Attosecond Transient Absorption Below the Excited States

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theoretical Model
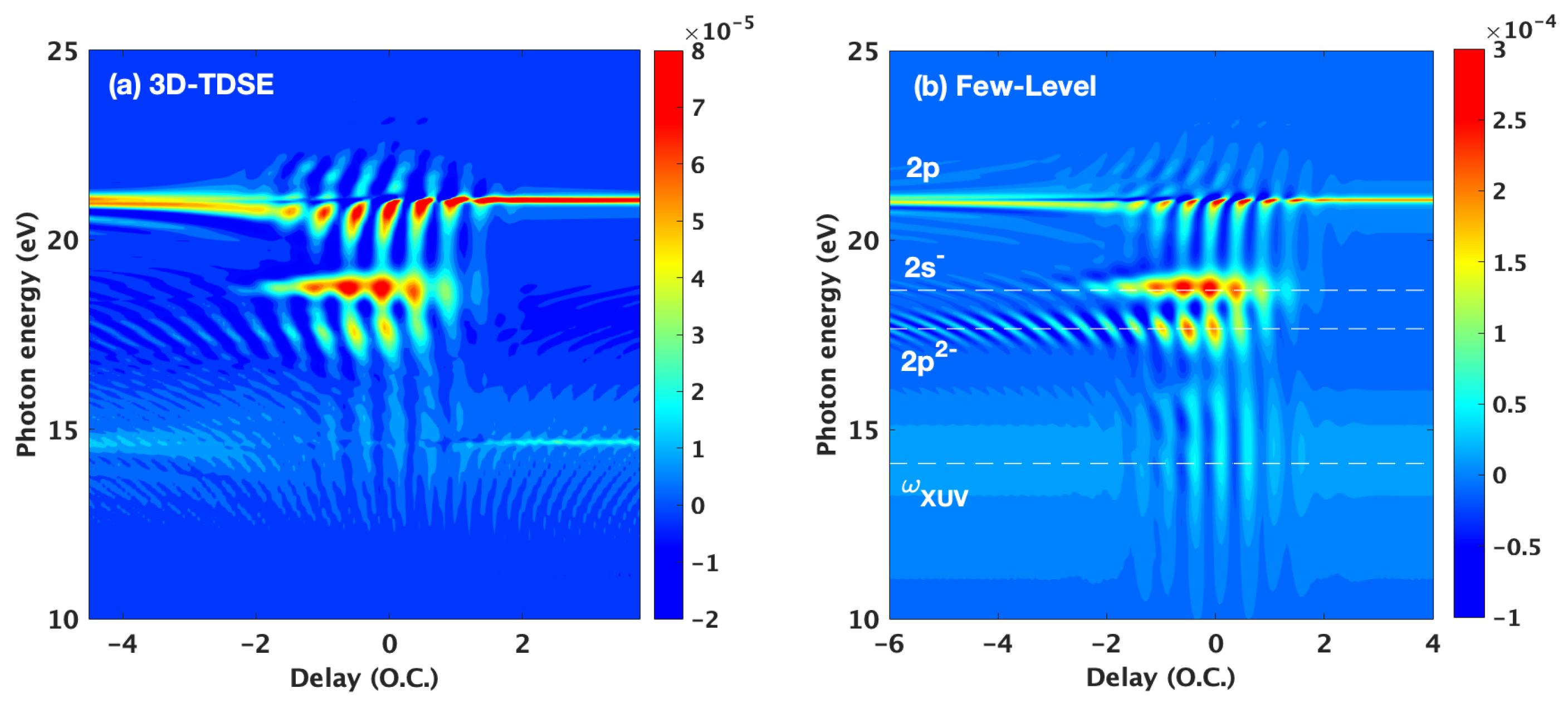
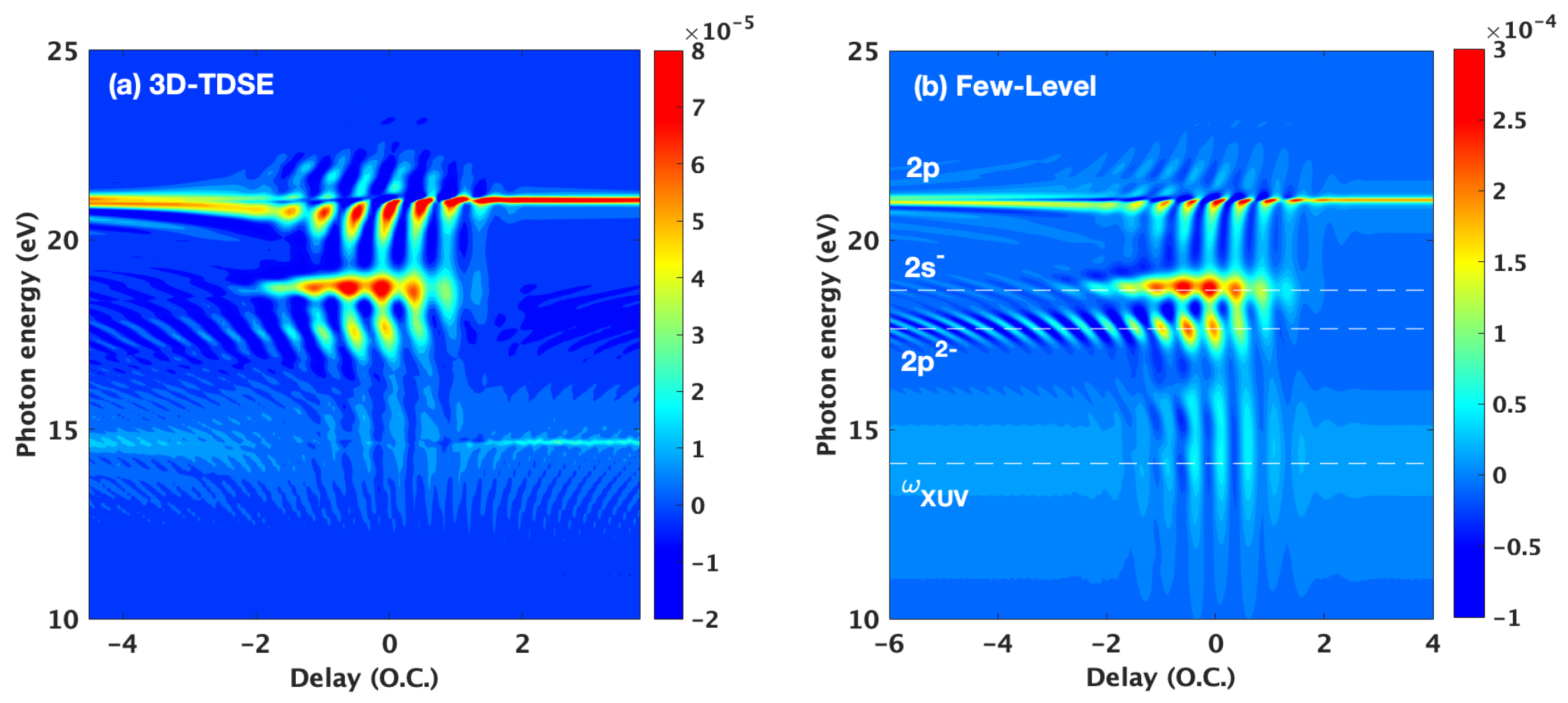
3. Results and Discussion
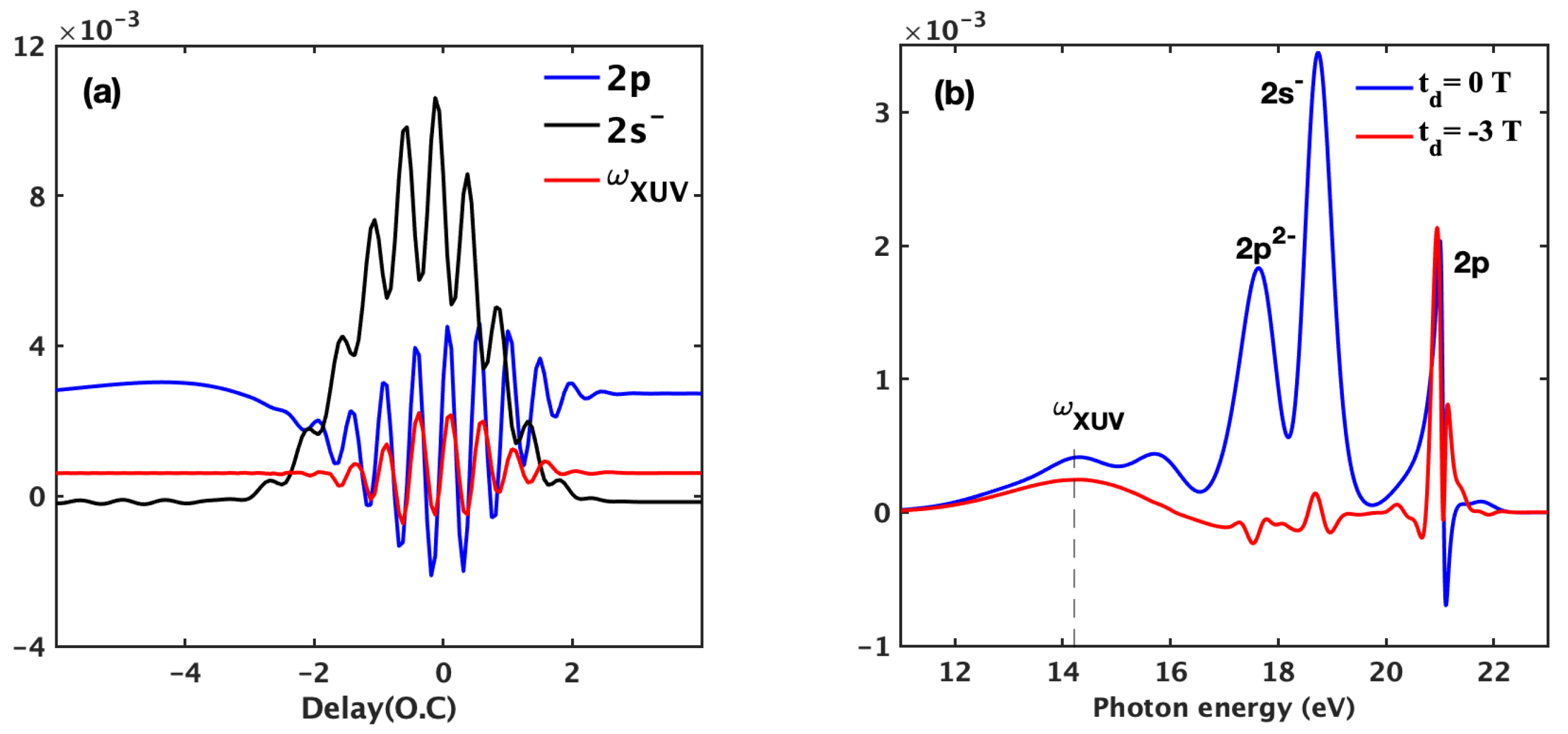
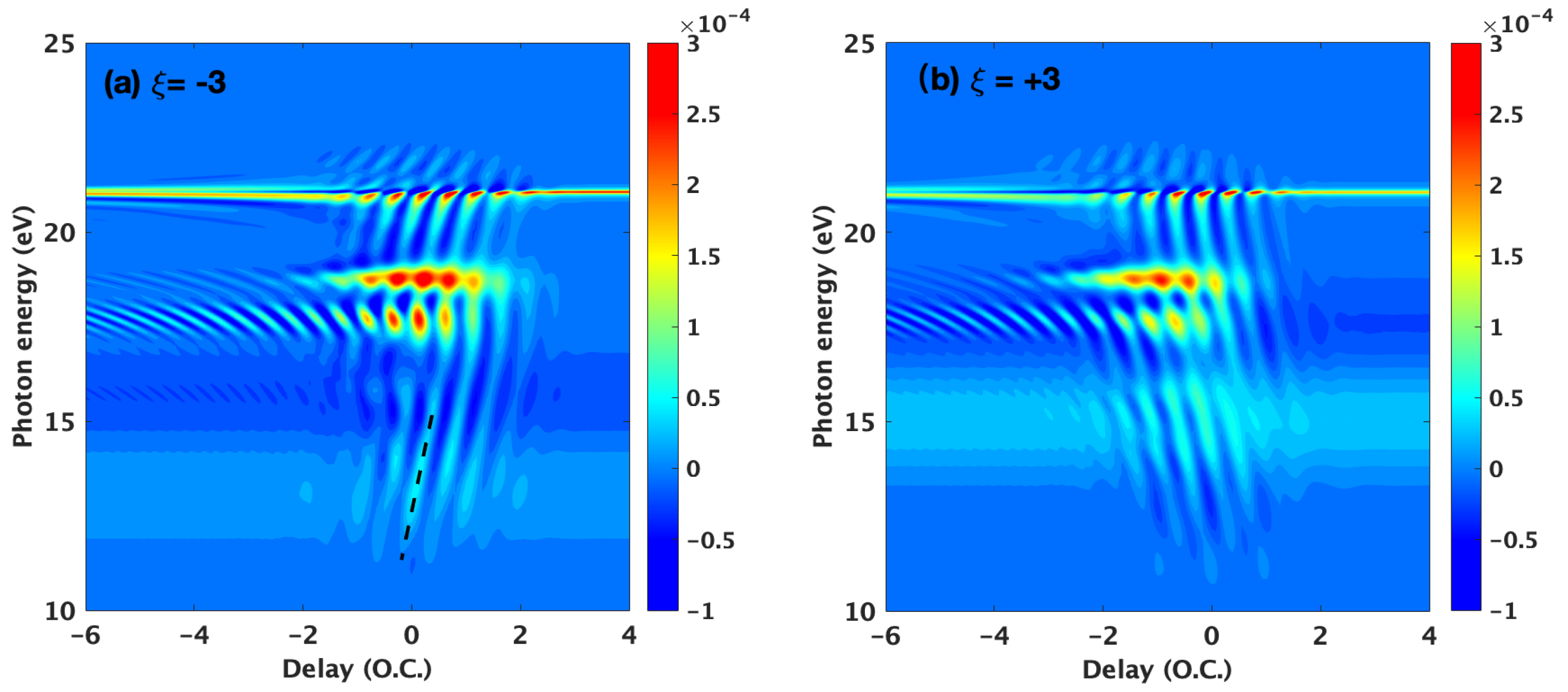
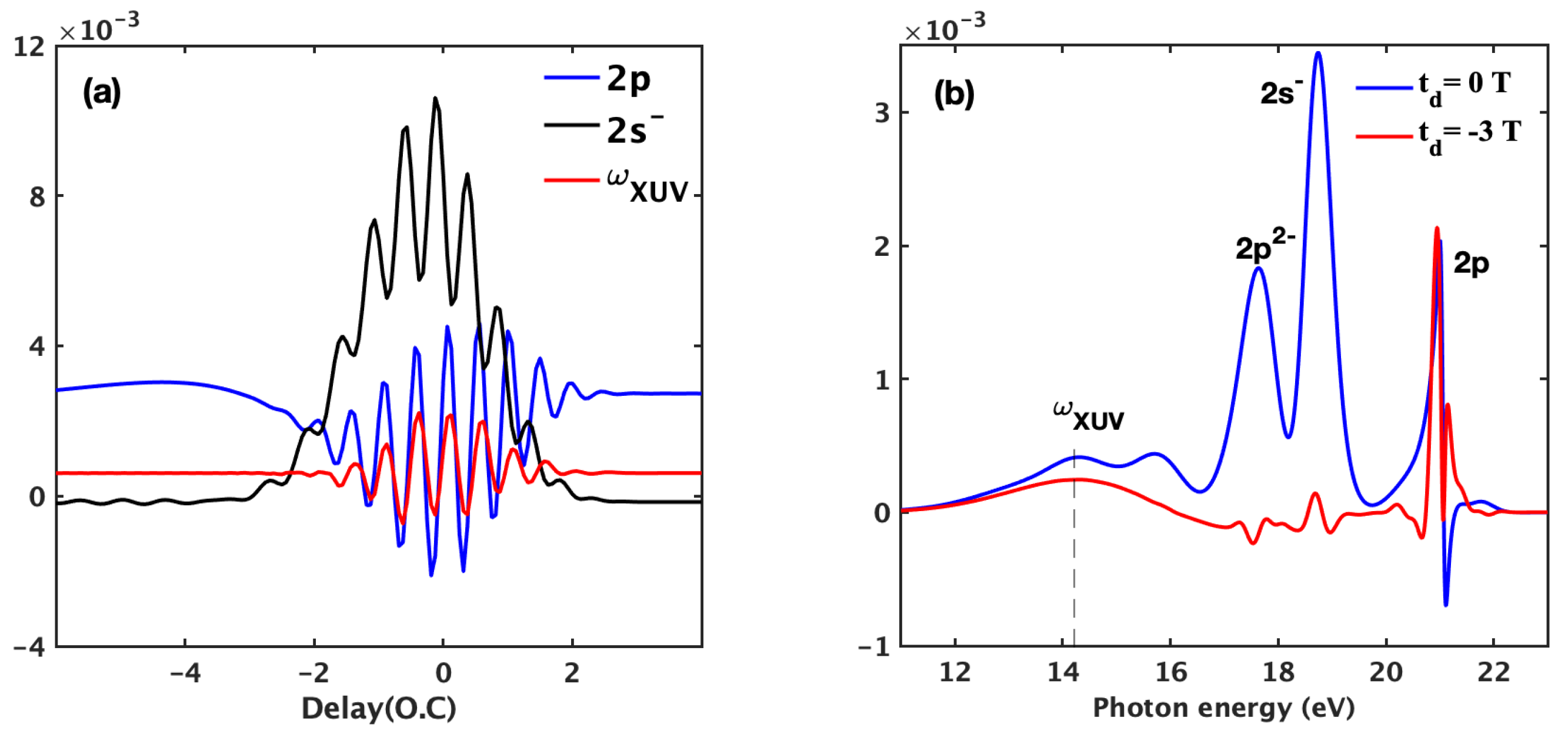
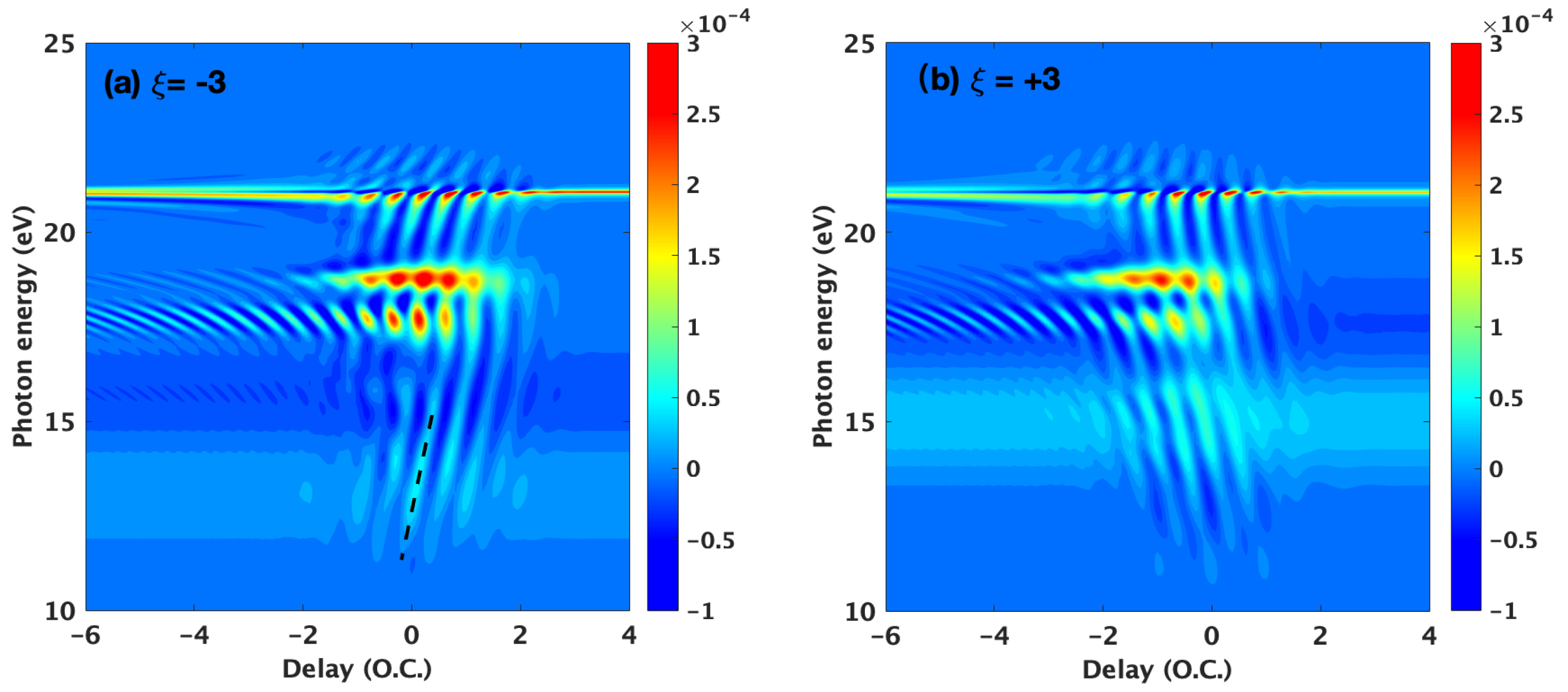
3.1. Basic Features of the ATA Spectrum below the Excited States
3.2. Time-Dependent Perturbation Theory
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATA | Attosecond transient absorption |
| XUV | Extreme ultraviolet |
| IR | Infrared |
| ATAS | Attosecond transient absorption spectroscopy |
| 3D-TDSE | Three-dimentional time-dependent Schrödinger equation |
| SAE | Single-active electron |
| O.C. | Optical cycle |
| LIS | Laser-induced state |
References
- Loh, Z.H.; Greene, C.H.; Leone, S.R. Femtosecond induced transparency and absorption in the extreme ultraviolet by coherent coupling of the He 2s2p (1Po) and 2p2 (1Se) double excitation states with 800 nm light. Chem. Phys. 2008, 350, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Goulielmakis, E.; Loh, Z.H.; Wirth, A.; Santra, R.; Rohringer, N.; Yakovlev, V.S.; Zherebtsov, S.; Pfeifer, T.; Azzeer, A.M.; Kling, M.F.; et al. Real-time observation of valence electron motion. Nature 2010, 466, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chini, M.; Chen, S.; Zhang, C.H.; He, F.; Cheng, Y.; Wu, Y.; Thumm, U.; Chang, Z. Attosecond time-resolved autoionization of argon. Phys. Rev. Lett. 2010, 105, 143002. [Google Scholar] [CrossRef] [PubMed]
- Holler, M.; Schapper, F.; Gallmann, L.; Keller, U. Attosecond Electron Wave-Packet Interference Observed by Transient Absorption. Phys. Rev. Lett. 2011, 106, 123601. [Google Scholar] [CrossRef]
- Chen, S.; Schafer, K.J.; Gaarde, M.B. Transient absorption of attosecond pulse trains by laser-dressed helium. Opt. Lett. 2012, 37, 2211–2213. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Kaldun, A.; Raith, P.; Meyer, K.; Laux, M.; Evers, J.; Keitel, C.H.; Greene, C.H.; Pfeifer, T. Lorentz meets Fano in spectral line shapes: A universal phase and its laser control. Science 2013, 340, 716–720. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.; Kaldun, A.; Argenti, L.; Raith, P.; Meyer, K.; Laux, M.; Zhang, Y.; Blättermann, A.; Hagstotz, S.; Ding, T.; et al. Reconstruction and control of a time-dependent two-electron wave packet. Nature 2014, 516, 374–378. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.R.; Bernhardt, B.; Warrick, E.R.; Wu, M.; Chen, S.; Gaarde, M.B.; Schafer, K.J.; Neumark, D.M.; Leone, S.R. Attosecond transient absorption probing of electronic superpositions of bound states in neon: Detection of quantum beats. New J. Phys. 2014, 16, 113016. [Google Scholar] [CrossRef] [Green Version]
- Sabbar, M.; Timmers, H.; Chen, Y.J.; Pymer, A.K.; Loh, Z.H.; Sayres, S.G.; Pabst, S.; Santra, R.; Leone, S.R. State-resolved attosecond reversible and irreversible dynamics in strong optical fields. Nat. Physics 2017, 13, 472–478. [Google Scholar] [CrossRef]
- Reimann, J.; Schlauderer, S.; Schmid, C.; Langer, F.; Baierl, S.; Kokh, K.; Tereshchenko, O.; Kimura, A.; Lange, C.; Güdde, J.; et al. Subcycle observation of lightwave-driven Dirac currents in a topological surface band. Nature 2018, 562, 396–400. [Google Scholar] [CrossRef]
- Ott, C.; Aufleger, L.; Ding, T.; Rebholz, M.; Magunia, A.; Hartmann, M.; Stooß, V.; Wachs, D.; Birk, P.; Borisova, G.D.; et al. Strong-Field Extreme-Ultraviolet Dressing of Atomic Double Excitation. Phys. Rev. Lett. 2019, 123, 163201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, R.; Jiménez-Galán, Á.; Amorim, B.; Smirnova, O.; Ivanov, M. Topological strong-field physics on sub-laser-cycle timescale. Nat. Photonics 2019, 13, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Chacón, A.; Kim, D.; Zhu, W.; Kelly, S.P.; Dauphin, A.; Pisanty, E.; Maxwell, A.S.; Picón, A.; Ciappina, M.F.; Kim, D.E.; et al. Circular dichroism in higher-order harmonic generation: Heralding topological phases and transitions in Chern insulators. Phys. Rev. B 2020, 102, 134115. [Google Scholar] [CrossRef]
- Baykusheva, D.; Chacón, A.; Lu, J.; Bailey, T.P.; Sobota, J.A.; Soifer, H.; Kirchmann, P.S.; Rotundu, C.; Uher, C.; Heinz, T.F.; et al. All-Optical Probe of Three-Dimensional Topological Insulators Based on High-Harmonic Generation by Circularly Polarized Laser Fields. Nano Lett. 2021, 21, 8970–8978. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, S.; Camp, S.; Schafer, K.J.; Gaarde, M.B. Theory of strong-field attosecond transient absorption. J. Phys. B-At. Mol. Opt. Phys. 2016, 49, 062003. [Google Scholar] [CrossRef]
- Harkema, N.; Bækhøj, J.E.; Liao, C.T.; Gaarde, M.B.; Schafer, K.J.; Sandhu, A. Controlling attosecond transient absorption with tunable, non-commensurate light fields. Opt. Lett. 2018, 43, 3357–3360. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, C.; Zheng, Y.; Zeng, Z.; Li, R. Attosecond chirp effect on the transient absorption spectrum of laser-dressed helium atom. Opt. Express 2017, 25, 7707–7718. [Google Scholar] [CrossRef]
- Stephen, R.L.; McCurdy, C.W.; Joachim, B.; Lorenz, S.C.; Zenghu, C.; Nirit, D.; Johannes, F.; Chris, H.G.; Misha, I.; Reinhard, K.; et al. What will it take to observe processes in’real time’? Nat. Photonics 2014, 8, 162–166. [Google Scholar]
- Chu, W.C.; Zhao, S.F.; Lin, C. Laser-assisted-autoionization dynamics of helium resonances with single attosecond pulses. Phys. Rev. A 2011, 84, 033426. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chini, M.; Cheng, Y.; Wu, Y.; Tong, X.M.; Chang, Z. Subcycle laser control and quantum interferences in attosecond photoabsorption of neon. Phys. Rev. A 2013, 87, 063413. [Google Scholar] [CrossRef] [Green Version]
- Chew, A.; Douguet, N.; Cariker, C.; Li, J.; Lindroth, E.; Ren, X.; Yin, Y.; Argenti, L.; Hill, W.T., III; Chang, Z. Attosecond transient absorption spectrum of argon at the L2,3 edge. Phys. Rev. A 2018, 97, 031407. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.; Stooß, V.; Birk, P.; Borisova, G.; Ott, C.; Pfeifer, T. Attosecond precision in delay measurements using transient absorption spectroscopy. Opt. Lett. 2019, 44, 4749–4752. [Google Scholar] [CrossRef] [PubMed]
- Mercouris, T.; Komninos, Y.; Nicolaides, C.A. Ultrafast time delay as a control parameter in resonant ionization by two XUV ultrashort pulses. J. Phys. At. Mol. Opt. Phys. 2020, 53, 095603. [Google Scholar] [CrossRef]
- Bækhøj, J.E.; Yue, L.; Madsen, L.B. Nuclear-motion effects in attosecond transient-absorption spectroscopy of molecules. Phys. Rev. A 2015, 91, 043408. [Google Scholar] [CrossRef] [Green Version]
- Sommer, A.; Bothschafter, E.; Sato, S.; Jakubeit, C.; Latka, T.; Razskazovskaya, O.; Fattahi, H.; Jobst, M.; Schweinberger, W.; Shirvanyan, V.; et al. Attosecond nonlinear polarization and light–matter energy transfer in solids. Nature 2016, 534, 86–90. [Google Scholar] [CrossRef]
- Oguri, K.; Mashiko, H.; Suda, A.; Gotoh, H. Lightwave-driven electronic phenomena in solids observed by attosecond transient absorption spectroscopy. In Progress in Ultrafast Intense Laser Science XIV; Springer: Cham, Switzerland, 2018; pp. 219–238. [Google Scholar]
- Hanus, V.; Csajbók, V.; Pápa, Z.; Budai, J.; Márton, Z.; Kiss, G.Z.; Sándor, P.; Paul, P.; Szeghalmi, A.; Wang, Z.; et al. Light-field-driven current control in solids with pJ-level laser pulses at 80 MHz repetition rate. Optica 2021, 8, 570–576. [Google Scholar] [CrossRef]
- Chini, M.; Zhao, B.; Wang, H.; Cheng, Y.; Hu, S.; Chang, Z. Subcycle ac stark shift of helium excited states probed with isolated attosecond pulses. Phys. Rev. Lett. 2012, 109, 073601. [Google Scholar] [CrossRef] [Green Version]
- Chini, M.; Wang, X.; Cheng, Y.; Wu, Y.; Zhao, D.; Telnov, D.A.; Chu, S.I.; Chang, Z. Sub-cycle oscillations in virtual states brought to light. Sci. Rep. 2013, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Bell, M.J.; Beck, A.R.; Mashiko, H.; Neumark, D.M.; Leone, S.R. Intensity dependence of light-induced states in transient absorption of laser-dressed helium measured with isolated attosecond pulses. J. Mod. Opt. 2013, 60, 1506–1516. [Google Scholar] [CrossRef]
- Reduzzi, M.; Hummert, J.; Dubrouil, A.; Calegari, F.; Nisoli, M.; Frassetto, F.; Poletto, L.; Chen, S.; Wu, M.; Gaarde, M.B.; et al. Polarization control of absorption of virtual dressed states in helium. Phys. Rev. A 2015, 92, 033408. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Li, Y.; Wang, X.; Yuan, J.; Zhao, Z. Analysis of interference in attosecond transient absorption in adiabatic condition. Phys. Rev. A 2015, 92, 033412. [Google Scholar] [CrossRef] [Green Version]
- Rørstad, J.J.; Bækhøj, J.E.; Madsen, L.B. Analytic modeling of structures in attosecond transient-absorption spectra. Phys. Rev. A 2017, 96, 013430. [Google Scholar] [CrossRef] [Green Version]
- Ravn, N.S.; Madsen, L.B. Nonadiabatic effects in attosecond transient absorption spectroscopy. arXiv 2021, arXiv:2103.01582. [Google Scholar]
- Wu, M.; Chen, S.; Gaarde, M.B.; Schafer, K.J. Time-domain perspective on Autler-Townes splitting in attosecond transient absorption of laser-dressed helium atoms. Phys. Rev. A 2013, 88, 043416. [Google Scholar] [CrossRef]
- Chini, M.; Wang, X.; Cheng, Y.; Chang, Z. Resonance effects and quantum beats in attosecond transient absorption of helium. J. Phys. B-At. Mol. Opt. Phys. 2014, 47, 124009. [Google Scholar] [CrossRef]
- Seres, E.; Seres, J.; Namba, S.; Afa, J.; Serrat, C. Attosecond sublevel beating and nonlinear dressing on the 3d-to-5p and 3p-to-5s core-transitions at 91.3 eV and 210.4 eV in krypton. Opt. Express 2017, 25, 31774–31788. [Google Scholar] [CrossRef] [PubMed]
- Eberly, J.; Grobe, R.; Law, C.; Su, Q. Numerical experiments in strong and super-strong fields. In Atoms in Intense Laser Fields; Academic Press, Inc.: New York, NY, USA, 1992; pp. 301–334. [Google Scholar]
- Schafer, K.J.; Kulander, K.C. High harmonic generation from ultrafast pump lasers. Phys. Rev. Lett. 1997, 78, 638. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Nisoli, M. Complete characterization of a coherent superposition of atomic states by asymmetric attosecond photoionization. Phys. Rev. A 2012, 85, 053423. [Google Scholar] [CrossRef]
- Liu, C.; Zeng, Z.; Li, R.; Xu, Z.; Nisoli, M. Attosecond photoionization for reconstruction of bound-electron wave packets. Phys. Rev. A 2014, 90, 013403. [Google Scholar] [CrossRef]
- Chen, S.; Wu, M.; Gaarde, M.B.; Schafer, K.J. Laser-imposed phase in resonant absorption of an isolated attosecond pulse. Phys. Rev. A 2013, 88, 033409. [Google Scholar] [CrossRef]
- Chang, Z. Fundamentals of Attosecond Optics; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, J.; Wang, X.; Wang, M.; Zhou, C.; Ruan, S. Attosecond Transient Absorption Below the Excited States. Photonics 2022, 9, 269. https://doi.org/10.3390/photonics9040269
Xue J, Wang X, Wang M, Zhou C, Ruan S. Attosecond Transient Absorption Below the Excited States. Photonics. 2022; 9(4):269. https://doi.org/10.3390/photonics9040269
Chicago/Turabian StyleXue, Jinxing, Xinliang Wang, Meng Wang, Cangtao Zhou, and Shuangchen Ruan. 2022. "Attosecond Transient Absorption Below the Excited States" Photonics 9, no. 4: 269. https://doi.org/10.3390/photonics9040269
APA StyleXue, J., Wang, X., Wang, M., Zhou, C., & Ruan, S. (2022). Attosecond Transient Absorption Below the Excited States. Photonics, 9(4), 269. https://doi.org/10.3390/photonics9040269