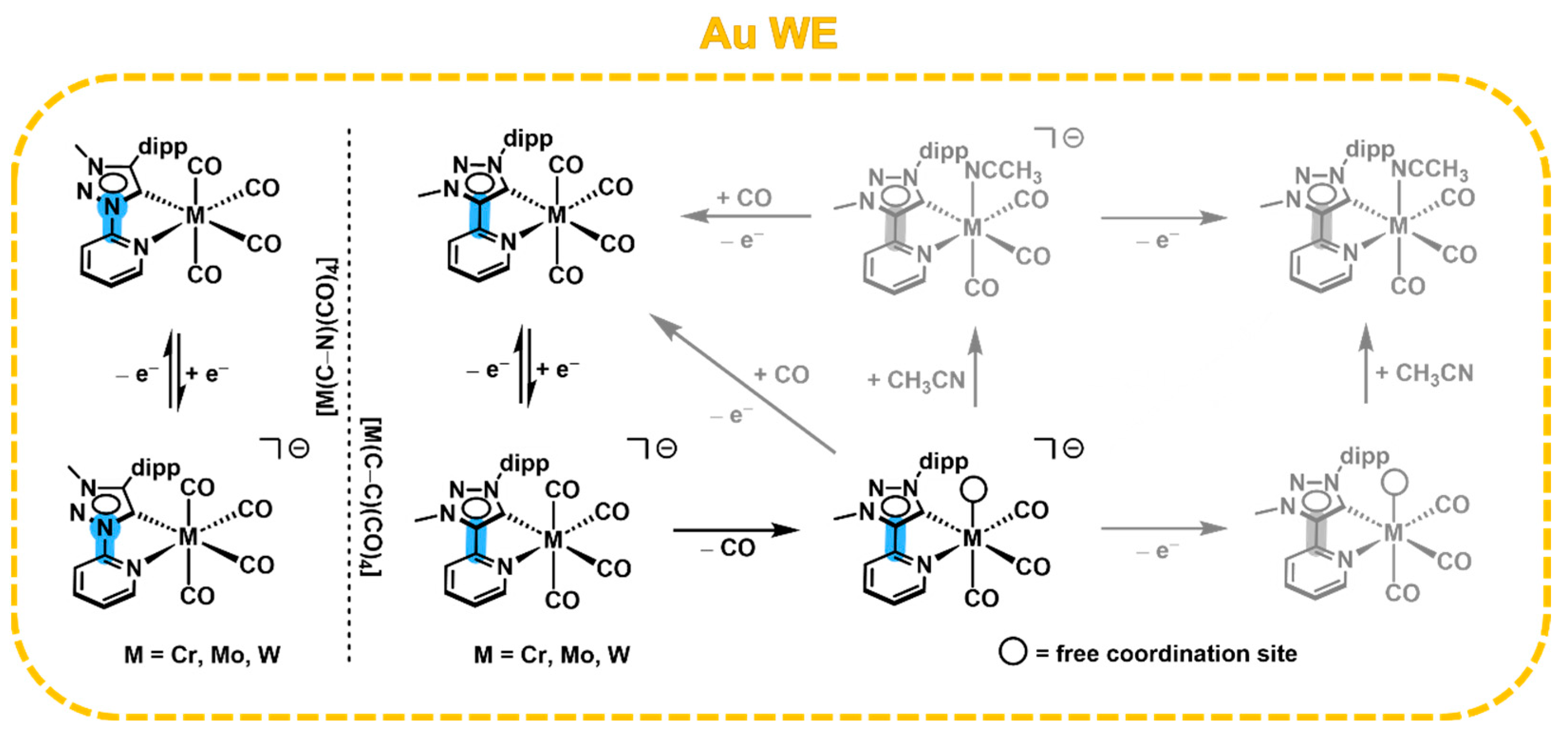
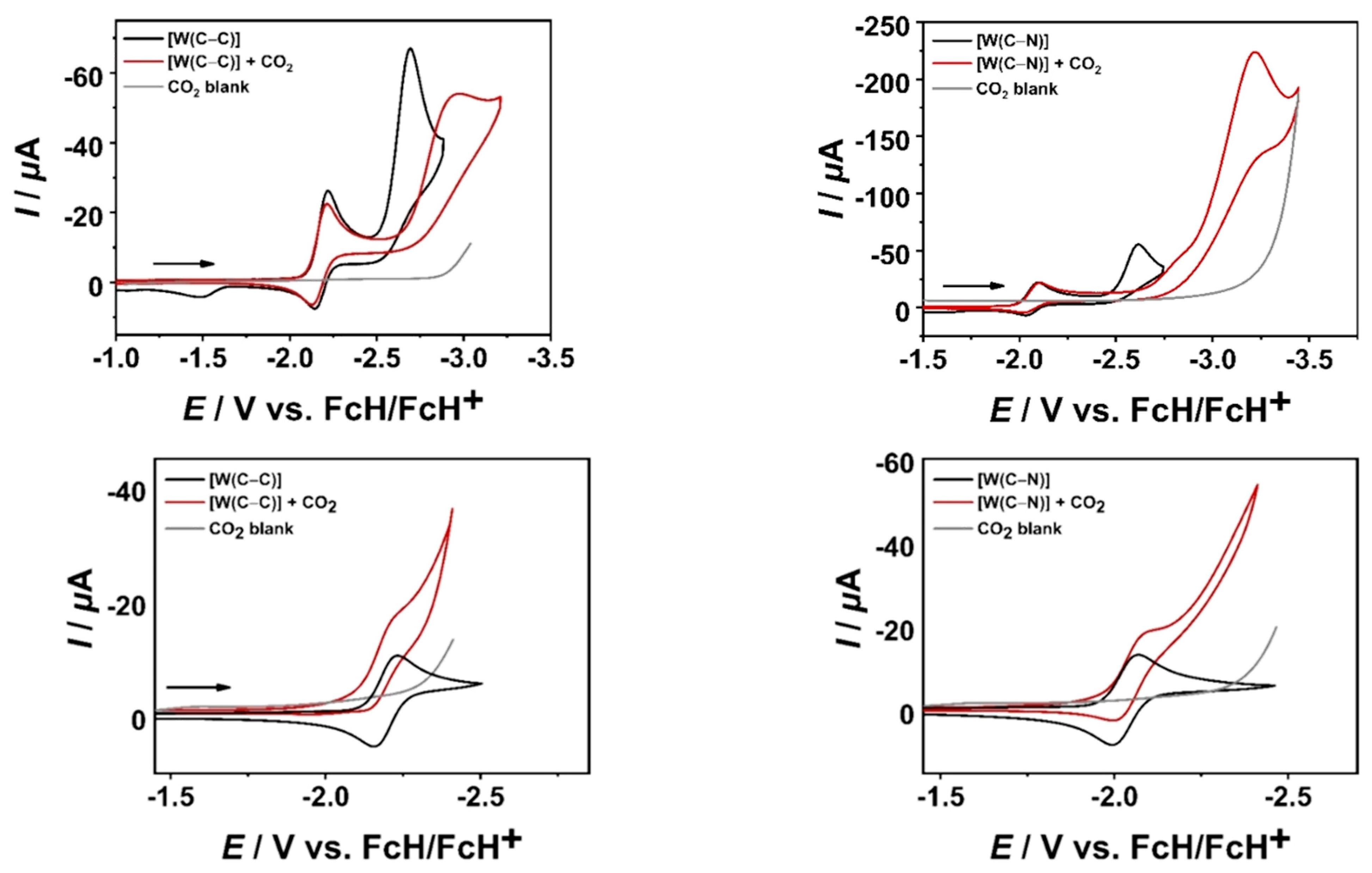
2.4. Cyclic Voltammetry with a Au WE and Electrochemical CO2 Reduction
3. Conclusions
4. Experimental Section
4.1. General Procedures, Materials, and Instrumentation
4.2. Electrochemistry
4.3. Spectroelectrochemistry
4.4. Calculations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Breugst, M.; Reissig, H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Maity, R.; Sarkar, B. Chemistry of Compounds Based on 1,2,3-Triazolylidene-Type Mesoionic Carbenes. JACS Au 2022, 2, 22–57. [Google Scholar] [CrossRef]
- Mathew, P.; Neels, A.; Albrecht, M. 1,2,3-Triazolylidenes as Versatile Abnormal Carbene Ligands for Late Transition Metals. J. Am. Chem. Soc. 2008, 130, 13534–13535. [Google Scholar] [CrossRef] [PubMed]
- Schweinfurth, D.; Hettmanczyk, L.; Suntrup, L.; Sarkar, B. Metal Complexes of Click-Derived Triazoles and Mesoionic Carbenes: Electron Transfer, Photochemistry, Magnetic Bistability, and Catalysis. Z. Anorg. Allg. Chem. 2017, 643, 554–584. [Google Scholar] [CrossRef]
- Crabtree, R.H. Abnormal, mesoionic and remote N-heterocyclic carbene complexes. Coord. Chem. Rev. 2013, 257, 755–766. [Google Scholar] [CrossRef]
- Donnelly, K.F.; Petronilho, A.; Albrecht, M. Application of 1,2,3-triazolylidenes as versatile NHC-type ligands: Synthesis, properties, and application in catalysis and beyond. Chem. Commun. 2013, 49, 1145–1159. [Google Scholar] [CrossRef]
- Bolje, A.; Hohloch, S.; Košmrlj, J.; Sarkar, B. RuII, IrIII and OsII mesoionic carbene complexes: Efficient catalysts for transfer hydrogenation of selected functionalities. Dalton Trans. 2016, 45, 15983–15993. [Google Scholar] [CrossRef] [PubMed]
- Bolje, A.; Košmrlj, J. A Selective Approach to Pyridine Appended 1,2,3-Triazolium Salts. Org. Lett. 2013, 15, 5084–5087. [Google Scholar] [CrossRef]
- Hohloch, S.; Suntrup, L.; Sarkar, B. Arene–Ruthenium(II) and −Iridium(III) Complexes with “Click”-Based Pyridyl-triazoles, Bis-triazoles, and Chelating Abnormal Carbenes: Applications in Catalytic Transfer Hydrogenation of Nitrobenzene. Organometallics 2013, 32, 7376–7385. [Google Scholar] [CrossRef]
- Kralj, J.; Bolje, A.; Polančec, D.S.; Steiner, I.; Gržan, T.; Tupek, A.; Stojanović, N.; Hohloch, S.; Urankar, D.; Osmak, M.; et al. Half-Sandwich Ir(III) and Os(II) Complexes of Pyridyl-Mesoionic Carbenes as Potential Anticancer Agents. Organometallics 2019, 38, 4082–4092. [Google Scholar] [CrossRef]
- Sabater, S.; Müller-Bunz, H.; Albrecht, M. Carboxylate-Functionalized Mesoionic Carbene Precursors: Decarboxylation, Ruthenium Bonding, and Catalytic Activity in Hydrogen Transfer Reactions. Organometallics 2016, 35, 2256–2266. [Google Scholar] [CrossRef]
- Saha, S.; Yadav, S.; Reshi, N.U.D.; Dutta, I.; Kunnikuruvan, S.; Bera, J.K. Electronic Asymmetry of an Annelated Pyridyl–Mesoionic Carbene Scaffold: Application in Pd(II)-Catalyzed Wacker-Type Oxidation of Olefins. ACS Catal. 2020, 10, 11385–11393. [Google Scholar] [CrossRef]
- Suntrup, L.; Stein, F.; Klein, J.; Wilting, A.; Parlane, F.G.L.; Brown, C.M.; Fiedler, J.; Berlinguette, C.P.; Siewert, I.; Sarkar, B. Rhenium Complexes of Pyridyl-Mesoionic Carbenes: Photochemical Properties and Electrocatalytic CO2 Reduction. Inorg. Chem. 2020, 59, 4215–4227. [Google Scholar] [CrossRef]
- Hansmann, M.M.; Antoni, P.W.; Pesch, H. Stable Mesoionic N-Heterocyclic Olefins (mNHOs). Angew. Chem. 2020, 132, 5831–5836. [Google Scholar] [CrossRef]
- Liang, Q.; Song, D. Recent advances of mesoionic N-heterocyclic olefins. Dalton Trans. 2022, 51, 9191–9198. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wu, Y.; Huang, L.; Hu, C.; Yan, X. Synthesis, Characterization and Photophysical Properties of Mesoionic N-Heterocyclic Imines. Chem. Asian J. 2022, 17, e202200281. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Neuman, N.I.; Walter, R.R.M.; Ringenberg, M.R.; Sarkar, B. Mesoionic Imines (MIIs): Strong Donors and Versatile Ligands for Transition Metals and Main Group Substrates. Angew. Chem. Int. Ed. Engl. 2022, 61, e202200653. [Google Scholar] [CrossRef] [PubMed]
- Suntrup, L.; Klenk, S.; Klein, J.; Sobottka, S.; Sarkar, B. Gauging Donor/Acceptor Properties and Redox Stability of Chelating Click-Derived Triazoles and Triazolylidenes: A Case Study with Rhenium(I) Complexes. Inorg. Chem. 2017, 56, 5771–5783. [Google Scholar] [CrossRef] [PubMed]
- Collin, J. Electrochemical Reduction of Carbon Dioxide Mediated by Molecular Catalysts. Coord. Chem. Rev. 1989, 93, 245–268. [Google Scholar] [CrossRef]
- Gonell, S.; Massey, M.D.; Moseley, I.P.; Schauer, C.K.; Muckerman, J.T.; Miller, A.J.M. The Trans Effect in Electrocatalytic CO2 Reduction: Mechanistic Studies of Asymmetric Ruthenium Pyridyl-Carbene Catalysts. J. Am. Chem. Soc. 2019, 141, 6658–6671. [Google Scholar] [CrossRef] [PubMed]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine). J. Chem. Soc., Chem. Commun. 1984, 6, 328–330. [Google Scholar] [CrossRef]
- Smieja, J.M.; Kubiak, C.P. Re(bipy-tBu)(CO)3Cl-improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies. Inorg. Chem. 2010, 49, 9283–9289. [Google Scholar] [CrossRef]
- Todorova, T.K.; Huan, T.N.; Wang, X.; Agarwala, H.; Fontecave, M. Controlling Hydrogen Evolution during Photoreduction of CO2 to Formic Acid Using Rh(R-bpy)(Cp*)Cl+ Catalysts: A Structure-Activity Study. Inorg. Chem. 2019, 58, 6893–6903. [Google Scholar] [CrossRef] [PubMed]
- Windle, C.D.; Perutz, R.N. Advances in molecular photocatalytic and electrocatalytic CO2 reduction. Coord. Chem. Rev. 2012, 256, 2562–2570. [Google Scholar] [CrossRef]
- Clark, M.L.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Electrocatalytic CO2 reduction by M(bpy-R)(CO)4 (M = Mo, W.; R = H, tBu) complexes. Electrochemical, spectroscopic, and computational studies and comparison with group 7 catalysts. Chem. Sci. 2014, 5, 1894–1900. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Sordello, F.; Minero, C.; Nencini, L.; Fiedler, J.; Gobetto, R.; Nervi, C. Electrochemical Reduction of CO2 by M(CO)4 (diimine) Complexes (M = Mo, W): Catalytic Activity Improved by 2,2′-Dipyridylamine. ChemElectroChem 2015, 2, 1372–1379. [Google Scholar] [CrossRef]
- Franco, F.; Pinto, M.F.; Royo, B.; Lloret-Fillol, J. A Highly Active N-Heterocyclic Carbene Manganese(I) Complex for Selective Electrocatalytic CO2 Reduction to CO. Angew. Chem. Int. Ed. 2018, 57, 4603–4606. [Google Scholar] [CrossRef]
- Friães, S.; Realista, S.; Gomes, C.S.B.; Martinho, P.N.; Royo, B. Click-Derived Triazoles and Triazolylidenes of Manganese for Electrocatalytic Reduction of CO2. Molecules 2021, 26, 6325. [Google Scholar] [CrossRef]
- Gonell, S.; Lloret-Fillol, J.; Miller, A.J.M. An Iron Pyridyl-Carbene Electrocatalyst for Low Overpotential CO2 Reduction to CO. ACS Catal. 2021, 11, 615–626. [Google Scholar] [CrossRef]
- Grice, K.A.; Saucedo, C. Electrocatalytic Reduction of CO2 by Group 6 M(CO)6 Species without “Non-Innocent” Ligands. Inorg. Chem. 2016, 55, 6240–6246. [Google Scholar] [CrossRef]
- Huang, C.; Liu, J.; Huang, H.-H.; Ke, Z. Recent progress in electro- and photo-catalytic CO2 reduction using N-heterocyclic carbene transition metal complexes. Polyhedron 2021, 203, 115147. [Google Scholar] [CrossRef]
- Machan, C.W.; Stanton, C.J.; Vandezande, J.E.; Majetich, G.F.; Schaefer, H.F.; Kubiak, C.P.; Agarwal, J. Electrocatalytic Reduction of Carbon Dioxide by Mn(CN)(2,2′-bipyridine)(CO)3: CN Coordination Alters Mechanism. Inorg. Chem. 2015, 54, 8849–8856. [Google Scholar] [CrossRef]
- Neri, G.; Donaldson, P.M.; Cowan, A.J. The Role of Electrode-Catalyst Interactions in Enabling Efficient CO2 Reduction with Mo(bpy)(CO)4 As Revealed by Vibrational Sum-Frequency Generation Spectroscopy. J. Am. Chem. Soc. 2017, 139, 13791–13797. [Google Scholar] [CrossRef] [PubMed]
- Sieh, D.; Lacy, D.C.; Peters, J.C.; Kubiak, C.P. Reduction of CO2 by Pyridine Monoimine Molybdenum Carbonyl Complexes: Cooperative Metal-Ligand Binding of CO2. Chem. Eur. J. 2015, 21, 8497–8503. [Google Scholar] [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Tory, J.; Setterfield-Price, B.; Dryfe, R.A.W.; Hartl, F. [M(CO)]4 (2,2′-bipyridine)] (M = Cr, Mo, W) Complexes as Efficient Catalysts for Electrochemical Reduction of CO2 at a Gold Electrode. ChemElectroChem 2015, 2, 213–217. [Google Scholar] [CrossRef]
- Reda, T.; Plugge, C.M.; Abram, N.J.; Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 10654–10658. [Google Scholar] [CrossRef] [PubMed]
- Bens, T.; Walter, R.R.M.; Beerhues, J.; Schmitt, M.; Krossing, I.; Sarkar, B. The Best of Both Worlds: Combining the Power of MICs and WCAs to generate Stable and Crystalline CrI-tetracarbonyl Complexes with π-Accepting Ligands. Chem. Eur. J. 2023, 29, e202301205. [Google Scholar] [CrossRef]
- Bens, T.; Boden, P.; Di Martino-Fumo, P.; Beerhues, J.; Albold, U.; Sobottka, S.; Neuman, N.I.; Gerhards, M.; Sarkar, B. Chromium(0) and Molydenum(0) Complexes with a Pyridyl-Mesoionic Carbene Ligand: Structural, (Spectro)electrochemical, Photochemical, and Theoretical Investigations. Inorg. Chem. 2020, 59, 15504–15513. [Google Scholar] [CrossRef]
- Boden, P.; Di Martino-Fumo, P.; Bens, T.; Steiger, S.; Albold, U.; Niedner-Schatteburg, G.; Gerhards, M.; Sarkar, B. NIR-Emissive Chromium(0), Molybdenum(0), and Tungsten(0) Complexes in the Solid State at Room Temperature. Chem. Eur. J. 2021, 27, 12959–12964. [Google Scholar] [CrossRef] [PubMed]
- Boden, P.J.; Di Martino-Fumo, P.; Bens, T.; Steiger, S.T.; Marhöfer, D.; Niedner-Schatteburg, G.; Sarkar, B. Mechanistic and Kinetic Investigations of ON/OFF (Photo)Switchable Binding of Carbon Monoxide by Chromium(0), Molybdenum(0) and Tungsten(0) Carbonyl Complexes with a Pyridyl-Mesoionic Carbene Ligand. Chem. Eur. J. 2022, 28, e202201038. [Google Scholar] [CrossRef] [PubMed]
- Bens, T.; Marhöfer, D.; Boden, P.; Steiger, S.T.; Suntrup, L.; Niedner-Schatteburg, G.; Sarkar, B. A Different Perspective on Tuning the Photophysical and Photochemical Properties: The Influence of Constitutional Isomers in Group 6 Carbonyl Complexes with Pyridyl-Mesoionic Carbenes. Inorg. Chem. 2023, 62, 16182–16195. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Cameron, L.; Poland, E.M.; Yu, L.-J.; Moggach, S.A.; Fuller, R.O.; Huang, H.; Sun, J.; Thickett, S.C.; Massi, M.; et al. Photoactive Metal Carbonyl Complexes Bearing N-Heterocyclic Carbene Ligands: Synthesis, Characterization, and Viability as Photoredox Catalysts. Inorg. Chem. 2022, 61, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Bens, T.; Walter, R.R.M.; Beerhues, J.; Lücke, C.; Gabler, J.; Sarkar, B. Isolation, Characterization and Reactivity of Key Intermediates Relevant to Reductive (Electro)catalysis with Cp*Rh Complexes Containing Pyridyl-MIC (MIC=Mesoionic Carbene) Ligands. Chem. Eur. J. 2024, 30, e202302354. [Google Scholar] [CrossRef] [PubMed]
- Bohnenberger, J.; Schmitt, M.; Feuerstein, W.; Krummenacher, I.; Butschke, B.; Czajka, J.; Malinowski, P.J.; Breher, F.; Krossing, I. Completing the triad: Synthesis and full characterization of homoleptic and heteroleptic carbonyl and nitrosyl complexes of the group VI metals. Chem. Sci. 2020, 11, 3592–3603. [Google Scholar] [CrossRef] [PubMed]
- Vlček, A., Jr. Highlights of the spectroscopy, photochemistry and electrochemistry of [M(CO)4(α-diimine)] complexes, M=Cr, Mo, W. Coord. Chem. Rev. 2002, 230, 225–242. [Google Scholar] [CrossRef]
- Huheey, J.E.; Keiter, E.A.; Keiter, R.L. Huheey—Anorganische Chemie; Walter de Gruyter: Berlin, Germany; Boston, MA, USA, 2008; ISBN 978-3-11-030433-6. [Google Scholar]
- Kaim, W.; Fiedler, J. Spectroelectrochemistry: The best of two worlds. Chem. Soc. Rev. 2009, 38, 3373–3382. [Google Scholar] [CrossRef]
- Crawley, M.R.; Kadassery, K.J.; Oldacre, A.N.; Friedman, A.E.; Lacy, D.C.; Cook, T.R. Rhenium(I) Phosphazane Complexes for Electrocatalytic CO2 Reduction. Organometallics 2019, 38, 1664–1676. [Google Scholar] [CrossRef]
- Knebel, W.J.; Angelici, R.J. Kinetic and Equilibrium Studies of Bi- and Tridentate Chelate Ring-Opening Reactions of Metal Carbonyl Complexes. Inorg. Chem. 1974, 13, 632–637. [Google Scholar] [CrossRef]
- Knebel, W.J.; Angelici, R.J. Mechanism of Chelate Ring-Opening in Metal Carbonyl Complexes. Inorg. Chem. 1974, 13, 627–631. [Google Scholar] [CrossRef]
- Lee, C.-C.; Ke, W.-C.; Chan, K.-T.; Lai, C.-L.; Hu, C.-H.; Lee, H.M. Nickel(II) Complexes of Bidentate N-Heterocyclic Carbene/Phosphine Ligands: Efficient Catalysts for Suzuki Coupling of Aryl Chlorides. Chem. Eur. J. 2007, 13, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; McCarthy, B.D.; Dempsey, J.L. On decomposition, degradation, and voltammetric deviation: The electrochemist’s field guide to identifying precatalyst transformation. Chem. Soc. Rev. 2019, 48, 2927–2945. [Google Scholar] [CrossRef] [PubMed]
- Budavari, S. (Ed.) The Merck-Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 11th ed.; 3. print; Centennial ed.; Merck & Co., Inc.: Rahway, NJ, USA, 1991; ISBN 091191028X. [Google Scholar]
- Krejčik, M.; Daněk, M.; Hartl, F. Simple construction of an infrared optically transparent thin-layer electrochemical cell: Applications to the redox reactions of ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-di-t-butyl-catecholat)−. J. Electroanal. Chem. Interfacial 1991, 317, 179. [Google Scholar] [CrossRef]
- Klein, J.; Stuckmann, A.; Sobottka, S.; Suntrup, L.; van der Meer, M.; Hommes, P.; Reissig, H.-U.; Sarkar, B. Ruthenium Complexes with Strongly Electron-Donating Terpyridine Ligands: Effect of the Working Electrode on Electrochemical and Spectroelectrochemical Properties. Chem. Eur. J. 2017, 23, 12314–12325. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phy. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phy. 1998, 109, 392–399. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phy. 2010, 132, 154104-1–154104-19. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phy. 2011, 135, 144105-1–144105-11. [Google Scholar] [CrossRef]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef]
- Neese, F.; Olbrich, G. Efficient use of the resolution of the identity approximation in time-dependent density functional calculations with hybrid density functionals. Chem. Phy. Lett. 2002, 362, 170–178. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Petrenko, T.; Kossmann, S.; Neese, F. Efficient time-dependent density functional theory approximations for hybrid density functionals: Analytical gradients and parallelization. J. Chem. Phy. 2011, 134, 054116-1–054116-14. [Google Scholar] [CrossRef] [PubMed]
- Vahtras, O.; Almlöf, J.; Feyereisen, M.W. Integral approximations for LCAO-SCF calculations. Chem. Phy. Lett. 1993, 213, 514–518. [Google Scholar] [CrossRef]
- Whitten, J.L. Coulombic potential energy integrals and approximations. J. Chem. Phy. 1973, 58, 4496–4501. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb. Chem. Phy. Lett. 1995, 242, 652–660. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phy. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Zhurko, G.A. Chemcraft-Graphical Program for Visualization of Quantum Chemistry Computations; Version 1.8; Chemcraft: Ivanovo, Russia, 2023. [Google Scholar]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. Red./V ΔEp | 2. Red./V | 1. Ox./V ΔEp | |||
|---|---|---|---|---|---|
| [Cr(C–C)(CO)4] [41] | 2.26 | 0.07 | 2.80 | 0.21 | 0.07 |
| [Cr(C–N)(CO)4] [42] | 2.16 | 0.07 | 2.79 | 0.17 | 0.07 |
| [Mo(C–C)(CO)4] | 2.21 | 0.07 | 2.70 | 0.07 a | |
| [Mo(C–N)(CO)4] [42] | 2.10 | 0.08 | 2.68 | 0.08 a | |
| [W(C–C)(CO)4] | 2.19 | 0.08 | 2.69 | 0.07 a | |
| [W(C–N)(CO)4] | 2.05 | 0.06 | 2.65 | 0.12 a | |
| [Cr(C–C)(CO)4]− | [Mo(C–C)(CO)4]− | [W(C–C)(CO)4]− | [W(C–N)(CO)4]− | |
|---|---|---|---|---|
| g | 2.0030 | 2.0033 | 2.0028 | 2.0032 |
| AM | 5.90 | 1.97 | 13.10 | 2.65 |
| AN1 | 17.90 | 10.37 | 10.39 | 16.92 |
| AN2 | 17.80 | 4.95 | 5.60 | 14.22 |
| AN3 | 11.00 | 16.10 | 5.60 | 6.47 |
| AN4 | 9.60 | 9.60 | 4.53 | 5.82 |
| AH1 | 11.70 | 12.00 | 4.16 | 16.20 |
| AH2 | 9.50 | 11.90 | 20.27 | 13.77 |
| AH3 | 3.00 | 10.50 | 20.27 | 13.22 |
| AH4 | 2.00 | 14.45 | 15.36 | 7.19 |
| AH5 | - | - | 1.01 | 3.19 |
| AH6 | - | - | 1.01 | 3.19 |
| AH7 | - | - | 0.63 | - |
| AH8 | - | - | 1.81 | - |
| AH9 | - | - | 0.49 | - |
| AH10 | - | - | 0.50 | - |
| lwpp a/mT | [0 0.123] | [0 0.121] | [0 0.054] | [0 0.161] |
| [Cr(C–C)(CO)4] [41] | 1998 | 1890 | 1875 (sh) | 1822 | 1896 |
| [Cr(C–N)(CO)4] [42] | 1998 | 1890 | 1878 (sh) | 1830 | 1899 |
| [Mo(C–C)(CO)4] | 2004 | 1894 | 1876 (sh) | 1827 | 1900 |
| [Mo(C–N)(CO)4] [42] | 2006 | 1896 | 1876 (sh) | 1830 | 1902 |
| [W(C–C)(CO)4] | 1998 | 1882 | 1870 (sh) | 1826 | 1894 |
| [W(C–N)(CO)4] | 2000 | 1884 | 1873 (sh) | 1830 | 1897 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bens, T.; Sarkar, B. Investigations of the Influence of Two Pyridyl-Mesoionic Carbene Constitutional Isomers on the Electrochemical and Spectroelectrochemical Properties of Group 6 Metal Carbonyl Complexes. Inorganics 2024, 12, 46. https://doi.org/10.3390/inorganics12020046
Bens T, Sarkar B. Investigations of the Influence of Two Pyridyl-Mesoionic Carbene Constitutional Isomers on the Electrochemical and Spectroelectrochemical Properties of Group 6 Metal Carbonyl Complexes. Inorganics. 2024; 12(2):46. https://doi.org/10.3390/inorganics12020046
Chicago/Turabian StyleBens, Tobias, and Biprajit Sarkar. 2024. "Investigations of the Influence of Two Pyridyl-Mesoionic Carbene Constitutional Isomers on the Electrochemical and Spectroelectrochemical Properties of Group 6 Metal Carbonyl Complexes" Inorganics 12, no. 2: 46. https://doi.org/10.3390/inorganics12020046
APA StyleBens, T., & Sarkar, B. (2024). Investigations of the Influence of Two Pyridyl-Mesoionic Carbene Constitutional Isomers on the Electrochemical and Spectroelectrochemical Properties of Group 6 Metal Carbonyl Complexes. Inorganics, 12(2), 46. https://doi.org/10.3390/inorganics12020046