
A Previously Unrecognized Granulomatous Variant of Gamma-Delta T-Cell Lymphoma




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
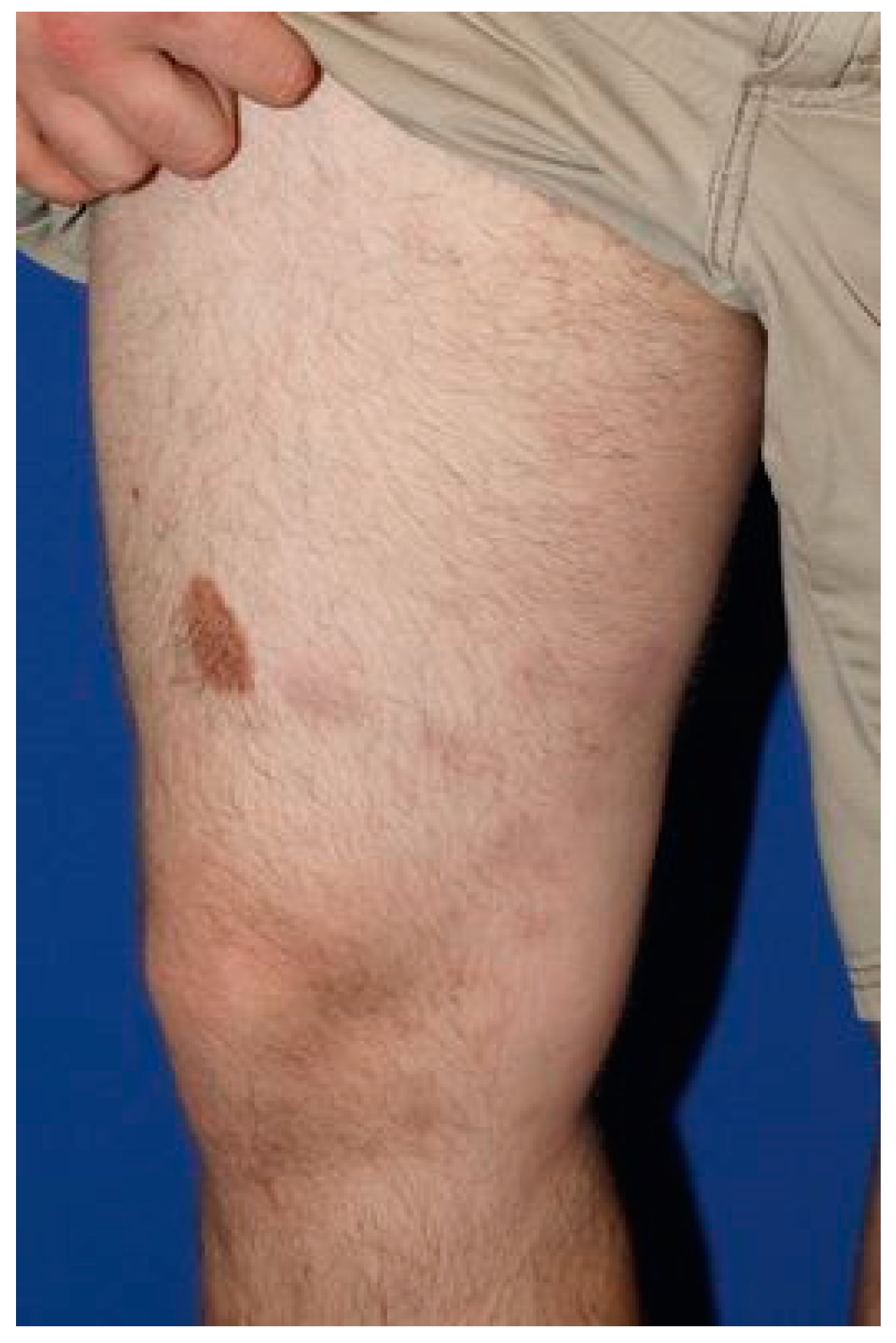
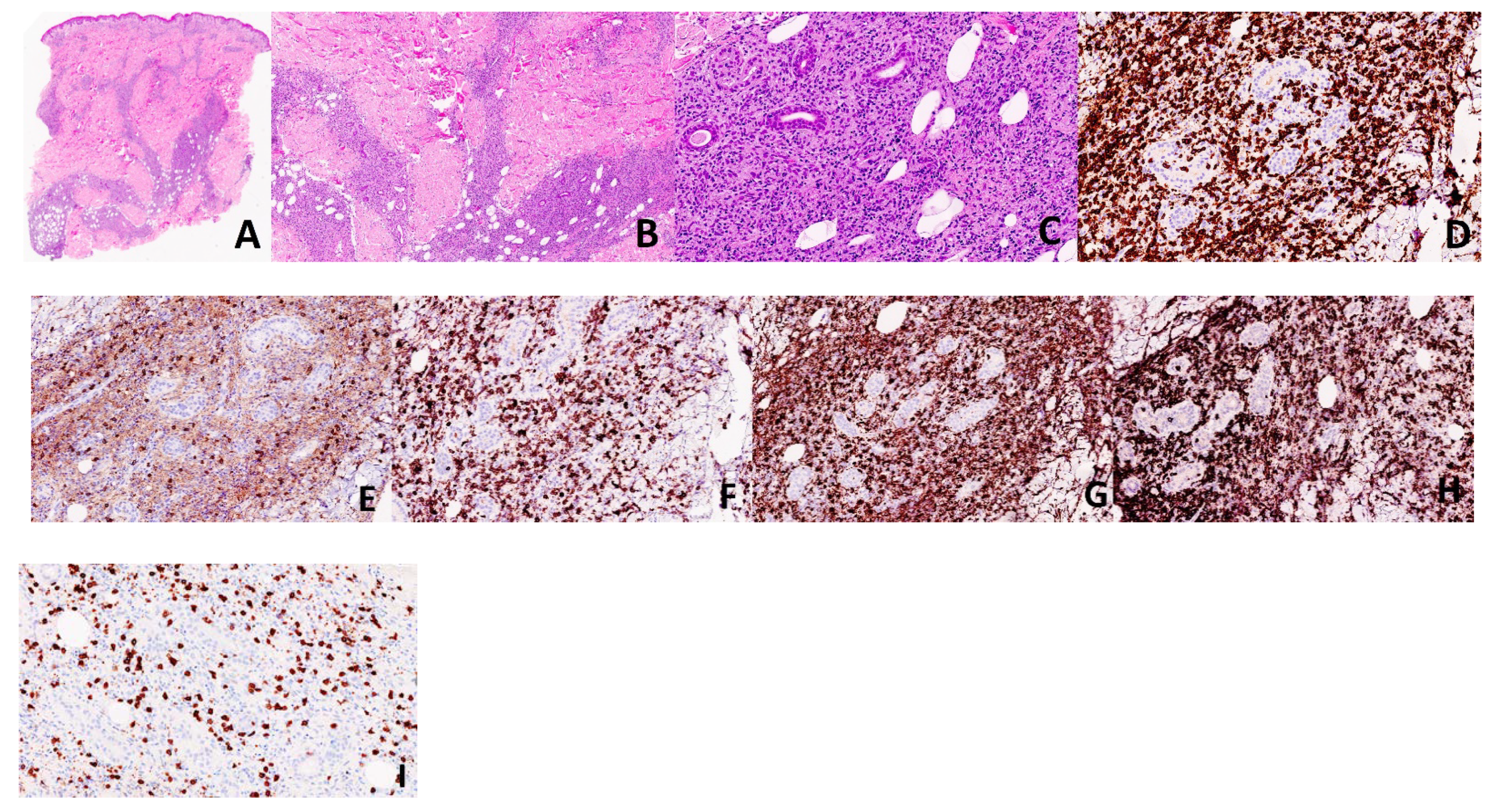
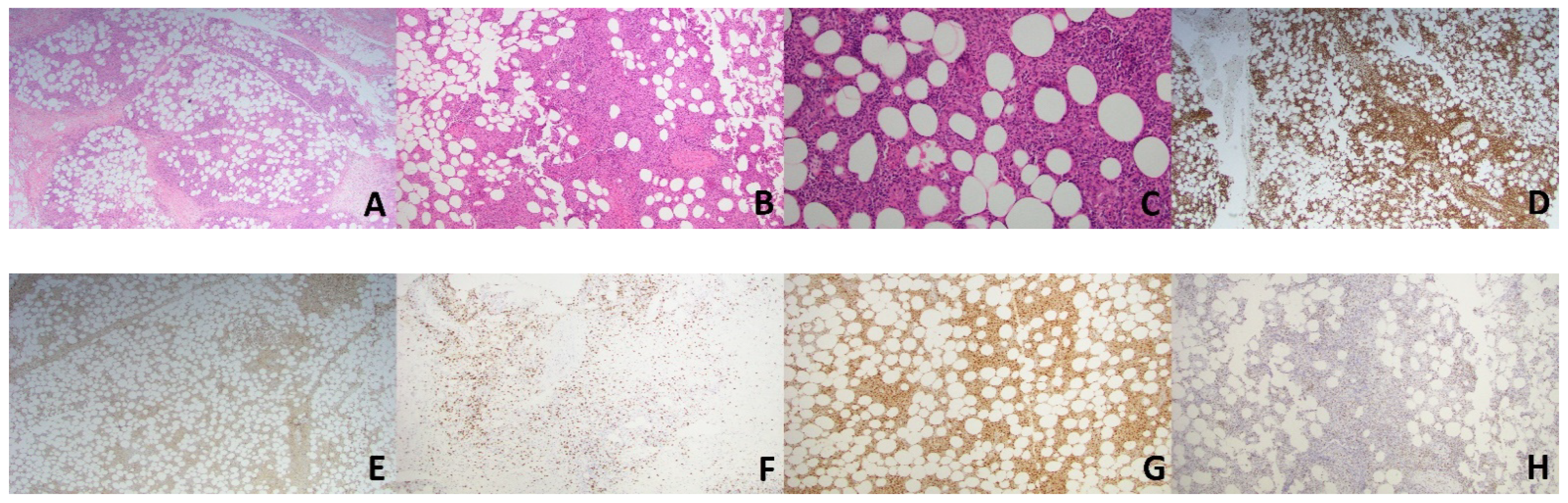
2. Case Presentation
2.1. Case 1
2.2. Case 2
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Koch, R.; Jaffe, E.S.; Mensing, C.; Zeis, M.; Schmitz, N.; Sander, C.A. Cutaneous gamma/delta T-cell lymphoma. J. Dtsch. Dermatol. Ges. 2009, 7, 1065–1067. [Google Scholar] [CrossRef]
- Burg, G.; Kempf, W.; Cozzio, A.; Feit, J.; Willemze, R.; Jaffe, E.S.; Dummer, R.; Berti, E.; Cerroni, L.; Chimenti, S.; et al. WHO/EORTC classification of cutaneous lymphomas 2005: Histological and molecular aspects. J. Cutan. Pathol. 2005, 32, 647–674. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Ohno, T.; Nakamine, H.; Oka, K.; Matsuzuka, F.; Miwa, H.; Shiku, H.; Kimura, N.; Nanba, K.; Kita, K. Gamma delta T-cell lymphoma: A clinicopathologic study of 6 cases including extrahepatosplenic type. Int. J. Hematol. 1999, 69, 186–195. [Google Scholar] [PubMed]
- Gibson, J.F.; Kapur, L.; Sokhn, J.; Xu, M.; Foss, F.M. A fatal case of primary cutaneous gamma–delta T-cell lymphoma complicated by HLH and cardiac amyloidosis. Clin. Case Rep. 2014, 3, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Namiki, T.; Ueno, M.; Iikawa, M.; Tokoro, S.; Nishizawa, A.; Yamamoto, K.; Miura, K.; Yokozeki, H. A Case of Primary Cutaneous Gamma-Delta T-Cell Lymphoma with Pautrier Microabscess. Ann. Dermatol. 2017, 29, 229–232. [Google Scholar] [CrossRef]
- Guizzardi, M.; Hendrickx, I.A.G.; Mancini, L.L.; Monti, M. Cytotoxic gamma/delta subcutaneous panniculitis-like T-cell lymphoma: Report of a case with pulmonary involvement unresponsive to therapy. J. Eur. Acad. Dermatol. Venereol. 2003, 17, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Wang, X. Indolent Primary Cutaneous gamma/delta T-Cell Lymphoma Localized to the Subcutaneous Panniculus and Its Association with Atypical Lymphocytic Lobular Panniculitis. Am. J. Clin. Pathol. 2012, 138, 50–56. [Google Scholar] [CrossRef]
- Endly, D.C.; Weenig, R.H.; Peters, M.S.; Viswanatha, D.S.; Comfere, N.I. Indolent course of cutaneous gamma-delta T-cell lymphoma. J. Cutan. Pathol. 2013, 40, 896–902. [Google Scholar] [CrossRef]
- Merrill, E.; Agbay, R.; Miranda, R.N.; Aung, P.P.; Tetzlaff, M.T.; Young, K.H.; Curry, J.L.; Nagarajan, P.; Ivan, D.; Prieto, V.G.; et al. Primary Cutaneous T-Cell Lymphomas Showing Gamma-Delta (gammadelta) Phenotype and Predominantly Epidermotropic Pattern are Clinicopathologically Distinct from Classic Primary Cutaneous gammadelta T-Cell Lymphomas. Am. J. Surg. Pathol. 2017, 41, 204–215. [Google Scholar] [CrossRef]
- Guitart, J.; Weisenburger, D.D.; Subtil, A.; Kim, E.; Wood, G.; Duvic, M.; Olsen, E.; Junkins-Hopkins, J.; Rosen, S.; Sundram, U.; et al. Cutaneous gammadelta T-cell lymphomas: A spectrum of presentations with overlap with other cytotoxic lymphomas. Am. J. Surg. Pathol. 2012, 36, 1656–1665. [Google Scholar] [CrossRef]
- Khallaayoune, M.; Grange, F.; Condamina, M.; Szablewski, V.; Guillot, B.; Dereure, O. Primary Cutaneous Gamma-Delta T-cell Lymphoma: Not an Aggressive Disease in All Cases. Acta Derm. Venereol. 2020, 100, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Von Dücker, L.; Fleischer, M.; Stutz, N.; Thieme, M.; Witte, M.; Zillikens, D.; Sadik, C.D.; Terheyden, P. Primary Cutaneous Gamma-Delta T-Cell Lymphoma with Long-Term Indolent Clinical Course Initially Mimicking Lupus Erythematosus Profundus. Front. Oncol. 2020, 10, 133. [Google Scholar] [CrossRef]
- Willemze, R.; Jansen, P.M.; Cerroni, L.; Berti, E.; Santucci, M.; Assaf, C.; Canninga-van Dijk, M.R.; Carlotti, A.; Geerts, M.-L.; Hahtola, S.; et al. Subcutaneous panniculitis-like T-cell lymphoma: Definition, classification, and prognostic factors: An EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008, 111, 838–845. [Google Scholar] [CrossRef]
- Jones, D.; Vega, F.; Sarris, A.H.; Medeiros, L.J. CD4− CD8− ‘Double-Negative’ Cutaneous T-Cell Lymphomas Share Common Histologic Features and an Aggressive Clinical Course. Am. J. Surg. Pathol. 2002, 26, 225–231. [Google Scholar] [CrossRef]
- Toro, J.R.; Liewehr, D.J.; Pabby, N.; Sorbara, L.; Raffeld, M.; Steinberg, S.M.; Jaffe, E.S. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood 2003, 101, 3407–3412. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pinilla, S.M.; Ortiz-Romero, P.L.; Monsalvez, V.; Tomás, I.E.; Almagro, M.; Sevilla, A.; Camacho, G.; Longo, M.I.; Pulpillo, A.; Diaz-Perez, J.; et al. TCR-gamma Expression in Primary Cutaneous T-cell Lymphomas. Am. J. Surg. Pathol. 2013, 37, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Sacks, E.L.; Donaldson, S.S.; Gordon, J.; Dorfman, R.F. Epithelioid granulomas associated with Hodgkin’s disease: Clinical correla-tions in 55 previously untreated patients. Cancer 1978, 41, 562–567. [Google Scholar] [CrossRef]
- Kadin, M.E.; Donaldson, S.S.; Dorfman, R.F. Isolated Granulomas in Hodgkin’s Disease. N. Engl. J. Med. 1970, 283, 859–861. [Google Scholar] [CrossRef]
- Scarabello, A.; Leinweber, B.; Ardigò, M.; Rütten, A.; Feller, A.C.; Kerl, H.; Cerroni, L. Cutaneous lymphomas with prominent granulomatous reaction: A potential pitfall in the histopathologic diagnosis of cuta-neous T- and B-cell lymphomas. Am. J. Surg. Pathol. 2002, 26, 1259–1268. [Google Scholar] [CrossRef]
- Prescott, R.; Banerjee, S.; Cross, P. Subcutaneous T-cell lymphoma with florid granulomatous panniculitis. Histopathology 2007, 20, 535–537. [Google Scholar] [CrossRef]
- Lee, C.; Hsi, A.; Lazova, R. Subcutaneous Panniculitis-Like T-Cell Lymphoma with Granulomas as the Predominant Feature. Am. J. Dermatopathol. 2019, 41, 667–670. [Google Scholar] [CrossRef]
- Lewis, D.J.; Falck, B.A.; Kantrow, S.M.; Pozadzides, J.V.; Hinojosa, T.; Huang, S.; Diwan, A.H.; Prieto, V.G.; Duvic, M. Necrotizing Gran-ulomatous Dermatitis and Panniculitis Masquerading as T Cell Lymphoma. Skinmed 2019, 17, 406–408. [Google Scholar]
- Caudron, A.; Bouaziz, J.; Battistella, M.; Sibon, D.; Lok, C.; Leclech, C.; Ortonne, N.; Molinier-Frenkel, V.; Bagot, M. Two Atypical Cases of Cutaneous Gamma/Delta T-Cell Lymphomas. Dermatology 2011, 222, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Shalom, G.; Gurion, R.; Benharroch, D. Primary cutaneous gamma/delta T-cell lymphoma. An atypical case with bone marrow granulomas. J. Dermatol. Case Rep. 2015, 9, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.F.; Sanchez, N.P.; Flynn, T.C.; Sanchez, J.L.; Mihm, M.C., Jr.; Soter, N.A. Erythema nodosum leprosum: Nature and extent of the cutaneous microvascular alterations. J. Am. Acad. Dermatol. 1986, 14, 59–69. [Google Scholar] [CrossRef]
- Tsunematsu, S.; Natsuizaka, M.; Fujita, H.; Otsuka, N.; Terashita, K.; Sato, F.; Kobayashi, T.; Nakai, M.; Tsukuda, Y.; Horimoto, H.; et al. Hepatosplenic Gamma-delta T-cell Lymphoma Associated with Epstein-Barr Virus. Intern. Med. 2014, 53, 2079–2082. [Google Scholar] [CrossRef]
- Cerroni, L. Skin Lymphoma; Wiley: Hoboken, NJ, USA, 2020. [Google Scholar]
- Yu, W.W.; Hsieh, P.P.; Chuang, S.S. Cutaneous EBV-positive gammadelta T-cell lymphoma vs. extranodal NK/T-cell lymphoma: A case report and literature review. J. Cutan. Pathol. 2012, 40, 310–316. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Meyer, C.; Bonneville, M. Gammadelta T Cells: First Line of Defense and Beyond. Annu. Rev. Immunol. 2014, 32, 121–155. [Google Scholar] [CrossRef]
- Tripodo, C.; Iannitto, E.; Florena, A.M.; Pucillo, C.; Piccaluga, P.P.; Franco, V.; Pileri, S.A. Gamma-delta T-cell lymphomas. Nat. Rev. Clin. Oncol. 2009, 6, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.; Doukas, P.G.; Escala, M.E.M.; Ringbloom, K.G.; Shih, D.J.H.; Yang, J.; Tegtmeyer, K.; Park, J.; Thomas, J.J.; Selli, M.E.; et al. Cellular origins and genetic landscape of cutaneous gamma delta T cell lymphomas. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Serre, K.; Norell, H. Gammadelta T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef]
- Girardi, M. Immunosurveillance and Immunoregulation by gammadelta T Cells. J. Investig. Dermatol. 2006, 126, 25–31. [Google Scholar] [CrossRef]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. Gammadelta T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef]
- Hao, J.; Wu, X.; Xia, S.; Li, Z.; Wen, T.; Zhao, N.; Wu, Z.; Wang, P.; Zhao, L.; Yin, Z. Current progress in gammadelta T-cell biology. Cell. Mol. Immunol. 2010, 7, 409–413. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Moser, B. Professional Antigen-Presentation Function by Human gammadelta T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.; Willimann, K.; Bioley, G.; Levy, N.; Eberl, M.; Luo, M.; Tampé, R.; Levy, F.; Romero, P.; Moser, B. Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses. Proc. Natl. Acad. Sci. USA 2009, 106, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.P.; Tabor, L.; Sun, X.; Woolard, M.D.; Simecka, J.W. Depletion of CD8+T Cells Exacerbates CD4+Th Cell-Associated Inflammatory Lesions During Murine Mycoplasma Respiratory Disease. J. Immunol. 2002, 168, 3493–3501. [Google Scholar] [CrossRef] [PubMed]
- Ismaili, J.; Olislagers, V.; Poupot, R.; Fournie, J.J.; Goldman, M. Human gamma delta T Cells Induce Dendritic Cell Maturation. Clin. Immunol. 2002, 103, 296–302. [Google Scholar] [CrossRef]
- Petrasca, A.; Doherty, D.G. Human Vdelta2(+) gammadelta T Cells Differentially Induce Maturation, Cytokine Production, and Alloreactive T Cell Stim-ulation by Dendritic Cells and B Cells. Front. Immunol. 2014, 5, 650. [Google Scholar] [CrossRef]
- Burg, G.; Dummer, R.; Wilhelm, M.; Nestle, F.; Ott, M.M.; Feller, A.; Hefner, H.; Lanz, U.; Schwinn, A.; Wiede, J. A Subcutaneous delta-positive T-Cell Lymphoma That Produces Interferon Gamma. N. Engl. J. Med. 1991, 325, 1078–1081. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pukhalskaya, T.; Smoller, B.R.; Menke, D.M.; Sokumbi, O. A Previously Unrecognized Granulomatous Variant of Gamma-Delta T-Cell Lymphoma. Dermatopathology 2021, 8, 221-228. https://doi.org/10.3390/dermatopathology8020027
Pukhalskaya T, Smoller BR, Menke DM, Sokumbi O. A Previously Unrecognized Granulomatous Variant of Gamma-Delta T-Cell Lymphoma. Dermatopathology. 2021; 8(2):221-228. https://doi.org/10.3390/dermatopathology8020027
Chicago/Turabian StylePukhalskaya, Tatsiana, Bruce R. Smoller, David M. Menke, and Olayemi Sokumbi. 2021. "A Previously Unrecognized Granulomatous Variant of Gamma-Delta T-Cell Lymphoma" Dermatopathology 8, no. 2: 221-228. https://doi.org/10.3390/dermatopathology8020027
APA StylePukhalskaya, T., Smoller, B. R., Menke, D. M., & Sokumbi, O. (2021). A Previously Unrecognized Granulomatous Variant of Gamma-Delta T-Cell Lymphoma. Dermatopathology, 8(2), 221-228. https://doi.org/10.3390/dermatopathology8020027