
Theoretical Investigation of Inorganic Particulate Matter: The Case of Water Adsorption on a NaCl Particle Model Studied Using Grand Canonical Monte Carlo Simulations

 , , ,
, , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
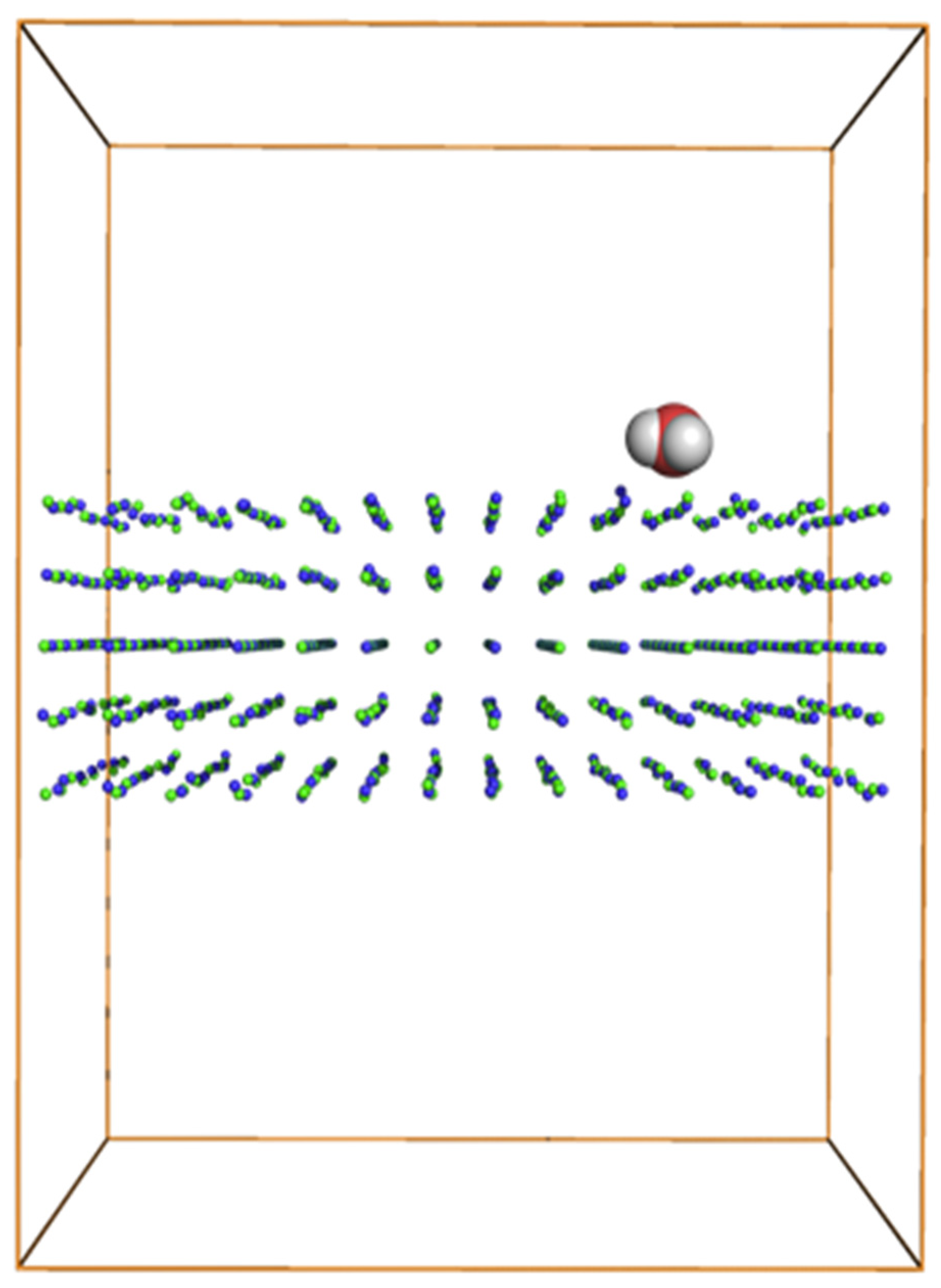
2. Results and Discussion
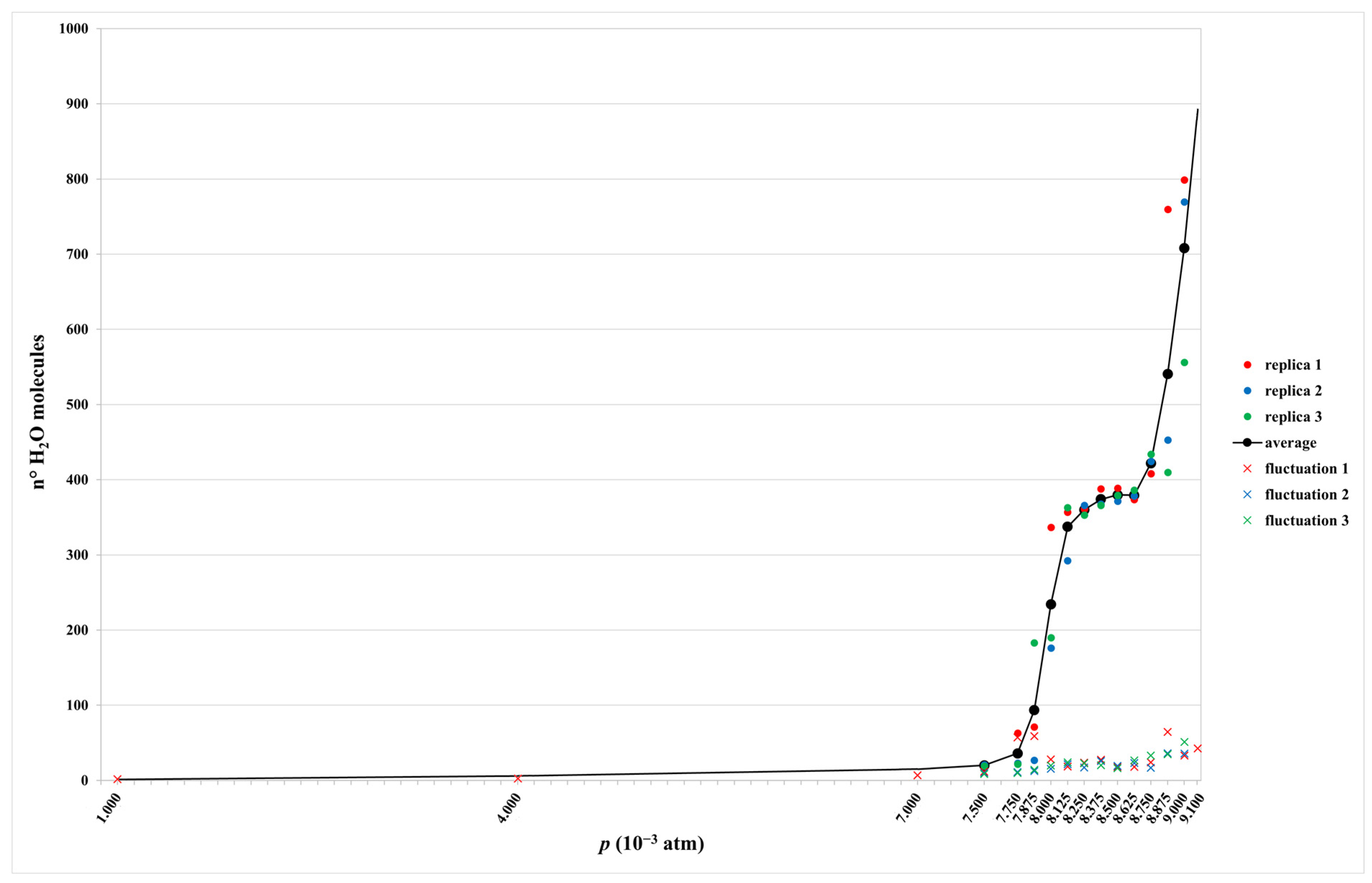
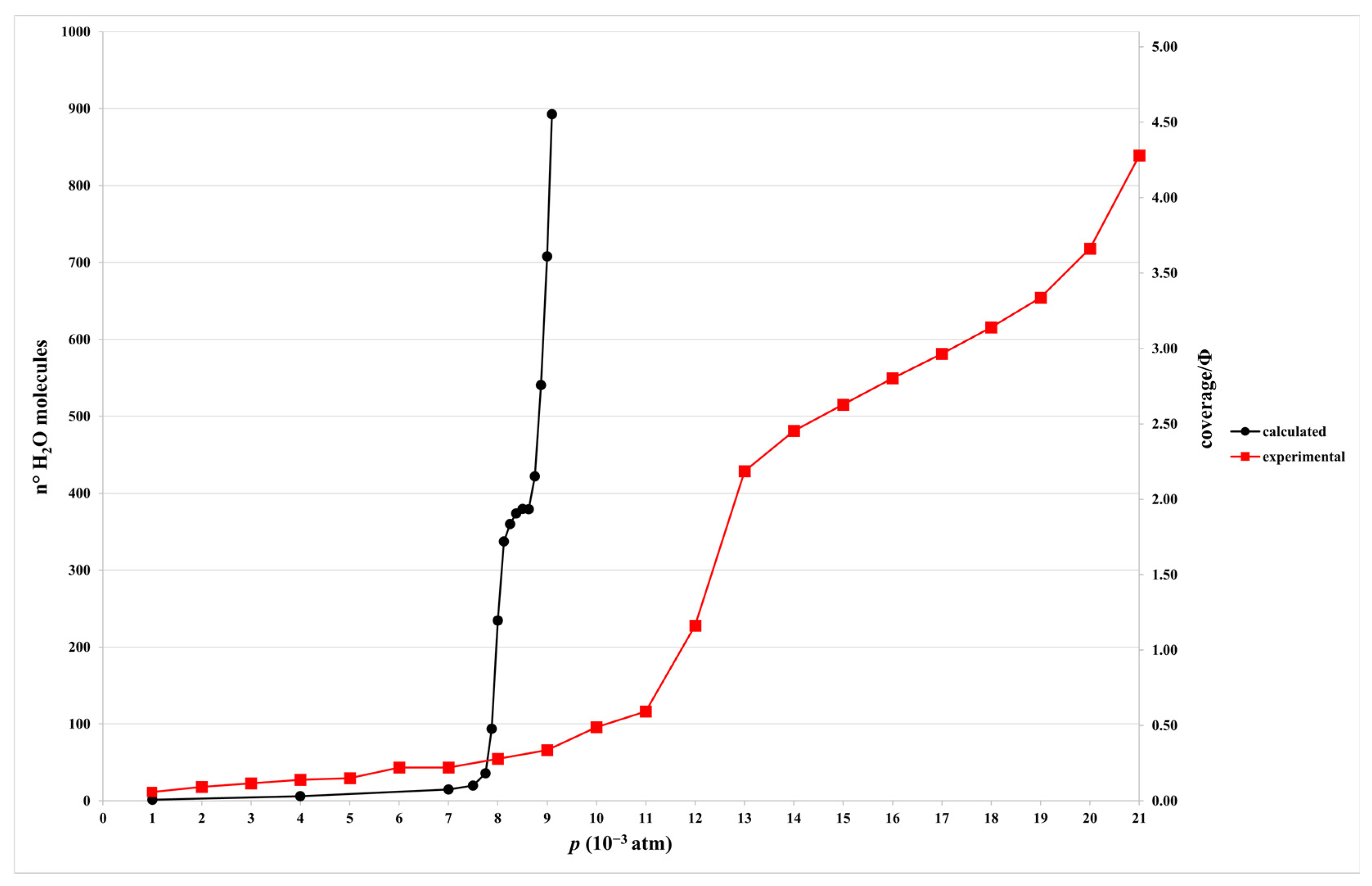
2.1. Isotherm
2.2. Identification of Water Aggregate Structures
- The mean number (and standard error) of islands identified in each simulation, together with the mean number (and standard error) of water molecules contained in each island.
- The mean number (and standard error) of fully covered surfaces identified in each simulation, and the average number of water molecules on each surface. For example, a value equal to 1 will indicate that, on average, only one of the two NaCl surfaces is completely covered.
- The standard error of the mean () is obtained as the ratio between the standard deviation associated with the set of values and the square root of the number of averaged values: .
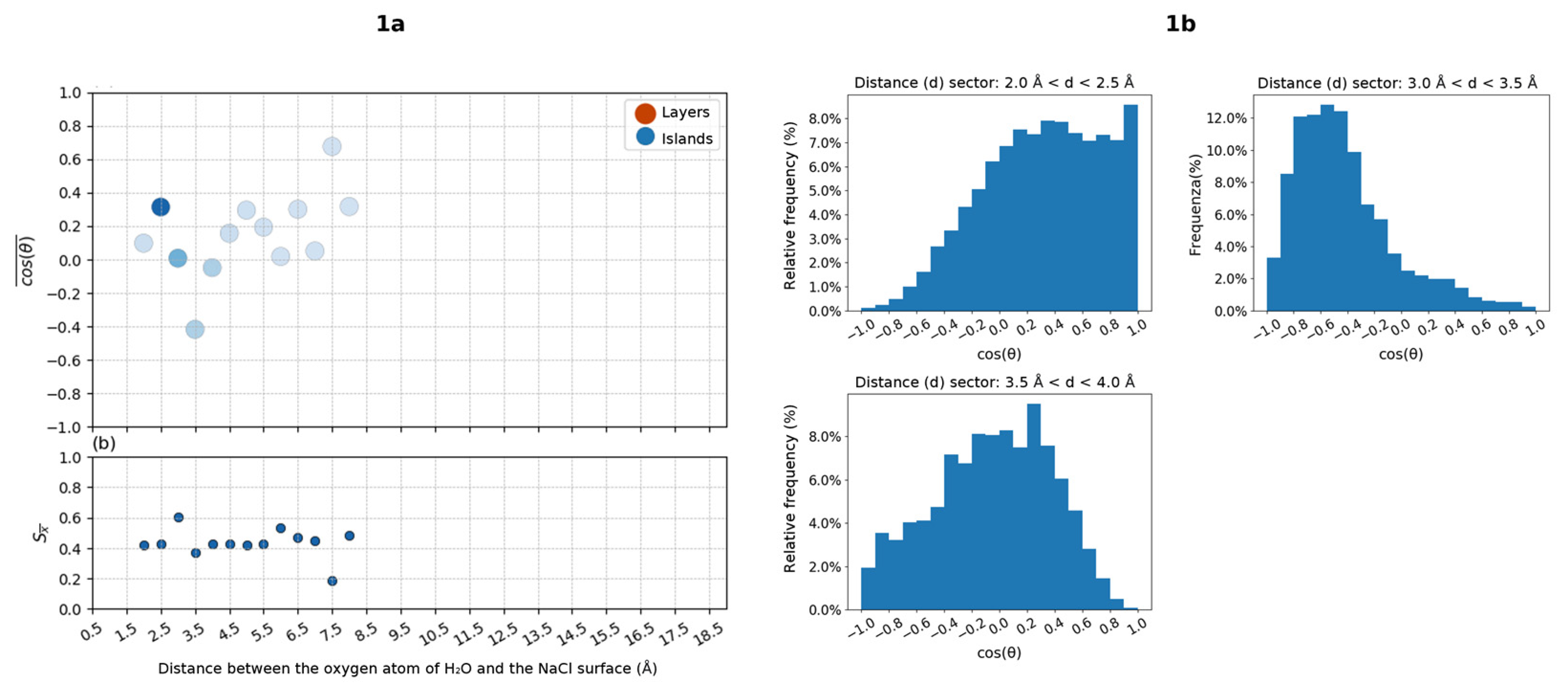
2.3. Orientation of Water Molecules as a Function of Distance from the NaCl Surface
- A plot of the mean cos(θ) value of the water molecules as a function of the distance from the surface. In the plot, cos(θ) values of water molecules belonging to islands or layers are differently colored. The intensity of the color associated with each point of the graph is proportional to the number of averaged values. Each point of the graph, and thus each average value of cos(θ), has been obtained as the average of the orientations of all the water molecules that—during the analysis of the frames—have been classified as belonging to a certain “distance sector” from the NaCl surface. Each “distance sector” includes water molecules that have a distance value from the surface between two extremes separated by a step of 0.5 Å. Each average value will therefore be associated with a distance value from the surface, on the abscissa axis of the graph, which will indicate the upper limit of the “distance sector”. For example, a value of cos(θ), associated with a distance of 0.5 Å from the surface, is the average of the orientations of all water molecules characterized by a distance from the surface between 0 and 0.5 Å.
- For each “distance sector”, a histogram is generated, which represents the distribution of orientations in the entire simulation. The histogram is obtained by dividing the range of cos(θ) values, which go from −1 to +1, into intervals of 0.2 units. The intervals (on the x axis) define the basis of adjacent rectangles, whose height is the percentage frequency associated with the number of orientations identified in the specific interval. The frequencies shown are therefore relative frequencies, obtained from the ratio between the number of water molecules for which an orientation consistent with a certain interval has been determined, and the total number of water molecules belonging to the “distance sector”. The histogram is then normalized, and the sum of the heights of all the rectangles gives a percentage of 100%.
3. Methods
3.1. Grand Canonical Monte Carlo Simulations
3.2. Cluster Analysis
3.3. Analysis of the Influence of NaCl Surface on Orientation of Adsorbed H2O Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Putaud, J.-P.; Van Dingenen, R.; Alastuey, A.; Bauer, H.; Birmili, W.; Cyrys, J.; Flentje, H.; Fuzzi, S.; Gehrig, R.; Hansson, H.C.; et al. A European Aerosol Phenomenology–3: Physical and Chemical Characteristics of Particulate Matter from 60 Rural, Urban, and Kerbside Sites across Europe. Atmos. Environ. 2010, 44, 1308–1320. [Google Scholar] [CrossRef]
- Zhang, B. The Effect of Aerosols to Climate Change and Society. J. Geosci. Environ. Prot. 2020, 8, 55–78. [Google Scholar] [CrossRef]
- Andreae, M.O.; Jones, C.D.; Cox, P.M. Strong Present-Day Aerosol Cooling Implies a Hot Future. Nature 2005, 435, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Watson-Parris, D.; Smith, C.J. Large Uncertainty in Future Warming Due to Aerosol Forcing. Nat. Clim. Chang. 2022, 12, 1111–1113. [Google Scholar] [CrossRef]
- Ramanathan, V.C.; Crutzen, P.J.; Kiehl, J.T.; Rosenfeld, D. Aerosols, Climate, and the Hydrological Cycle. Science 2001, 294, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Boucher, O. Atmospheric Aerosols: Properties and Climate Impacts; Springer: Berlin/Heidelberg, Germany, 2015; ISBN 9789401796491. [Google Scholar]
- Charlson, R.J.; Langner, J.; Rodhe, H.; Leovy, C.B.; Warren, S.G. Perturbation of the Northern Hemisphere Radiative Balance by Backscattering from Anthropogenic Sulfate Aerosols. Tellus A Dyn. 1991, 43, 152. [Google Scholar]
- Andrews, E.; Ogren, J.A.; Bonasoni, P.; Marinoni, A.; Cuevas, E.; Rodríguez, S.; Sun, J.Y.; Jaffe, D.A.; Fischer, E.V.; Baltensperger, U.; et al. Climatology of Aerosol Radiative Properties in the Free Troposphere. Atmos. Res. 2011, 102, 365–393. [Google Scholar] [CrossRef]
- Stevens, B.; Feingold, G. Untangling Aerosol Effects on Clouds and Precipitation in a Buffered System. Nature 2009, 461, 607–613. [Google Scholar] [CrossRef]
- Carslaw, K.S.; Lee, L.A.; Reddington, C.L.; Pringle, K.J.; Rap, A.; Forster, P.M.; Mann, G.W.; Spracklen, D.V.; Woodhouse, M.T.; Regayre, L.A.; et al. Large Contribution of Natural Aerosols to Uncertainty in Indirect Forcing. Nature 2013, 503, 67–71. [Google Scholar] [CrossRef]
- Lohmann, U.; Feichter, J. Global Indirect Aerosol Effects: A Review. Atmos. Chem. Phys. 2005, 5, 715–737. [Google Scholar] [CrossRef]
- Davidson, C.I.; Phalen, R.F.; Solomon, P.A. Airborne Particulate Matter and Human Health: A Review. Aerosol Sci. Technol. 2007, 39, 737–749. [Google Scholar] [CrossRef]
- Lighty, J.S.; Veranth, J.M.; Sarofim, A.F. Combustion Aerosols: Factors Governing Their Size and Composition and Implications to Human Health. J. Air Waste Manag. Assoc. 2011, 50, 1565–1618. [Google Scholar] [CrossRef] [PubMed]
- Arden Pope, C., III; Burnett, R.T.; Thurston, G.D.; Thun, M.J.; Calle, E.E.; Krewski, D.; Godleski, J.J. Cardiovascular Mortality and Long-Term Exposure to Particulate Air Pollution. Circulation 2004, 109, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.A.; Neil Alexis, C.B.; Leonard Bernstein, I.; Nel, A.; Peden, D.; Diaz-Sanchez, D.; Tarlo, S.M.; Brock Williams, P.; Bernstein, J.A. Health Effects of Air Pollution. J. Allergy Clin. Immunol. 2004, 114, 1116–1123. [Google Scholar] [CrossRef]
- Tang, M.; Chan, C.K.; Li, Y.J.; Su, H.; Ma, Q.; Wu, Z.; Zhang, G.; Wang, Z.; Ge, M.; Hu, M.; et al. A Review of Experimental Techniques for Aerosol Hygroscopicity Studies. Atmos. Chem. Phys. 2019, 19, 12631–12686. [Google Scholar] [CrossRef]
- Tang, M.; Cziczo, D.J.; Grassian, V.H. Interactions of Water with Mineral Dust Aerosol: Water Adsorption, Hygroscopicity, Cloud Condensation, and Ice Nucleation. Chem. Rev. 2016, 116, 4205–4259. [Google Scholar] [CrossRef]
- Valsaraj, K.T. A Review of the Aqueous Aerosol Surface Chemistry in the Atmospheric Context. Open J. Phys. Chem. 2012, 2, 58–66. [Google Scholar] [CrossRef]
- Sandu, I.; Chirazi, M.; Canache, M.; Sandu, I.G.; Alexianu, M.T.; Sandu, A.V.; Vasilache, V. Research on Nacl Saline Aerosols I. Natural and Artificial Sources and Their Implications. J. Environ. Eng. Landsc. Manag. 2010, 9, 881–888. [Google Scholar] [CrossRef]
- Sandu, I.; Chirazi, M.; Sandu, I.G. Research on Nacl Saline Aerosols II. New Artificial Halochamber Characteristics. Environmentalist 2010, 9, 1105–1113. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Yang, X.; Li, W.; Xue, L.; Wang, T.; Chen, J.; Wang, W. Others Mixed Chloride Aerosols and Their Atmospheric Implications: A Review. Aerosol Air Qual. Res. 2017, 17, 878–887. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J. The Tropospheric Chemistry of Sea Salt: A Molecular-Level View of the Chemistry of NaCl and NaBr. Chem. Rev. 2003, 103, 4801–4822. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.W.; Li, W.J.; Zhang, D.Z.; Zhang, J.C.; Lin, Y.T.; Shen, X.J.; Sun, J.Y.; Chen, J.M.; Zhang, X.Y.; Zhang, Y.M.; et al. Sea Salt Aerosols as a Reactive Surface for Inorganic and Organic Acidic Gases in the Arctic Troposphere. Atmos. Chem. Phys. 2015, 15, 11341–11353. [Google Scholar] [CrossRef]
- David, D.; Weis, G.E.E. Water Content and Morphology of Sodium Chloride Aerosol Particles. J. Geophys. Res. 1999, 104, 21275–21285. [Google Scholar]
- Davies, J.A.; Cox, R.A. Kinetics of the Heterogeneous Reaction of HNO3 with NaCl: Effect of Water Vapor. J. Phys. Chem. A 1998, 102, 7631–7642. [Google Scholar] [CrossRef]
- Allen, H.C.; Laux, J.M.; Vogt, R.; Finlayson-Pitts, B.J.; Hemminger, J.C. Water-Induced Reorganization of Ultrathin Nitrate Films on NaCl: Implications for the Tropospheric Chemistry of Sea Salt Particles. J. Phys. Chem. 1996, 100, 6371–6375. [Google Scholar] [CrossRef]
- Barraclough, P.B.; Hall, P.G. The Adsorption of Water Vapour by Lithium Fluoride, Sodium Fluoride and Sodium Chloride. Surf. Sci. 1974, 46, 393–417. [Google Scholar] [CrossRef]
- Peters, S.J.; Ewing, G.E. Thin Film Water on NaCl(100) under Ambient Conditions: An Infrared Study. Langmuir 1997, 13, 6345–6348. [Google Scholar] [CrossRef]
- Foster, M.C.; Ewing, G.E. Adsorption of Water on the NaCl(001) Surface. II. An Infrared Study at Ambient Temperatures. J. Chem. Phys. 2000, 112, 6817–6826. [Google Scholar] [CrossRef]
- Engkvist, O.; Stone, A.J. Adsorption of Water on NaCl(001). I. Intermolecular Potentials and Low Temperature Structures. J. Chem. Phys. 1999, 110, 12089–12096. [Google Scholar] [CrossRef]
- Engkvist, O.; Stone, A.J. Adsorption of Water on the NaCl(001) Surface. III. Monte Carlo Simulations at Ambient Temperatures. J. Chem. Phys. 2000, 112, 6827–6833. [Google Scholar] [CrossRef]
- Verdaguer, A.; Sacha, G.M.; Luna, M.; Ogletree, D.F.; Salmeron, M. Initial Stages of Water Adsorption on NaCl (100) Studied by Scanning Polarization Force Microscopy. J. Chem. Phys. 2005, 123, 124703. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Meng, S.; Wang, E.G. Water Adsorption on a NaCl (001) Surface: A Density Functional Theory Study. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 74, 245409. [Google Scholar] [CrossRef]
- Guo, J.; Meng, X.; Chen, J.; Peng, J.; Sheng, J.; Li, X.-Z.; Xu, L.; Shi, J.-R.; Wang, E.; Jiang, Y. Real-Space Imaging of Interfacial Water with Submolecular Resolution. Nat. Mater. 2014, 13, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Vlasov, V.P.; Muslimov, A.E.; Kanevsky, V.M. Water Adsorption on the (001) Surface of NaCl. J. Surf. Investig. X-ray Synchrotron Neutron Tech. 2020, 14, 1040–1043. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation; Elsevier Science & Technology: Amsterdam, The Netherlands, 2002; ISBN 9786611019747. [Google Scholar]
- Purton, J.A.; Crabtree, J.C.; Parker, S.C. DL_MONTE: A General Purpose Program for Parallel Monte Carlo Simulation. Mol. Simul. 2013, 39, 1240–1252. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., 3rd. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Schubert, E.; Sander, J.; Ester, M.; Kriegel, H.P.; Xu, X. DBSCAN Revisited, Revisited. ACM Trans. Database Syst. 2017, 42, 1–21. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizza, F.; Rovaletti, A.; Carbone, G.; Miyake, T.; Greco, C.; Cosentino, U. Theoretical Investigation of Inorganic Particulate Matter: The Case of Water Adsorption on a NaCl Particle Model Studied Using Grand Canonical Monte Carlo Simulations. Inorganics 2023, 11, 421. https://doi.org/10.3390/inorganics11110421
Rizza F, Rovaletti A, Carbone G, Miyake T, Greco C, Cosentino U. Theoretical Investigation of Inorganic Particulate Matter: The Case of Water Adsorption on a NaCl Particle Model Studied Using Grand Canonical Monte Carlo Simulations. Inorganics. 2023; 11(11):421. https://doi.org/10.3390/inorganics11110421
Chicago/Turabian StyleRizza, Fabio, Anna Rovaletti, Giorgio Carbone, Toshiko Miyake, Claudio Greco, and Ugo Cosentino. 2023. "Theoretical Investigation of Inorganic Particulate Matter: The Case of Water Adsorption on a NaCl Particle Model Studied Using Grand Canonical Monte Carlo Simulations" Inorganics 11, no. 11: 421. https://doi.org/10.3390/inorganics11110421
APA StyleRizza, F., Rovaletti, A., Carbone, G., Miyake, T., Greco, C., & Cosentino, U. (2023). Theoretical Investigation of Inorganic Particulate Matter: The Case of Water Adsorption on a NaCl Particle Model Studied Using Grand Canonical Monte Carlo Simulations. Inorganics, 11(11), 421. https://doi.org/10.3390/inorganics11110421