Abstract
α-Pinene is an intriguing monoterpene as it has been reported to undergo the self-disproportionation of enantiomers (SDE) phenomenon via gas chromatography (GC), the only compound to decisively demonstrate this. Examples of the SDE involving the gaseous phase—sublimation aside—are extremely rare. Attempts to replicate the GC results were unsuccessful, though the authors argued convincingly for the difficulty of observing the phenomenon. However, we could effect for α-pinene SDE via evaporation off silica gel and by foam fractionation—albeit the SDE magnitude for both was only very slight—to confirm that α-pinene can undergo the SDE for processes involving a gaseous phase and thus validate the plausibility of the GC report. The indications are that the molecular associations responsible for the various SDE observations of α-pinene occur not in the gaseous phase or the bulk phase but rather in two-dimensional (2D) adsorbed monolayers and are not based on conventional functional group-based intermolecular interactions and instead are, most likely, as a result of homo- and heterochiral packing differences in the 2D monolayers—a well-known 2D chiral-based association packing effect. These are also the first reports of the occurrence of the SDE using an adsorptive bubble separation process (foam fractionation) and involving a gaseous phase other than sublimation, GC, and distillation.
1. Introduction
The self-disproportionation of enantiomers (SDE) phenomenon [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16] occurs whenever any physicochemical process is used to fractionate a scalemic sample (an unequal mixture of two enantiomers of a compound) resulting in an uneven distribution of the enantiomers across the fractions due to molecular associations yielding differential formation of homo- and heterochiral associates [1,2,3,4]. The SDE manifests itself in all manner of physicochemical processes, the most well-known being recrystallization, and also includes processes involving the gaseous phase, such as sublimation [12,13,14,15]. Astonishingly, the SDE has even been observed by distillation (SDEvD) with two confirmed cases in the literature (Figure 1), viz. isopropyl 3,3,3-trifluorolactate (1) [17] and methyl N-trifluoroacetyl valate (2) [18]. This is truly remarkable as the energies involved in the distillation process are generally considered to be too great [19] to allow the formation of the molecular associations necessary for the manifestation of the SDE, or at least a sufficient amount of them for the differentiation between homo- and heterochiral associates to result in the SDE.
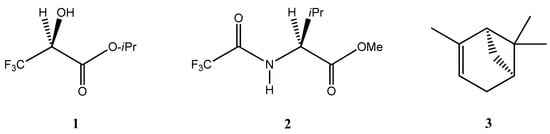
Figure 1.
The structures of isopropyl 3,3,3-trifluorolactate (1) [17] and methyl N-trifluoroacetyl valate (2) [18] that both exhibit SDEvD and α-pinene (3) that have been reported [20] to undergo SDEvGC.
The monoterpene α-pinene (3) is an intriguing compound as it has been reported [20], perhaps even more remarkably, to undergo SDE via gas chromatography (GC), the only known example to decisively demonstrate this. Again, it could be considered that not only would the energies involved in the GC process preclude observation of the SDE phenomenon, but in addition is the fact that GC is an intrinsically dilute system compared to distillation, thus reducing even more the formation of molecular associates and thereby further reducing the probability of observing the SDE phenomenon. Moreover, it could generally be assumed that molecular association in hydrocarbons is minimal and would be insufficient to affect any type of SDE occurrence, let alone for an intrinsically SDE-inhibitory process such as GC.
We were greatly intrigued by the SDEvGC result, and simply out of scientific curiosity, we attempted to replicate the SDEvGC, ostensibly under what we considered to be identical conditions and using an identical column, but were unsuccessful. This is not to question the integrity of the previous work, as the authors provided a credible explanation for why the SDEvGC phenomenon would be difficult to observe. Undeterred, we sought to examine the behavior of α-pinene (3) by other methods to test if it can undergo SDE by other processes or to see if it can exhibit substantial molecular association by other related chiral-based phenomena and thus lend credence to the SDEvGC report.
2. Results and Discussion
2.1. Attempted Replication of SDE via GC (SDEvGC)
The previous SDEvGC results [20] are even more surprising when the structure of α-pinene (3) is considered. It is a simple hydrocarbon with a single, double bond, thus lacking functional groups capable of forming strong intermolecular interactions, a prerequisite generally considered necessary for the occurrence of the SDE phenomenon, at least for processes involving condensed states. Hence, the amount of molecular association would be presumed to be too low to affect the SDE to an observable extent as the hydrocarbon intermolecular interactions would not be expected to be strong enough, and any molecular associates that are formed would be easily disrupted under GC conditions. Yet, α-pinene (3) exhibits SDEvGC, a result which would be a surprise even for compounds with much greater prospects for intermolecular interaction leading to reasonable levels of molecular association and thus the SDE. In addition to GC being considered a process too energetic to allow intermolecular interactions leading to the SDE, the highly dilute state of the analyte during GC analysis is also likely to limit the formation of the necessary molecular associates, at least in the gaseous phase.
In our attempts to replicate the SDEvGC results [20], we ostensibly used the same conditions (stationary phase, column dimensions, oven temperature, sample loading, and gas velocity), but we were unable to replicate the observations as reported for their most pronounced result for a sample of α-pinene (3) with the same enantiomeric excess (ee) despite repeated attempts and with subsequent multiple variations of the conditions. The authors of the SDEvGC report [20] have given a convincing and compelling argument for why the SDEvGC would be difficult to observe, viz. that there is an extremely narrow window for observing the SDE between conditions that are too dilute to be effective for the SDE and more concentrated conditions leading to mass overload resulting in consequent broadening of the peaks and thereby obscuring observation of the SDE. Of note, this is in contrast to liquid chromatography, where higher concentrations greatly enhance the SDE, and the window for observation of the SDE is consequently much wider. Clearly, the manner in which the intermolecular interactions occur, giving rise to the SDE, must differ between the two chromatographic methods based on this behavior. Accordingly, it may only require minor alterations in the applied conditions for the SDEvGC to be completely quashed, and it may simply, for example, be that the GC column used in the previous work [20] was in better condition than that used in this work—or vice versa—and as a consequence we were unable to reproduce the SDEvGC phenomenon to the extent that it was observable. Since it has been concluded [21] that the SDEvGC is theoretically conceivable, we sought other means to observe the SDE of α-pinene (3) for physicochemical processes involving the gaseous phase and thus validate the plausibility of SDEvGC with experimental observations. We also wished to determine if the potential SDE observations were based on conventional functional group-based intermolecular interactions, which ought to be observed by other processes such as distillation and spectroscopic methods such as infrared (IR) and nuclear magnetic resonance (NMR), or if the SDEvGC was based on fundamentally different intermolecular interactions.
2.2. NMR and IR Spectroscopic Analysis
The underlying physicochemical interaction giving rise to the SDE is the preferential formation of homo- versus heterochiral associates, and the effect of this dynamic equilibrium in solution or the neat liquid state can also be apparent in certain spectroscopies with either a change in concentration or sample ee, e.g., IR, optical rotation (OR), and NMR, where either distinct bands can arise or signals can shift (IR [17,22] and NMR [22,23,24,25]) or the magnitude of the metric can change (OR [26,27]). Particular cases of these phenomena have even been dubbed with explicit names, e.g., the Horeau effect [26,27] for OR and the self-induced diastereomeric anisochronism (SIDA) phenomenon [22,23,24,25] in solution-state NMR.
Thus, since SIDA and analogous phenomena in other spectroscopies are related to the phenomenon of SDE as they are similarly based on intermolecular interactions leading to an association, we examined α-pinene (3) by NMR and IR to see if we could detect enantiodiscriminatory-based molecular associations. The IR of neat (R)-, (S)-, and (rac)-α-pinene (3) samples failed to reveal differences between the three samples, thus likely ruling out molecular association in the bulk phase as the basis for the observed SDEvGC. This is in contrast to the case of isopropyl 3,3,3-trifluorolactate (1), where the SDEvD has been attributed [17,28,29] to a molecular association in the bulk phase as opposed to a molecular association in the gaseous phase, either exclusively or in conjunction with the bulk phase.
For α-pinene (3) in CDCl3 at concentrations of 10 mg/mL and 250 mg/mL, SIDA effects could not be discerned between samples of the S enantiomer and an S-enriched scalemate (33% ee) in the 1H NMR spectra at 25 °C. Similarly, for the 1H NMR spectra of samples in the gas phase at 25 °C, differences due to SIDA could not be discerned between samples of the R and S enantiomers and an S-enriched scalemate (33% ee). For neat samples of the S enantiomer, an S-enriched scalemate (50% ee), and the racemate, differences due to SIDA could not be discerned in either the 1H NMR spectra or in the 13C NMR spectra at 23 °C. Similarly for these samples at −41 °C, differences due to SIDA could not be discerned in the 1H NMR spectra. However, SIDA could just be discerned in the 13C NMR spectra by the splitting of one carbon signal in the spectrum of the scalemic sample. For samples of the S enantiomer, an S-enriched scalemate (50% ee), and the racemate at concentrations of 68% v/v in CDCl3, differences due to SIDA could not be discerned in either the 1H NMR spectra or in the 13C NMR spectra at 23 °C, and neither in the 1H NMR spectra at −41 °C. Curiously, SIDA could be seen rather more easily for the 68% v/v concentrated scalemic sample in comparison to the neat scalemic sample in the 13C NMR spectrum at −41 °C by the splitting of nine of the ten carbon signals. The low-temperature 13C NMR spectra exhibiting the SIDA phenomena are presented in the Supplementary Materials.
The difficulty in observing the feint SIDA effects meant that a comparison of enantiopure and racemic samples was unable to provide a definitive indication of SIDA due to variations in the conditions obscuring chemical shift changes that could be attributed to SIDA effects and only for the scalemic samples could SIDA be definitively identified. This difficulty to observe SIDA—only just observed at 11.75 T for signals of 13C nuclei from neat or extremely concentrated samples at low temperature—indicates that the molecular association of α-pinene (3) molecules is very weak and quite limited in the bulk phase.
2.3. Attempted SDE via Distillation (SDEvD)
We also attempted the SDEvD of α-pinene (3) with the consideration that the intermolecular interactions responsible for the SDEvGC might also be operative under distillation conditions. Thus despite little expectation of observing the SDEvD for α-pinene (3), we nevertheless distilled a scalemic sample of (R)-α-pinene (3) (36% ee) under reduced pressure using a Vigreux column to provide a modicum of theoretical plates. Chiral GC analysis confirmed that the ee’s of the collected fractions were unchanged within the limits of measurement from the starting material. One may view disparagingly the folly of attempting to observe the SDE for α-pinene (3) under such conditions but given not only the reported occurrences of SDEvD for (1) and (2)—albeit compounds structurally very different in comparison to α-pinene (3) and much more likely to be disposed towards SDEvD—in addition to the fact that SDEvGC has been observed for α-pinene (3), it is not beyond the realm of possibility. In any event, it can be construed that the lack of observation of SDEvD for α-pinene (3) suggests that any molecular association in the bulk phase is unable to result in SDEvD and, thus, in line with the IR and NMR of neat samples (vide supra), also suggests ruling out molecular association in the bulk phase as the basis for the observed SDEvGC. An obvious method to attempt next would be to use a distillation technique with a much greater number of available theoretical plates, e.g., spinning-band distillation. Unfortunately, a spinning-band distillation apparatus was unavailable for us during this work. However, the use of a spinning-band distillation apparatus to observe SDEvD may also be in vain, given consideration of the results above for IR and NMR and the results presented below.
2.4. SDE via Evaporation (SDEvE)
Our next attempt at observing the SDE for α-pinene (3) was to simply allow a scalemic sample of (S)-α-pinene (3) (53% ee) to evaporate (SDEvE) at ambient temperature and pressure from an open vial. Again there was no observable indication of the SDE occurring. Reasoning that it is unlikely that molecular association in the gas phase is responsible for the SDEvGC and, therefore, the SDEvGC must be a result of molecular association in the condensed phase in the GC, i.e., the stationary phase, we sought to mimic the GC conditions by providing a similar condensed state for the evaporation, viz. allowing the α-pinene (3) to evaporate off reverse-phase silica gel since it is akin to the 95% methylpolysiloxane stationary phase used in the SDEvGC report.
Thus, by depositing a small amount of a scalemic sample of (S)-α-pinene (3) (47.8% ee) onto reverse-phase silica gel in an open vial and then covering it with more reverse-phase silica gel and allowing for near complete evaporation of the α-pinene (3), slight enantioenrichment of the retained α-pinene (3) to 50.3% ee was observed, an SDE magnitude (Δee) [3] of −2.7% (the negative sign indicates that the racemic portion is the more mobile [30,31,32,33,34]). Although the Δee is very small, this is for a “one pot” system with only a limited number of theoretical plates, and thus, for capillary GC with tens of thousands of theoretical plates, the resultant Δee is, conceivably, likely to be greatly enhanced. The more retained portion (the enantiomeric excess portion, hence leading to enantioenrichment) indicates that for evaporation off reverse-phase silica gel, the homochiral association is the more stable adsorbed state. Interestingly, the retention of the enantiomeric excess portion (i.e., the mobility order) is opposite to what is observed for SDEvGC [20], where the excess enantiomer portion elutes first. It is not out of the ordinary that different conditions can result in inversion of the association preference; for example, even due to slight changes in the conditions, a reversal of elution order is not an uncommon observation in SDE via (liquid) chromatography (SDEvC) [30,31,32,33,34].
Since the SDEvE was observed for reverse-phase silica gel, we also tested normal-phase silica gel for which enantiodepletion of the remaining α-pinene (3) was observed (Δee, 2.9%). Thus, an opposite mobility order was observed for normal-phase silica gel in comparison to reverse-phase silica gel. As already stated, a reversal of elution order in liquid chromatography due to a change of stationary phase is not unusual [30,31,32,33,34]. Molecular sieves were also tested for which slight enantioenrichment of the remaining α-pinene (3) was observed (Δee, −1.4%), though in this case, the α-pinene (3) was accompanied by substantial amounts of isomerized products.
Since the evaporation of α-pinene (3) from the liquid state failed to produce an SDE effect while the evaporation of α-pinene (3) off silica gel did, it is suggestive that molecular association in the bulk phase is insufficient to provide differential formation of homo- and heterochiral associates, whereas the evaporation of α-pinene (3) from silica gel which leads to an SDE effect might be due to the differential formation of homo- and heterochiral associates as two dimensional (2D) adsorbed monolayers [35,36,37,38] (Figure 2). It is worth pointing out that the preferential 2D chiral-based association packing of chiral compounds adsorbed on achiral surfaces is a well-known phenomenon [35,36,37,38] in respect of the SDEvE results and, indeed, can account for them. Notably, an observable SDE arising from the preferential 2D chiral-based association packing of chiral compounds adsorbed on achiral surfaces has never been demonstrated. It is also worth noting that Olbrycht has argued strongly that the SDE in liquid chromatography is determined almost exclusively by the formation of homo- and heterochiral associates on already adsorbed molecules rather than the formation of homo- and heterochiral associates in the bulk liquid phase [7].
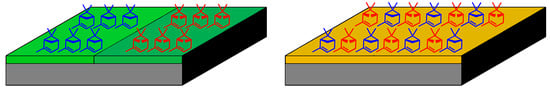
Figure 2.
Homochiral (left) and heterochiral (right) enantiodiscriminatory-based 2D-adsorbed monolayers [35,36,37,38] as a possible explanation for the SDEvE, SDE via adsorptive bubble separation (SDEvABS), and SDEvGC of scalemic α-pinene (3).
2.5. SDE via Adsorptive Bubble Separation (SDEvABS)
We next turned our attention to foam fractionation. Adsorptive bubble separation (ABS) methods, or specifically at the molecular level, foam fractionation, are well-known and widely used in many industries for many different types of separations [39,40,41,42,43,44,45]. We reasoned that 2D-adsorbed monolayers of α-pinene (3) could form on the surfaces of the foam bubbles, and thus, SDE could occur as a result of enantiodiscriminatory-based evaporation. Indeed, we were able to observe an SDE starting from a scalemic sample of (S)-α-pinene (3) (65% ee), albeit the Δee was again quite small and comparable to what was observed by SDEvE. Additionally, though it was difficult, yet still evident, to discern the enantioenrichment of the collected evaporated fractions, the enantiodepletion (Δee, 2.2%) of the α-pinene (3) remaining in the flask was apparent. The enantiodepletion of the flask contents—and thus, the mobility order is the same as the elution order observed in SDEvGC—indicates that heterochiral association is the more stable adsorbed species under these conditions. Though the SDEvABS provided an opposite mobility order to SDEvE using reverse-phase silica gel, it is likely to be due simply to a difference in the molecular association energetic preference between the two processes stemming from slight changes in the conditions as noted above rather than due to a fundamentally different means of molecular association giving rise to the SDE.
Since there is a relatively large glass surface area in the system, an attempt was also made to affect the SDEvE using the same apparatus without foam and allowing the flow of air to transport the α-pinene (3) through the column. However, SDE was not observed across the collected fractions, and clearly, the foam is pivotal for providing a large surface for monolayer formation.
2.6. Context of the SDE
It is important to note that the SDE always occurs to some degree [2,3,4,46,47] with the application of any physicochemical process to a scalemate, and thus, if the sample is fractionated, it is simply a question of whether the SDE magnitude is sufficient to affect the results, either by unwittingly perturbing them deleteriously or intentionally affecting a beneficial outcome. The evaporation results, therefore, despite the low Δee’s observed, represent a concern since processes allowing for evaporation may lead to workers unwittingly altering the ee of scalemic volatile liquid compounds, e.g., as may be encountered in natural product isolation or asymmetric synthesis. These results thus reinforce the cautionary forewarning of the dangers of unwittingly altering the ee of a sample by the application of a physicochemical process [2,3,4,46,47] as even seemingly benign practices such as leaving samples open on the bench [12], storing [5,12,15,48], and rotavaping [6,49] have led to the alteration of sample ee’s. Thus there is always a need to test for the SDE [2,3,4,46,47]. However, from these results, potentially useful applications may arise in terms of unconventional enantiopurification methods [2,3,4], for example, for ABS processes [44,45,50]. Thus we hope these results will come to the attention of practitioners as greater work is necessary to mechanistically rationalize the results and their impact in the fields of SDE, enantiopurification, 2D chiral-based association packing, etc.
3. Conclusions
Despite not being able to replicate the SDEvGC results of Zenkevich and Pavlovskii [20], we were nevertheless able to affect the SDE of α-pinene (3), albeit to only a minor degree, using a similar process to the SDEvGC conditions—evaporation off reverse-phase silica gel—thereby validating the plausibility of SDEvGC for α-pinene (3). Furthermore, we were also able to affect the SDE of α-pinene (3) using a different process, foam fractionation, but one which also involves a gaseous phase and for which the SDE is likely to be due to molecular association similar to that occurring in SDEvE. The mobility order is dependent on the applied conditions, with various mobility orders observed here, and is likely to be due to the molecular association’s energetic preferences rather than any fundamentally different means of molecular association. The indications from this work are that the molecular associations responsible for the various SDE observations of α-pinene occur not in the gaseous phase or the bulk phase but rather in 2D-adsorbed monolayers and are not based on conventional functional group-based intermolecular interactions and instead are, most likely, as a consequence of homo- and heterochiral packing differences in the 2D monolayers.
It is worth pointing out that while the preferential 2D-chiral-based association packing of chiral compounds adsorbed on achiral surfaces is a well-known phenomenon [35,36,37,38] and can indeed account for them, it has never been demonstrated that the phenomenon is able to generate an observable SDE. Of note, these are the first reports of the occurrence of the SDE using an adsorptive bubble separation process (foam fractionation) and another process involving a gaseous phase (SDEvE) other than sublimation, GC, or distillation. Moreover, in addition to reference [20] and these SDEs, this is the first time that the SIDA phenomenon has been observed for a hydrocarbon, and the occurrence of the SDEs for the hydrocarbon α-pinene (3) is thus further proof that the SDE always occurs.
4. Experimental
4.1. Materials
Both enantiomers of α-pinene (3) were used as received: (R)-α-pinene (3) was purchased from Sigma–Aldrich (97% and 80% ee) while (S)-α-pinene (3) was purchased from Acros Organics (98% and 90% ee). Racemic and scalemic samples of α-pinene (3) were prepared by mixing appropriate amounts of the two enantiomers. Reverse-phase silica gels (LiChrosorb RP-8 and RP-2, both 10 μm particle size) were purchased from Merck, normal-phase silica gel (Kieselgel 60, 0.2–0.5 mm) from Carl Roth, molecular sieves (6 mm diameter) from Sigma–Aldrich, and sodium dodecyl sulfate (SDS) from Fluka (99%).
4.2. GC
GC analysis was conducted using an Agilent Technologies 7890A GC system coupled to an Agilent Technologies MSD 5975C for acquiring EI mass spectra in positive mode (70 eV ionization energy). For achiral analysis, the GC was equipped with an Agilent HP-5MS column (30 m length × 0.25 mm id) with the stationary phase (0.25 μm film thickness) consisting of 5% phenylpolysiloxane–95% methylpolysiloxane. GC conditions: temperature program: 100 °C isothermal; flow rate, 1 mL/min; gas velocity, 36 cm/sec; injection volume, 1 μL; split ratio, 50:1; sample concentration, 1 μL/mL. For chiral analysis, the GC was equipped with an Agilent HP-CHIRAL-20β column (30 m length × 0.25 mm id) with stationary phase (0.25 μm film thickness) consisting of 35% phenylpolysiloxane–65% methylpolysiloxane containing 20% permethylated β-cyclodextrin. GC conditions: temperature program: 50 °C (1 min)–2 °C/min ramp–70 °C (4 min)–12 °C/min ramp–190 °C (10 min); flow rate, 0.5 mL/min; gas velocity, 26 cm/s; injection volume, 1 µL; split ratio, 100:1; sample concentration, 1 μL/mL.
4.3. NMR
NMR spectra were acquired at room temperature (either 25 or 23 °C) and −41 °C using Bruker Avance (11.75 T) and Avance III (9.4 T) NMR spectrometers operating at 600 and 400 MHz, respectively, and 150 and 100 MHz, respectively, for 1H nuclei and 13C nuclei, respectively. Samples of α-pinene (3) were acquired in CDCl3 (at concentrations of 10 mg/mL, 250 mg/mL, and 68% v/v), as neat liquids, and in the gas phase. Neat liquid samples were acquired using an insert containing the α-pinene (3) samples with CDCl3 in the outer region for lock and shimming, while for the gas-phase measurements, spectra were acquired unlocked, and shimming was first performed on a CDCl3 sample prior to replacing it with the gas-phase sample followed by additional shimming on the FID. The gas-phase samples were prepared by placing a small drop of the appropriate α-pinene (3) sample in the bottom of an NMR tube and sealing the tube tightly, allowing equilibration to occur over several hours prior to measurement. Tetramethyl silane contained in the CDCl3 was used for chemical shift referencing to evaluate any significant chemical shift changes between enantiopure, scalemic, and racemic α-pinene (3) samples, while gas phase samples also included a small amount of CHCl3 for chemical shift referencing. The 1H NMR spectra were processed without any applied line broadening or with a double exponential, while 13C NMR spectra were processed without any applied line broadening, the application of 1 Hz line broadening (single exponential decay), or with a double exponential as appropriate to closely observe resonating signals.
4.4. IR
IR spectra were recorded using a Bruker LUMOS instrument equipped with a germanium crystal for attenuated total reflectance (ATR) measurements. The minimum amount of each of (R)-, (S)-, and (rac)-α-pinene (3) sample was transferred to the open sampling surface of the instrument using a spatula, and the spectra rapidly acquired before the liquid evaporated.
4.5. Evaporation off Adsorbents
In a typical small-scale experiment, a scalemic sample of (S)-α-pinene (3) (~50% ee, 30 mg) was applied to a small amount of adsorbent in a vial and the wetted adsorbent then covered with more adsorbent (total amount of adsorbent: 240–280 mg for silica gels, 825 mg for molecular sieves). Samples were left open to the atmosphere for 1–5 days to allow the α-pinene (3) to evaporate, with the progress of evaporation monitored by weighing. After the majority of the α-pinene (3) had evaporated (85–95%), the adsorbent was transferred to a gravity filtration apparatus, and the remaining α-pinene (3) recovered by washing the adsorbent with CH2Cl2 (4 mL). The CH2Cl2 of the filtrate was then allowed to evaporate away at 2 °C, leaving the α-pinene (3) behind for analysis by chiral GC. Larger-scale experiments were also performed using 100–500 mg of α-pinene (3) with low ratios of silica gel to α-pinene (3), but evaporation took considerably longer, and only after considerable time was the bulk α-pinene (3) phase reduced to only monolayers.
4.6. Foam Fractionation
The apparatus used for the foam fractionation (Figure 3) and its operation were based on the design and methodology of Armstrong [44]. The quintessential part consists of a column (45 cm total length of bead bed and 2 cm id) packed with glass beads (6 mm diameter) which help to support the foam as well as break it up [44] and, importantly, form dry foam in the upper portions of the column [44,50]. Prior to the start of the process, a scalemic sample of (S)-α-pinene (3) (5 mL, 65% ee) was introduced into the pot holding 1.15 L of deionized water containing SDS (1.5 mM) as the foaming agent. Air was used to both generate and transport foam up the column by passing it into the aqueous solution through a sintered glass frit as a sparger, with the flow being regulated to produce a slow rise of the foam up the column. A steady state of reflux was attained by the spontaneous disintegration of the foam within the column and, additionally, for any column spillover, within a large bulb attached to the top of the column. After some time, α-pinene (3) was trapped by directing the gaseous outflow from the column into a small conical flask encased in a foam box packed with dry ice. From the collected fractions, small amounts of α-pinene (3) were taken for chiral GC analysis from the water–α-pinene (3) condensate using a Pasteur pipette or, if need be, assisted by the addition of a small amount of n-pentane.

Figure 3.
The apparatus (picture and schematic) used for the foam fractionation and its operation was based on the design and methodology of Armstrong [44].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/separations10070382/s1, Low-temperature 13C NMR spectra exhibiting the SIDA phenomenon.
Author Contributions
Conceptualization, A.W., V.A.S. and K.D.K.; methodology, K.D.K.; formal analysis, A.W., V.A.S. and K.D.K.; investigation, K.D.K.; resources, A.W. and K.D.K.; data curation, A.W., V.A.S. and K.D.K.; writing—original draft preparation, K.D.K.; writing—review and editing, A.W., V.A.S. and K.D.K.; supervision, K.D.K.; project administration, K.D.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available upon reasonable request from the corresponding author.
Acknowledgments
Visiting scholar Michael Russelle Alvarez is thanked for acquiring the NMR spectra, and Petra Krämer from the Organic Chemistry Institute, University of Heidelberg, is thanked for acquiring the IR spectra.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Soloshonok, V.A. Remarkable amplification of the self-disproportionation of enantiomers on achiral-phase chromatography columns. Angew. Chem. Int. Ed. 2006, 45, 766–769. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Roussel, C.; Kitagawa, O.; Sorochinsky, A.E. Self-disproportionation of enantiomers via achiral chromatography: A warning and an extra dimension in optical purifications. Chem. Soc. Rev. 2012, 41, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The self-disproportionation of enantiomers (SDE): A menace or an opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Wzorek, A.; Kołbus, A.; Urbaniak, M.; Han, J.; Soloshonok, V.A.; Klika, K.D. Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR. Symmetry 2021, 13, 543. [Google Scholar] [CrossRef]
- Abás, S.; Arróniz, C.; Molins, E.; Escolano, C. Access to the enantiopure pyrrolobenzodiazepine (PBD) dilactam nucleus via self-disproportionation of enantiomers. Tetrahedron 2018, 74, 867–871. [Google Scholar] [CrossRef]
- Olbrycht, M.; Gumieniak, J.; Mruc, P.; Balawejder, M.; Piątkowski, W.; Antos, D. Separation of non-racemic mixtures of enantiomers by achiral chromatography. J. Chromatogr. A 2023, 1693, 463977. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J.L.; Soloshonok, V.A. Optical Purifications via Self-Disproportionation of Enantiomers by Achiral Chromatography: Case Study of a Series of α-CF3-containing Secondary Alcohols. Chirality 2013, 25, 365–368. [Google Scholar] [CrossRef]
- Carman, R.M.; Klika, K.D. The Optical Fractionation of a Partially Racemic Natural Product by Chromatography over an Achiral Substrate. Aust. J. Chem. 1991, 44, 895–896. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Soloshonok, V.A. Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Berbasov, D.O. Self-Disproportionation of Enantiomers on Achiral Phase Chromatography. One More Example of Fluorine’s Magic Powers. Chim. Oggi-Chem. Today 2006, 24, 44–47. [Google Scholar]
- Soloshonok, V.A.; Ueki, H.; Yasumoto, M.; Mekala, S.; Hirschi, J.S.; Singleton, D.A. Phenomenon of Optical Self-Purification of Chiral Non-Racemic Compounds. J. Am. Chem. Soc. 2007, 129, 12112–12113. [Google Scholar] [CrossRef] [PubMed]
- Ueki, H.; Yasumoto, M.; Soloshonok, V.A. Rational application of self-disproportionation of enantiomers via sublimation—A novel methodological dimension for enantiomeric purifications. Tetrahedron Asymmetry 2010, 21, 1396–1400. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Carman, R.M.; Klika, K.D. Partially Racemic Compounds as Brushtail Possum Urinary Metabolites. Aust. J. Chem. 1992, 45, 651–657. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Klika, K.D. Terminology related to the phenomenon ‘self-disproportionation of enantiomers’ (SDE). Helv. Chem. Acta 2014, 97, 1583–1589. [Google Scholar] [CrossRef]
- Katagiri, T.; Yoda, C.; Furuhashi, K.; Ueki, K.; Kubota, T. Separation of an enantiomorph and its racemate by distillation: Strong chiral recognizing ability of trifluorolactates. Chem. Lett. 1996, 25, 115–116. [Google Scholar] [CrossRef]
- Koppenhoefer, B.; Trettin, U. Is it possible to affect the enantiomeric composition by a simple distillation process? Fresenius Z. Anal. Chem. 1989, 333, 750. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates, and Resolutions; Wiley & Sons, Inc.: New York, NY, USA, 1981. [Google Scholar]
- Zenkevich, I.G.; Pavlovskii, A.A. Effects of the Dynamic Modification of Stationary Phases by Sorbates in Gas Chromatography: The Possibility of Separating Enantiomers in Achiral Systems. Russ. J. Phys. Chem. A 2016, 90, 2110–2118. [Google Scholar] [CrossRef]
- Davankov, V.A. Can Enantiomers Be Separated in Achiral Chromatographic Systems. Russ. J. Phys. Chem. A 2016, 90, 2119–2121. [Google Scholar] [CrossRef]
- Baumann, A.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D.; Miller, A.K. Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon. Symmetry 2020, 12, 1106. [Google Scholar] [CrossRef]
- Szakács, Z.; Sánta, Z.; Lomoschitz, A.; Szántay, C., Jr. Self-induced recognition of enantiomers (SIRE) and its application in chiral NMR analysis. Trends Anal. Chem. 2018, 109, 180–197. [Google Scholar] [CrossRef]
- Nieminen, V.; Murzin, D.Y.; Klika, K.D. NMR and molecular modeling of the dimeric self-association of the enantiomers of 1,1′-bi-2-naphthol and 1-phenyl-2,2,2-trifluoroethanol in the solution state and their relevance to enantiomer self-disproportionation on achiral-phase chromatography (ESDAC). Org. Biomol. Chem. 2009, 7, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Storch, G.; Haas, M.; Trapp, O. Attracting Enantiomers: Chiral Analytes That Are Simultaneously Shift Reagents Allow Rapid Screening of Enantiomeric Ratios by NMR Spectroscopy. Chem. Eur. J. 2017, 23, 5414–5418. [Google Scholar] [CrossRef]
- Guetté, J.P.; Boucherot, D.; Horeau, A. Interactions diastereoisomeres d’enantiomeres en phase liquide—II. Tetrahedron Lett. 1973, 14, 465–468. [Google Scholar] [CrossRef]
- Guetté, J.P.; Horeau, A. Interactions diastereoisomeres d’antipodes en phase liquid. Tetrahedron 1974, 30, 1923–1931. [Google Scholar]
- Katagiri, T.; Takahashi, S.; Tsuboi, A.; Suzaki, M.; Uneyama, K. Discrimination of enantiomeric excess of optically active trifluorolactate by distillation: Evidence for a multi-center hydrogen bonding network in the liquid state. J. Fluor. Chem. 2010, 131, 517–520. [Google Scholar] [CrossRef]
- Katagiri, T.; Uneyama, K. Chiral recognition by multicenter single proton hydrogen bonding of trifluorolactates. Chem. Lett. 2001, 30, 1330–1331. [Google Scholar] [CrossRef]
- Wzorek, A.; Kamizela, A.; Sato, A.; Soloshonok, V.A. Self-Disproportionation of Enantiomers (SDE) via achiral gravity-driven column chromatography of N-fluoroacyl-1-phenylethylamines. J. Fluor. Chem. 2017, 196, 37–43. [Google Scholar] [CrossRef]
- Wzorek, A.; Klika, K.D.; Drabowicz, J.; Sato, A.; Aceña, J.L.; Soloshonok, V.A. The self-disproportionation of the enantiomers (SDE) of methyl n-pentyl sulfoxide via achiral, gravity-driven column chromatography: A case study. Org. Biomol. Chem. 2014, 12, 4738–4746. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Remarkable magnitude of the self-disproportionation of enantiomers (SDE) via achiral chromatography: Application to the practical-scale enantiopurification of β-amino acid esters. Amino Acids 2016, 48, 605–613. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A. Self-disproportionation of Enantiomers (SDE) of Chiral Nonracemic Amides via Achiral Chromatography. Isr. J. Chem. 2016, 56, 977–989. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A. Self-disproportionation of enantiomers via achiral gravity-driven column chromatography: A case study of N-acyl-α-phenylethylamines. J. Chromatgr. A 2016, 1467, 270–278. [Google Scholar] [CrossRef]
- Dutta, A.; Gellman, A.J. Enantiomer surface chemistry: Conglomerate versus racemate formation on surfaces. Chem. Soc. Rev. 2017, 46, 7787–7839. [Google Scholar] [CrossRef]
- Yun, Y.; Gellman, A.J. Competing Forces in Chiral Surface Chemistry: Enantiospecificity versus Enantiomer Aggregation. J. Phys. Chem. C 2016, 120, 27285–27295. [Google Scholar] [CrossRef]
- Yun, Y.; Gellman, A.J. Adsorption-induced auto-amplification of enantiomeric excess on an achiral surface. Nat. Chem. C 2015, 7, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, L.; Amabilino, D.B. Spontaneous resolution, whence and whither: From enantiomorphic solids to chiral liquid crystals, monolayers and macro- and supra-molecular polymers and assemblies. Chem. Soc. Rev. 2007, 36, 941–967. [Google Scholar] [CrossRef] [PubMed]
- Karger, B.L.; DeVivo, D.G. General Survey of Adsorptive Bubble Separation Processes. Sep. Sci. 1968, 3, 393–424. [Google Scholar] [CrossRef]
- Somasundaran, P. Foam Separation Methods. Sep. Purif. Methods 1972, 1, 117–198. [Google Scholar] [CrossRef]
- Somasundaran, P. Separation Using Foaming Techniques. Sep. Sci. 1975, 10, 93–109. [Google Scholar] [CrossRef]
- Pinfold, T.A. Adsorptive Bubble Separation Methods. Sep. Sci. 1970, 5, 379–384. [Google Scholar] [CrossRef]
- Tharapiwattananon, N.; Scamehorn, J.F.; Osuwan, S.; Harwell, J.H.; Haller, K.J. Surfactant Recovery from Water Using Foam Fractionation. Sep. Sci. Technol. 1996, 31, 1233–1258. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Zhou, E.Y.; Chen, S.; Le, K.; Tang, Y. Foam Floatation Enrichment of Enantiomers. Anal. Chem. 1994, 66, 4278–4282. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Schneiderheinze, J.M.; Hwang, Y.-S. Bubble Fractionation of Enantiomers from Solution Using Molecularly Imprinted Polymers as Collectors. Anal. Chem. 1998, 70, 3717–3719. [Google Scholar] [CrossRef]
- Han, J.; Dembinski, R.; Soloshonok, V.A.; Klika, K.D. A Call for a Change in Policy Regarding the Necessity for SDE Tests to Validate the Veracity of the Outcome of Enantioselective Syntheses, the Inherent Chiral State of Natural Products, and Other Cases Involving Enantioenriched Samples. Molecules 2021, 26, 3994. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D. Recommended Tests for the Self-Disproportionation of Enantiomers (SDE) to Ensure Accurate Reporting of the Stereochemical Outcome of Enantioselective Reactions. Molecules 2021, 26, 2757. [Google Scholar] [CrossRef]
- Flynn, A.J.; Ford, A.; Maguire, A.R. Localized Partitioning of Enantiomers in Solid Samples of Sulfoxides: Importance of Sampling Method in Determination of Enantiopurity. J. Org. Chem. 2020, 85, 10216–10221. [Google Scholar] [CrossRef] [PubMed]
- Doucet, H.; Fernandez, E.; Layzell, T.P.; Brown, J.M. The Scope of Catalytic Asymmetric Hydroboration/Oxidation with Rhodium Complexes of 1,1′-(2-Diarylphosphino-1-naphthyl)isoquinolines. Chem. Eur. J. 1999, 5, 1320–1330. [Google Scholar] [CrossRef]
- Dasarathy, D.; Ito, Y. Foam separation of Rhodamine-G and Evans Blue using a simple separatory bottle system. J. Chromatogr. A 2017, 1517, 215–218. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).