Challenges in 3D Live Cell Imaging



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. 3D Samples
3. Phenomena and Challenges
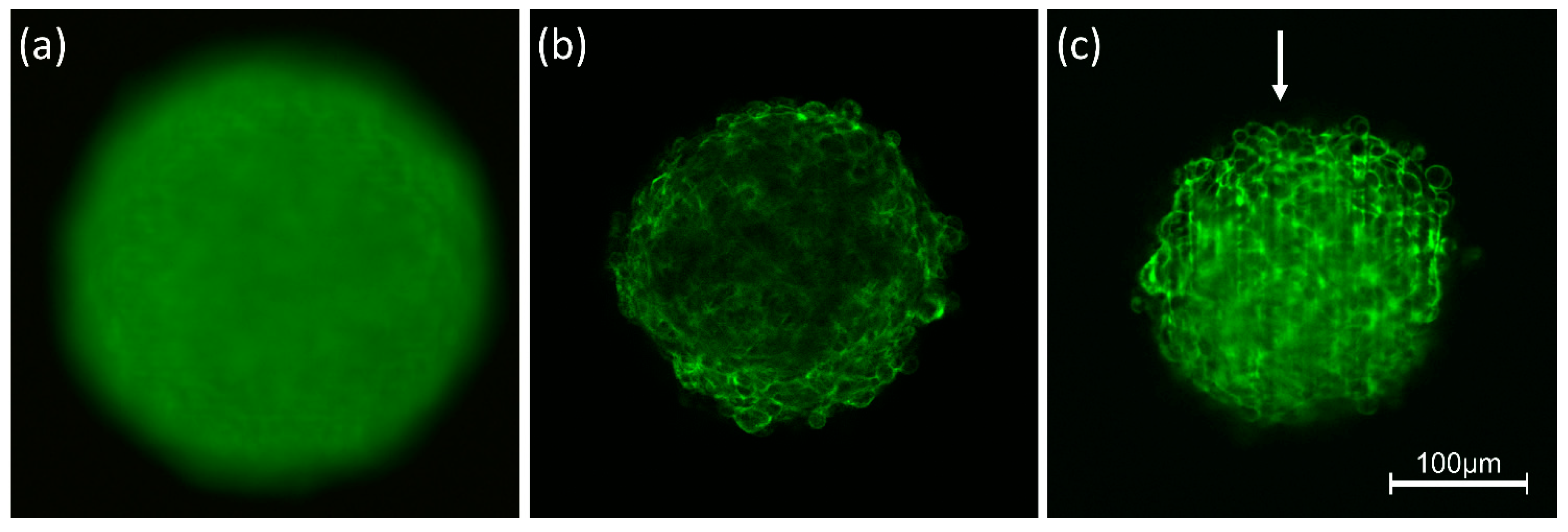
3.1. Light Scattering
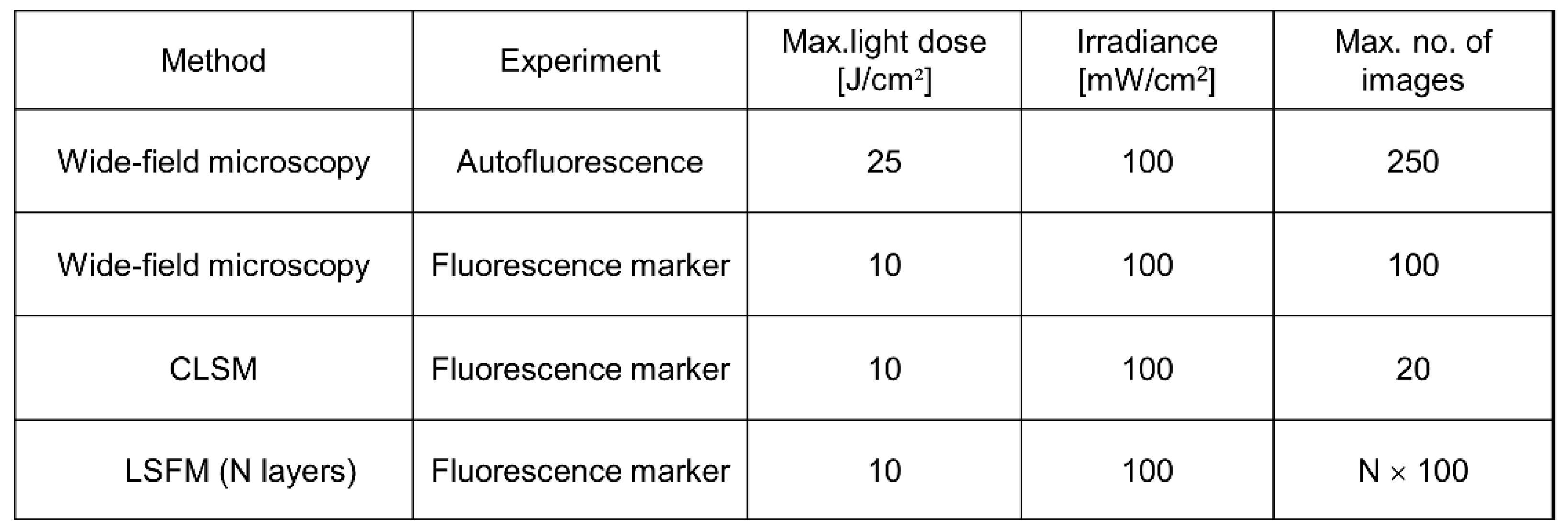
3.2. Phototoxicity, Photobleaching
3.3. “Big Data”
4. 3D Microscopy
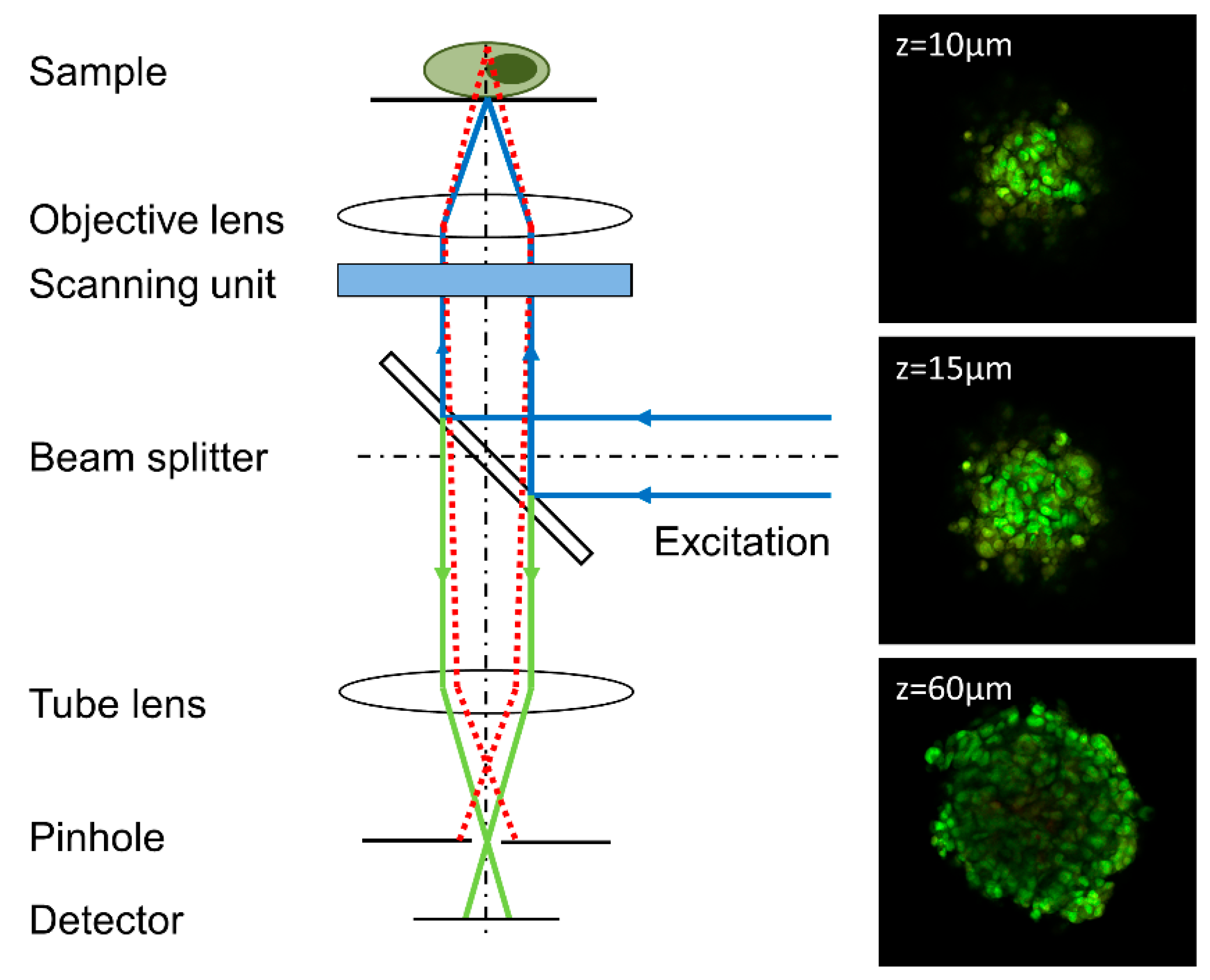
4.1. Confocal Laser Scanning Microscopy
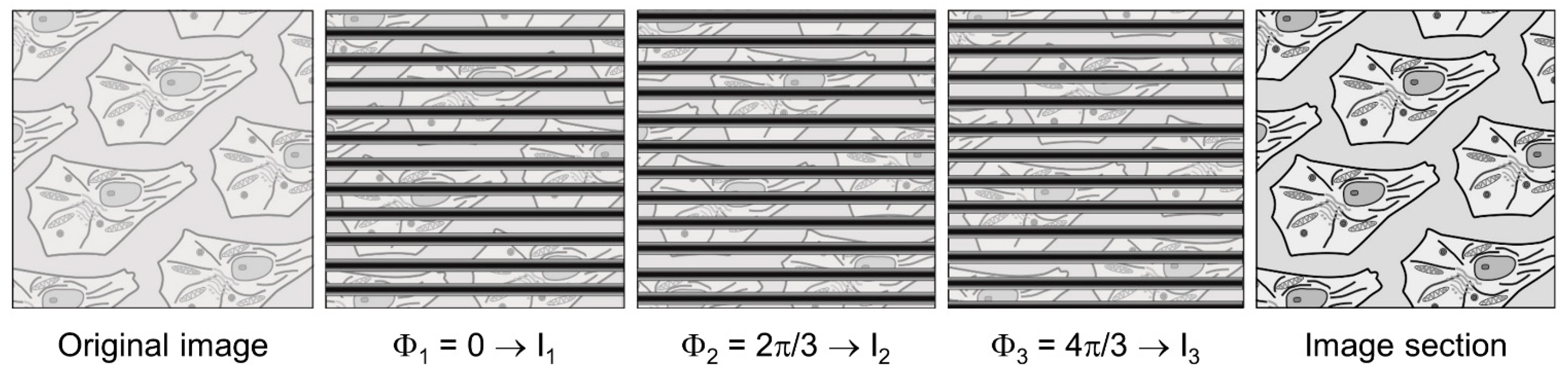
4.2. Widefield Microscopy
4.3. Deconvolution
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kunz-Schughart, L.A.; Freyer, J.P.; Hofstaedter, F.; Ebner, R. The Use of 3-D Cultures for High-Throughput Screening: The Multicellular Spheroid Model. J. Biomol. Screen. 2004, 9, 273–285. [Google Scholar] [CrossRef]
- Wittig, R.; Richter, V.; Wittig-Blaich, S.; Weber, P.; Strauss, W.S.L.; Bruns, T.; Dick, T.-P.; Schneckenburger, H. Biosensor-expressing spheroid cultures for imaging of drug-induced effects in three dimensions. J. Biomol. Screen. 2013, 18, 736–743. [Google Scholar] [CrossRef]
- Pawley, J. Handbook of Biological Confocal Microscopy, 3rd ed.; Springer: Boston, MA, USA, 1990. [Google Scholar] [CrossRef]
- Webb, R.H. Confocal optical microscopy. Rep. Prog. Phys. 1996, 59, 427–471. [Google Scholar] [CrossRef]
- Neil, M.A.; Juskaitis, R.; Wilson, T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett. 1997, 22, 1905–1907. [Google Scholar] [CrossRef]
- Pampaloni, F.; Chang, B.-J.; Stelzer, E.H.K. Light sheet-based fluorescence microscopy (LSFM) for the quantitative imaging of cells and tissues. Cell Tissue Res. 2015, 362, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Santi, P.A. Light sheet fluorescence microscopy: A review. J. Histochem. Cytochem. 2011, 59, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Schneckenburger, H.; Weber, P.; Wagner, M.; Schickinger, S.; Richter, V.; Bruns, T.; Strauss, W.S.L.; Wittig, R. Light exposure and cell viability in fluorescence microscopy. J. Microsc. 2012, 245, 311–318. [Google Scholar] [CrossRef]
- Bardsley, K.; Deegan, A.J.; El Haj, A.; Yang, Y. Current State-of-the-Art 3D Tissue Models and Their Compatibility with Live Cell Imaging. Adv. Exp. Med. Biol. 2017, 1035, 3–18. [Google Scholar] [CrossRef]
- Cambria, E.; Brunner, S.; Heusser, S.; Fisch, P.; Hitzl, W.; Ferguson, S.J.; Wuertz-Kozak, K. Cell-laden agarose-collagen composite hydrogels for mechanotransduction studies. Front. Bioeng. Biotechnol. 2020, 8, 346. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, G.; Couvreur, P.; Mura, S. Multicellular tumor spheroids: A relevant 3D model for the in vitro preclinical investigation of polymer nanomedicines. Polym. Chem. 2017, 8, 4947–4969. [Google Scholar] [CrossRef]
- Johnson, M.D.; Bryan, G.T.; Reznikoff, C.A. Serial cultivation of normal rat bladder epithelial cells in vitro. J. Urol. 1985, 133, 1076–1081. [Google Scholar] [CrossRef]
- Dusny, C.; Grünberger, A.; Probst, C.; Wiechert, W.; Kohlheyer, D.; Schmid, A. Technical bias of microcultivation environments on single cell physiology. Lab Chip 2015, 15, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef]
- Van Ineveld, R.L.; Ariese, H.C.R.; Wehrens, E.J.; Dekkers, J.F.; Rios, A.C. Single-Cell Resolution Three-Dimensional Imaging of Intact Organoids. J. Vis. Exp. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Abdullah, Z.; Brol, M.J.; Hellerbrand, C.; Fernandez, M.; Fiorotto, R.; Klein, S.; Königshofer, P.; Liedtke, C.; Lotersztajn, S.; et al. Recent Advances in Practical Methods for Liver Cell Biology: A Short Overview. Int. J. Mol. Sci. 2020, 21, 2027. [Google Scholar] [CrossRef]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Schneider, O.; Zeifang, L.; Fuchs, S.; Sailer, C.; Loskill, P. User-Friendly and Parallelized Generation of Human Induced Pluripotent Stem Cell-Derived Microtissues in a Centrifugal Heart-on-a-Chip. Tissue Eng. Part A 2019, 25, 786–798. [Google Scholar] [CrossRef]
- Glaser, A.K.; Reder, N.P.; Chen, Y.; McCarty, E.F.; Yin, C.; Wei, L.; Wang, Y.; True, L.D.; Liu, J.T.C. Light-sheet microscopy for slide-free non-destructive pathology of large clinical specimens. Nat. Biomed. Eng. 2017, 1, 0084. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.J.; Titus, S.A.; Wagner, A.K.; Simeonov, A. High-throughput fluorescence imaging approaches for drug discovery using in vitro and in vivo three-dimensional models. Expert Opin. Drug Discov. 2015, 10, 1347–1361. [Google Scholar] [CrossRef]
- Bruns, T.; Schickinger, S.; Schneckenburger, H. Sample holder for axial rotation of specimens in 3D Microscopy. J. Microsc. 2015, 260, 30–36. [Google Scholar] [CrossRef]
- Brunstin, A.; Mullaney, P.F. Differential Light-Scattering from Spherical Mammalian Cells. Biophys. J. 1974, 14, 439–453. [Google Scholar] [CrossRef]
- Mourant, J.R.; Johnson, T.M.; Doddi, V.; Freyer, J.P. Angular dependent light scattering from multicellular spheroids. J. Biomed. Opt. 2002, 7, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, C.S.; Sherwood, C.A.; Bigio, I.J. Wavelength-dependent backscattering measurements for quantitative real-time monitoring of apoptosis in living cells. J. Biomed. Opt. 2009, 14, 064013. [Google Scholar] [CrossRef]
- Mulvey, C.S.; Zhang, K.; Bobby Liu, W.H.; Waxman, D.J.; Bigio, I.J. Wavelength-dependent backscattering measurements for quantitative monitoring of apoptosis, part 2: Early spectral changes during apoptosis are linked to apoptotic volume decrease. J. Biomed. Opt. 2011, 16, 117002. [Google Scholar] [CrossRef]
- Bohren, C.F.; Huffman, D.R. Absorption and Scattering of Light by Small Particles; Wiley-Interscience Publications: New York, NY, USA, 1998. [Google Scholar] [CrossRef]
- König, K. Multiphoton microscopy in life sciences. J. Microsc. 2000, 200 Pt 2, 83–104. [Google Scholar] [CrossRef]
- Miller, D.R.; Jarrett, J.W.; Hassan, A.M.; Dunn, A.K. Deep Tissue Imaging with Multiphoton Fluorescence Microscopy. Curr. Opin. Biomed. Eng. 2017, 4, 32–39. [Google Scholar] [CrossRef]
- Costa, E.C.; Silva, D.N.; Moreira, A.F.; Correia, I.J. Optical clearing methods: An overview of the techniques used for the imaging of 3D spheroids. Biotechnol. Bioeng. 2019, 116, 2742–2763. [Google Scholar] [CrossRef]
- Sdobnov, A.Y.; Lademann, J.; Darvin, M.E.; Tuchin, V.V. Methods for Optical Skin Clearing in Molecular Optical Imaging in Dermatology. Biochemistry (Moscow) 2019, 84 (Suppl. S1), S144–S158. [Google Scholar] [CrossRef]
- Susaki, E.A.; Tainaka, K.; Perrin, D.; Yukinaga, H.; Kuno, A.; Ueda, H.R. Advanced CUBIC protocols for whole-brain and whole-body clearing and imaging. Nat. Protoc. 2015, 10, 1709–1727. [Google Scholar] [CrossRef]
- Costantini, I.; Cicchi, R.; Silvestri, L.; Vanzi, F.; Pavone, F.S. In-vivo and ex-vivo optical clearing methods for biological tissues: Review. Biomed. Opt. Express 2019, 10, 5251–5267. [Google Scholar] [CrossRef]
- Schneckenburger, H.; Richter, V.; Wagner, M. Live-Cell Optical Microscopy with Limited Light Doses; SPIE Spotlight Series; SPIE: Bellingham, WA, USA, 2018; Volume 42, p. 38. [Google Scholar] [CrossRef]
- Song, L.; Hennink, E.J.; Young, I.T.; Tanke, H.J. Photobleaching Kinetics of Fluorescein in Quantitative Fluorescence Microscopy. Biophys. J. 1995, 68, 2588–2600. [Google Scholar] [CrossRef]
- Moerner, W.E.; Shechtman, Y.; Wang, Q. Single-molecule spectroscopy and imaging over the decades. Faraday Discuss. 2015, 184, 9–36. [Google Scholar] [CrossRef]
- Ishikawa-Ankerhold, H.C.; Ankerhold, R.; Drummen, G.P. Advanced fluorescence microscopy techniques–FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012, 17, 4047–4132. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Snapp, E.L.; Phair, R.D. The Development and Enhancement of FRAP as a Key Tool for Investigating Protein Dynamics. Biophys. J. 2018, 115, 1146–1155. [Google Scholar] [CrossRef]
- Amat, F.; Höckendorf, B.; Wan, Y.; Lemon, W.C.; McDole, K.; Keller, P.J. Efficient processing and analysis of large-scale light-sheet microscopy data. Nat. Prot. 2015, 10, 1679–1696. [Google Scholar] [CrossRef]
- Reynaud, E.G.; Peychl, J.; Huisken, J.; Tomancak, P. Guide to light-sheet microscopy for adventurous biologists. Nat. Methods 2015, 12, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Allan, C.; Burel, J.M.; Moore, J.; Blackburn, C.; Linkert, M.; Loynton, S.; MacDonald, D.; Moore, W.J.; Neves, C.; Patterson, A.; et al. OMERO: Flexible, model-driven data management for experimental biology. Nat. Methods 2012, 9, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Orth, A.; Schaak, D.; Schonbrun, E. Microscopy, Meet Big Data. Cell Syst. 2017, 4, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Pietzsch, T.; Saalfeld, S.; Preibisch, S.; Tomancak, P. BigDataViewer: Visualization and processing for large image data sets. Nat. Methods 2015, 12, 481–483. [Google Scholar] [CrossRef]
- Berg, S.; Kutra, D.; Kroeger, T.; Straehle, C.N.; Kausler, B.X.; Haubold, C.; Schiegg, M.; Ales, J.; Beier, T.; Rudy, M.; et al. ilastik: Interactive machine learning for (bio)image analysis. Nat. Methods 2019, 16, 1226–1232. [Google Scholar] [CrossRef]
- Nakano, A. Spinning-disk confocal microscopy–A cutting-edge tool for imaging of membrane traffic. Cell Struct. Funct. 2002, 27, 349–355. [Google Scholar] [CrossRef]
- Huff, J. The Airyscan detector from ZEISS: Confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 2015, 12, i–ii. [Google Scholar] [CrossRef]
- Müller, C.B.; Enderlein, J. Image Scanning Microscopy. Phys. Rev. Lett. 2010, 104, 198101. [Google Scholar] [CrossRef]
- De Luca, G.M.; Breedijk, R.M.; Brandt, R.A.; Zeelenberg, C.H.; de Jong, B.E.; Timmermans, W.; Azar, L.N.; Hoebe, R.A.; Stallinga, S.; Manders, E.M. Re-scan confocal microscopy: Scanning twice for better resolution. Biomed. Opt. Express 2013, 4, 2644–2656. [Google Scholar] [CrossRef]
- Wildanger, D.; Medda, R.; Kastrup, L.; Hell, S.W. A compact STED microscope providing 3D nanoscale resolution. J. Microsc. 2009, 236, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Benninger, R.K.; Piston, D.W. Two-photon excitation microscopy for the study of living cells and tissues. Curr. Protoc. Cell Biol. 2013, 59, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, J.; Orlova, N.; Grewe, B.F. Wide. Fast. Deep: Recent Advances in Multiphoton Microscopy of In Vivo Neuronal Activity. J. Neurosci. 2019, 39, 9042–9052. [Google Scholar] [CrossRef]
- Schneckenburger, H.; Richter, V. Laser Scanning versus Wide-Field-Choosing the Appropriate Microscope in Life Sciences. Appl. Sci. 2021, 11, 733. [Google Scholar] [CrossRef]
- Heintzmann, R.; Cremer, C. Laterally modulated excitation microscopy: Improvement of resolution by using a diffraction grating. In Optical Biopsies and Microscopic Techniques III, Proceedings of the SPIE 3568, Stockholm, Sweden, 8–12 September 1998; SPIE: Bellingham, WA, USA, 1999; pp. 185–196. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef]
- Betzig, E. Excitation strategies for optical lattice microscopy. Opt. Express 2005, 13, 3021–3036. [Google Scholar] [CrossRef]
- Heintzmann, R. Saturated patterned excitation microscopy with two-dimensional excitation patterns. Micron 2003, 34, 283–291. [Google Scholar] [CrossRef]
- Bruns, T.; Bauer, M.; Bruns, S.; Meyer, H.; Kubin, D.; Schneckenburger, H. Miniaturized modules for light sheet microscopy with low chromatic aberration. J. Microsc. 2016, 264, 261–267. [Google Scholar] [CrossRef]
- Bruns, T.; Schickinger, S.; Schneckenburger, H. Single plane illumination module and micro-capillary approach for a wide-field microscope. J. Vis. Exp. 2014, 90, e51993. [Google Scholar] [CrossRef] [PubMed]
- Greger, K.; Swoger, J.; Stelzer, E.H.K. Basic building units and properties of a fluorescence single plane illumination microscope. Rev. Sci. Instrum. 2007, 78, 023705. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Bruns, S.; Bruns, T.; Weber, P.; Wagner, M.; Cremer, C.; Schneckenburger, H. Axial Tomography in Live Cell Laser Microscopy. J. Biomed. Opt. 2017, 22, 91505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Levoy, M.; Ng, R.; Adams, A.; Footer, M.; Horowitz, M. Light field microscopy. ACM Trans. Graph. 2006, 25, 924–934. [Google Scholar] [CrossRef]
- Prevedel, R.; Yoon, Y.G.; Hoffmann, M.; Pak, N.; Wetzstein, G.; Kato, S.; Schrödel, T.; Raskar, R.; Zimmer, M.; Boyden, E.S.; et al. Simultaneous whole-animal 3d imaging of neuronal activity using light-field microscopy. Nat. Methods 2014, 11, 727–730. [Google Scholar] [CrossRef]
- Wagner, N.; Beuttenmueller, F.; Norlin, N.; Gierten, J.; Boffi, J.C.; Wittbrodt, J.; Weigert, M.; Hufnagel, L.; Prevedel, R.; Kreshuk, A. Deep learning-enhanced light-field imaging with continuous validation. Nat. Methods 2021, 18, 557–563. [Google Scholar] [CrossRef]
- Wallace, W.; Schaefer, L.H.; Swedlow, J.R. A workingperson’s guide to deconvolution in light microscopy. Biotechniques 2001, 31, 1076–1078. [Google Scholar] [CrossRef]
- Verdaasdonk, J.S.; Stephens, A.D.; Haase, J.; Bloom, K. Bending the rules: Widefield microscopy and the Abbe limit of resolution. J. Cell. Physiol. 2014, 229, 132–138. [Google Scholar] [CrossRef]
- Becker, K.; Saghafi, S.; Pende, M.; Sabdyusheva-Litschauer, I.; Hahn, C.M.; Foroughipour, M.; Jährling, N.; Dodt, H.-U. Deconvolution of light sheet microscopy recordings. Sci. Rep. 2019, 9, 17625. [Google Scholar] [CrossRef]
- Guo, M.; Li, Y.; Su, Y.; Lambert, T.; Nogare, D.D.; Moyle, M.W.; Duncan, L.H.; Ikegami, B.; Santella, A.; Rey-Suarez, I. Rapid image deconvolution and multiview fusion for optical microscopy. Nat. Biotechnol. 2020, 38, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Sage, D.; Donati, L.; Soulez, F.; Fortun, D.; Schmit, G.; Seitz, A.; Guiet, R.; Vonesch, C.; Unser, M. DeconvolutionLab2: An open-source software for deconvolution microscopy. Methods 2017, 115, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198 Pt 2, 82–87. [Google Scholar] [CrossRef]
- Chakrova, N.; Rieger, B.; Stallinga, S. Deconvolution methods for structured illumination microscopy. J. Opt. Soc. Am. A 2016, 33, B12–B20. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, N.; Peckys, D.B. Live Cell Electron Microscopy Is Probably Impossible. ACS Nano 2016, 10, 9061–9063. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence lifetime imaging microscopy: Fundamentals and advances in instrumentation, analysis, and applications. J. Biomed. Opt. 2020, 25, 1–43. [Google Scholar] [CrossRef]
- Tregidgo, C.; Levitt, J.A.; Suhling, K. Effect of refractive index on the fluorescence lifetime of green fluorescent protein. J. Biomed. Opt. 2008, 13, 031218. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Lanzerstorfer, P.; Weghuber, J.; Schneckenburger, H. Probing small distances in live cell imaging. Photonics 2021, 8, 176. [Google Scholar] [CrossRef]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Viallet, P.M.; Vo-Dinh, T. Studying 3D subdomains of proteins at the nanometer scale using fluorescence spectroscopy. Methods Mol. Biol. 2005, 300, 165–189. [Google Scholar] [CrossRef]
- Huebsch, N.D.; Mooney, D.J. Fluorescent resonance energy transfer: A tool for probing molecular cell-biomaterial interactions in three dimensions. Biomaterials 2007, 28, 2424–2437. [Google Scholar] [CrossRef]
- Weber, P.; Schickinger, S.; Wagner, M.; Angres, B.; Bruns, T.; Schneckenburger, H. Monitoring of apoptosis in 3D cell cultures by FRET and light sheet fluorescence microscopy. Int. J. Mol. Sci. 2015, 16, 5375–5385. [Google Scholar] [CrossRef]
- Li, Q.; Lau, A.; Morris, T.J.; Guo, L.; Fordyce, C.B.; Stanley, E.F. A syntaxin 1, Galphao, and N-type calcium channel complex at a presynaptic nerve terminal: Analysis by quantitative immunocolocalization. J. Neurosci. 2004, 24, 4070–4081. [Google Scholar] [CrossRef]
- Manders, E.; Stap, J.; Brakenhoff, G.; Van Driel, R.; Aten, J.A. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992, 103, 857–862. [Google Scholar] [CrossRef]
- Obara, B.; Jabeen, A.; Fernandez, N.; Laissue, P.P. A novel method for quantified, superresolved, three-dimensional colocalisation of isotropic, fluorescent particles. Histochem. Cell Biol. 2013, 139, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Gilles, J.F.; Dos Santos, M.; Boudier, T.; Bolte, S.; Heck, N. DiAna, An ImageJ tool for object-based 3D co-localization and distance analysis. Methods 2017, 155, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Jaskolski, F.; Mulle, C.; Manzoni, O.J. An automated method to quantify and visualize colocalized fluorescent signals. J. Neurosci. Methods 2005, 146, 42–49. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneckenburger, H.; Richter, V. Challenges in 3D Live Cell Imaging. Photonics 2021, 8, 275. https://doi.org/10.3390/photonics8070275
Schneckenburger H, Richter V. Challenges in 3D Live Cell Imaging. Photonics. 2021; 8(7):275. https://doi.org/10.3390/photonics8070275
Chicago/Turabian StyleSchneckenburger, Herbert, and Verena Richter. 2021. "Challenges in 3D Live Cell Imaging" Photonics 8, no. 7: 275. https://doi.org/10.3390/photonics8070275
APA StyleSchneckenburger, H., & Richter, V. (2021). Challenges in 3D Live Cell Imaging. Photonics, 8(7), 275. https://doi.org/10.3390/photonics8070275